

Abstract
Objectives: The focus of this study was to determine the dedicator of cytokinesis 2 (DOCK2), extracellular signal‐regulated kinase 1/2 (ERK1/2), c‐Jun N‐terminal kinase‐1 (JNK) and Akt signals involved in CXCL13‐mediated prostate cancer (PCa) cell invasion and proliferation.
Materials and methods: Androgen‐sensitive (LNCaP), hormone‐refractory (PC3) cells and normal cells (RWPE‐1) were used to determine CXCL13‐mediated PCa cell invasion and proliferation. Immuno‐blotting, fast activated cell‐based (FACE) ELISA, caspase activity, cell invasion and proliferation assays were performed to ascertain some of the signalling events involved in PCa cell proliferation and invasion.
Results: Unlike androgen‐sensitive LNCaP cells, we report for the first time that the hormone‐refractory cell line, PC3, expresses DOCK2. CXCL13‐mediated LNCaP and PC3 cell invasion was regulated by Akt and ERK1/2 activation in a DOCK2‐independent fashion. CXCL13 also promoted LNCaP cell proliferation in a JNK‐dependent fashion even in the absence of DOCK2. In contrast, CXCL13 induced PC3 cell proliferation through JNK activation, which required DOCK2.
Conclusions: Our results show CXCL13‐mediated PCa cell invasion requires Akt and ERK1/2 activation and suggests a new role for DOCK2 in proliferation of hormone‐refractory CXCR5‐positive PCa cells.
Introduction
Androgen deprivation is an initial therapeutic intervention for treatment of metastatic prostate cancer (PCa) (1). However, this treatment becomes ineffective when tumours progress to androgen‐independence and acquire advanced metastatic potential (2, 3). The mechanisms underlying hormone‐independent growth of PCa cells involve alterations in the androgen receptor and related co‐regulators of transcription, as well as emergence of growth pathways that replace signals normally regulated by androgens in the prostatic epithelium (4). One potential alternative pathway that hormone‐refractory PCa cells take advantage of to survive and proliferate, is the use of chemokines as growth factors (4).
Chemokines have been shown to play a pivotal role in homing and directional migration of chemokine receptor‐bearing tumour cells to target organs where corresponding ligands are expressed, contributing to the process of metastasis (5, 6). In addition to their role in cell trafficking, chemokines promote tumour cell survival and serve as growth factors in the tumour microenvironment (7, 8). We have recently shown that PCa cell lines differentially express the chemokine CXCR5 and that its expression positively correlates with PCa progression (9). Indeed, PCa cell lines selectively expressed matrix metalloproteinases (MMPs) in a CXCR5‐dependent fashion to presumably support tumour cell invasion processes by degrading extracellular matrix (ECM) components. In a related study, we showed that serum CXCL13 levels positively correlated with prostatic disease and prostate‐specific antigen (PSA), as well as mediate PCa cell migration, integrin clustering and cell adhesion (10). Moreover, CXCR5/CXCL13 receptor/ligand pairs induced release of beta‐N‐acetylhexosaminidase by osteoblasts to support bone remodelling and endochondral ossification (11). These findings provided the rationale for this study, which further dissects the molecular mechanisms of CXCL13:CXCR5 interactions in PCa cell invasion and growth.
Chemokines’ binding to their cognate receptors sustains Akt activation and provides cells with a growth signal that allows them to withstand apoptotic stimuli (12). Stimulation of chemokine receptors also leads to phosphoinositide‐3 kinase (PI3K)‐dependent activation of mitogen‐activated protein (MAP) kinase extracellular signal–regulated kinase (ERK)‐1/2 through induction of Ras signalling, which contributes to PCa cell invasion and proliferation (13, 14). Alternatively, JNK is another mediator of cell survival, proliferation and tumour progression, and functions autonomously from the PI3K pathway (15). CXCL13 is capable of inducing phosphorylation of JNK in B‐lymphocytes (16), which might require DOCK2 as an upstream signalling molecule in certain cell types (17). To explain, DOCK2 is a novel member of the Caenorhabditis elegans Ced‐5, mammalian DOCK180, and Drosophila melanogaster MyoblastCity (CDM) family of scaffold proteins and has been shown to regulate cytoskeletal dynamics by activating Rac isoforms (18, 19) and directing lymphocyte and neutrophil chemotaxis (20, 21, 22). To our knowledge, the role of DOCK2 in PCa cell invasion and growth has not been studied previously. Here, we have examined expression of DOCK2 and its role in CXCR5‐mediated signal transduction for invasion, growth and survival of hormone‐responsive (LNCaP) and ‐refractory (PC3) PCa cell lines.
Materials and methods
Cell lines and culture
Prostate cancer cells (LNCaP and PC3) and normal prostatic epithelial cells (RWPE‐1) were obtained from the American Type Culture Collection (ATCC). RWPE‐1 cells were cultured in keratinocyte serum‐free media supplemented with bovine pituitary extract (0.05 mg/ml) and epidermal growth factor (5 ng/ml). Prostate cancer cells (LNCaP and PC3) were cultured in complete RPMI‐1640 supplemented with 10% foetal bovine serum (FBS) maintained in a cell culture incubator at 37 °C in humidified atmosphere with 5% CO2. Cell lines were serum‐starved for 16 h prior to assays.
Treatment of cells with siRNA against DOCK2
Cell lines (2 × 105 cells per well) were seeded in six‐well tissue culture plates in antibiotic‐free normal growth medium, supplemented with 10% FBS and cultured at 37 °C in humidified atmosphere with 5% CO2 until 70% confluence was achieved. Cells were then transfected with 2 μm (∼1 μg) of DOCK2 siRNA duplex or control siRNA for 8 h following the manufacturer’s protocol (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Transfection medium was replaced with normal growth medium, and cells were cultured for additional 24–72 h. Efficacy and duration of DOCK2 silencing were determined by western blot analysis (Fig. 1b).
Figure 1.
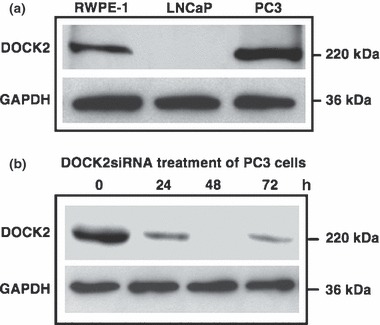
DOCK2 Expression by PCa cells. (a) Total cell lysates (60 μg) from RWPE‐1, LNCaP and PC3 cells were resolved on SDS–PAGE and subjected to immunoblotting using antibodies against DOCK2. GAPDH was used as loading control. (b) DOCK2 siRNA duplex (2 μm) was used for silencing DOCK2 following manufacturer’s protocol (Santa Cruz). Efficacy of DOCK2 silencing after 0, 24, 48 and 72 h was determined by western blot analysis.
Immunoblotting
Immunoblot analysis was conducted on total untreated cell lysates or lysates of RWPE‐1, LNCaP and PC3 cell lines treated with DOCK2 siRNA, control siRNA and/or 100 ng/ml of CXCL13 (PeproTech, Rocky Hill, NJ, USA). Cells were lysed in buffer containing 50 mm Tris–HCl, pH 7.4, 150 mm NaCl, NP‐40 (1%) supplemented with protease and phosphatase inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA). Protein concentration of cell lysates were determined using bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL, USA). Equal amounts (60 μg) of cell lysates were denatured by boiling in Laemmli buffer for 5 min, resolved on 4–15% gradient sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE), and transferred to nitrocellulose membranes using a semi‐dry transfer cell system (Bio‐Rad, Hercules, CA, USA). Membranes were blocked for 1 h at room temperature in 5% non‐fat milk in 1X Tris‐Tween buffered saline (TTBS; 30 mm Tris‐Base, 150 mm NaCl and 0.1% Tween 20), followed by washing with 1X TTBS. Primary antibodies against DOCK2, or phospho‐JNK‐Thr 183/Tyr 185 (Santa Cruz Biotechnology) were added to the membranes and incubated for 16 h at 4 °C in 5% non‐fat milk. Membranes were then washed and corresponding horseradish peroxidase (HRP)‐conjugated secondary antibodies were added for 1 h followed by additional washes. Immunoreactive proteins were visualized on autoradiographic films using chemiluminescent detection reagent (Pierce). Following development, membranes were stripped, blocked in 5% non‐fat milk for 1 h and reprobed with glyceraldehyde 3‐phosphate dehydrogenase (GAPDH; Ambion, Austin, TX, USA) to ensure equal loading.
Cell invasion assay
Cell invasion assay was performed using BD Biocoat™ tumour migration and Matrigel™ tumour invasion chambers (BD Biosciences, San Jose, CA, USA). Briefly, Matrigel inserts were hydrated for 2 h with 500 μl of DMEM at 37 °C with 5% CO2. CXCL13 (100 ng/ml) was added to the lower chambers containing serum‐free RPMI medium. LNCaP and PC3 cells were pre‐treated with isotype control antibody or anti‐human CXCR5 antibody (1 μg/ml; R&D Systems, Minneapolis, MN, USA), DOCK2 siRNA (2 μm), or control siRNA (2 μm) prior to harvest, and added to upper chambers in serum‐free RPMI medium at 10 000 cells per well. Cells were allowed to invade and migrate for 8 h at 37 °C with 5% CO2. Non‐migrating cells on upper surfaces of membranes were removed with cotton swabs. Cells that migrated to lower surfaces of membranes were fixed in methanol at room temperature for 5 min, stained with crystal violet for 2 min and washed in distilled water. Membranes were peeled and placed on glass slides. Cells were then counted using light microscopy at 40× magnification. Percentage cell invasion was calculated as follows: percentage of invasion = mean number of cells invading through MatrigelTM insert membrane, divided by mean number of cells migrating through control insert membrane multiplied by one‐hundred. Experiments were performed in triplicate and repeated three times.
Fast activated cell‐based (FACE) ELISA for Akt and ERK1/2
PCa cell lines were cultured and seeded in 96‐well plates at 5000 cells per well in complete RPMI supplemented with 10% FBS. Cells were serum‐starved overnight and pre‐treated with isotype control antibody or anti‐human CXCR5 antibody (1 μg/ml), DOCK2 siRNA or control siRNA (2 μm). Following treatment, cells were stimulated with (or without) CXCL13 (100 ng/ml) for 5 or 10 min. Next, fast‐activated cell‐based ELISA (FACE™; Active Motif, Carlsbad, CA, USA) assays were performed to measure modifications in levels of active and total Akt or ERK1/2 expression, by LNCaP and PC3 cell lines. Briefly, treated cells were fixed in 4% paraformaldehyde in phosphate‐buffered saline (137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4 and 1.4 mm KH2PO4, pH 7.4) for 20 min. Antibody blocking buffer was added for 1 h, followed by anti‐phospho‐Ser473‐specific Akt or anti‐phospho‐ERK1/2 primary antibody. The same procedure was repeated using antibodies against total Akt and ERK1/2. Cells were then washed, and HRP‐conjugated secondary antibody was added for 1 h. Chemiluminescence was read using SpectraMax® luminometer (Molecular Devices, Downingtown, PA, USA). After chemiluminescence readings were recorded, cells were stained with crystal violet for 30 min and absorbance was read on a microplate spectrophotometer at 595 nm. Measured OD595 reading indicates the relative number of cells in each well. Experiments were performed in triplicate and results show the ratio of active (phosphorylated) to total Akt or ERK1/2 normalized to cell density per well.
MTT cell proliferation assay
To assess the role of CXCL13:CXCR5‐mediated DOCK2 and JNK activation on PCa cell population growth and viability, RWPE‐1, LNCaP and PC3 cells were treated with isotype control antibody or anti‐human CXCR5 antibody (1 μg/ml), JNK inhibitor (AS601245; 10 μm; Sigma‐Aldrich, St Louis, MO, USA), DOCK2 siRNA or control siRNA, and proliferation was measured using the MTT [3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide] assay. Briefly, treated cells were seeded in 96‐well plates at density of 5000 cells per well per 100 μl media and allowed to adhere for 6 hours, followed by addition of CXCL13 (100 ng/ml) to corresponding wells. Plates in triplicates were then incubated for 24, 48 and 72 h. Each day, 3 h prior to end of incubation time, the plates were centrifuged at 200 g for 5 min to pellet potential floating cells, then media were aspirated out of wells and 100 μl of fresh media containing 10% volume/volume MTT was added to each well. Plates were returned to the incubator for additional 3 h. At the end of incubation time, media were aspirated out of wells, and 200 μl DMSO was added to each well to solubilize the formazan crystals. Plates were then read using a microplate spectrophotometer at 540 nm. Absorbance of formazan dye solution was in direct proportion to number of proliferating cells per well.
Caspase 3/7 activity assay
Effector caspase‐3 and ‐7 activities were measured using Caspase‐Glo 3/7 Assay (Promega, Madison, WI, USA) according to manufacturer’s protocol. Briefly, isotype control antibody or anti‐human CXCR5 antibody (1 μg/ml)‐, JNK inhibitor (1 μm)‐, DOCK2 siRNA (2 μm)‐, control siRNA (2 μm), or wortmannin (1 μm)‐treated RWPE‐1, LNCaP and PC3 cell lines were seeded in 96‐well plates as in the MTT cell viability assay. Plates were assayed at 24, 48 and 72 h by adding 100 μl Caspase‐Glo 3/7 reagent containing caspase 3/7 buffer and proluminescent caspase 3/7 substrate to each sample. After 1 h incubation at room temperature, luminescence of each sample was measured using a microplate‐reading luminometer. All samples were assayed in triplicate.
Statistical analysis
Migration and invasion, FACE™, MTT and caspase 3/7 assays were performed in triplicate and at least three separate studies with similar results were performed. Error bars represent ± standard error of means. Statistical analysis was performed using paired or unpaired t‐tests as appropriate. P‐value < 0.05 was considered statistically significant.
Results
Differential expression of DOCK2 by RWPE‐1, LNCaP and PC3 cells and its role in cell invasion
Western blot analyses of total prostate cell lysates showed that DOCK2 was expressed by RWPE‐1 and PC3, but not by LNCaP cells (Fig. 1a). We next examined functional relevance of DOCK2 expression by assessing its role in PCa cell invasion. Our results showed that treatment of PC3 cells with DOCK2 siRNA optimally inhibited its expression after 48 h with only moderate inhibition at other time points (Fig. 1b). Hence, the former schedule was used in subsequent experiments requiring siRNA‐mediated knockdown of DOCK2. This inhibition did not impair ability of PC3 cells to invade towards CXCL13 compared to the significant decrease in invasion of PCa cells treated with anti‐CXCR5 antibody (Fig. 2). As expected, DOCK2 inhibition had no effect on invasion potential of LNCaP cells, as these cells lack DOCK2. Therefore, we concluded that CXCL13‐mediated PCa cell invasion was not regulated by DOCK2.
Figure 2.
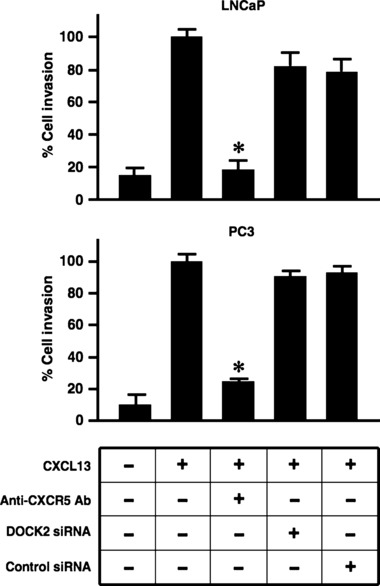
CXCL13:CXCR5 interaction promotes LNCaP and PC3 cell invasion independent of DOCK2. LNCaP and PC3 cells were tested for their ability to invade MatrigelTM matrix and migrate through an 8.0 μm porous membrane in the presence of CXCL13 (100 ng/ml), isotype control antibody or anti‐human CXCR5 antibody (1 μg/ml), DOCK2 or control siRNA (2 μm). Cells which invaded to the lower surfaces of the membrane were stained with crystal violet and counted using light microscopy at 40× magnification. Percentage cell invasion was calculated following manufacturer’s instructions (BD Biosciences). Error bars represent standard error of means of three independent experiments. *Significant differences (P < 0.05) relative to CXCL13‐treated cells (control).
CXCL13‐mediated signalling events that regulate Akt and ERK1/2 activation in PCa cells
Fast‐activated cell based ELISA (FACE) assay was performed to quantify Akt and ERK1/2 activation in PCa cells following CXCL13 stimulation. LNCaP and PC3 cell lines exhibited an increase in phosphorylated to total Akt and ERK1/2 ratios in response to CXCL13 stimulation (Fig. 3). Treatment with anti‐human CXCR5 antibody significantly abrogated CXCL13‐mediated Akt and ERK1/2 activation in both cell lines. As expected, DOCK2 siRNA had no effect on Akt and ERK1/2 activation in LNCaP cells, as these cells lack DOCK2 (Fig. 1a). However, DOCK2 siRNA pre‐treated PC3 cells showed comparable levels of phosphorylated to total Akt and ERK1/2 ratios as cells treated with CXCL13 alone, suggesting that CXCL13‐mediated Akt and ERK1/2 activation is DOCK2‐independent.
Figure 3.
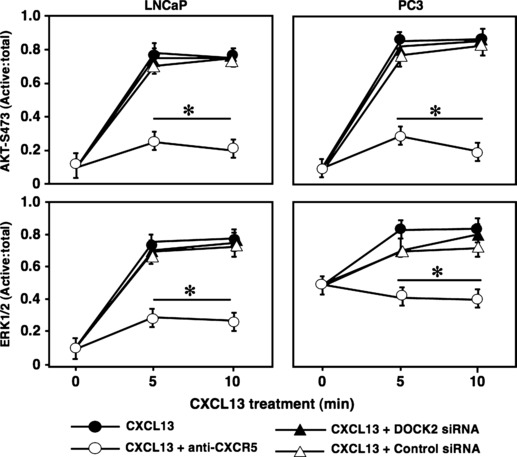
CXCL13 regulates activation of Akt and ERK1/2. FACE assays were performed to measure active and total Akt or ERK1/2 in LNCaP and PC3 cell lines. Cells were treated with isotype control antibody or anti‐human CXCR5 antibody (1 μg/ml), DOCK2 siRNA or control siRNA (2 μm), or JNK inhibitor (10 μm) in the presence of CXCL13 (100 ng/ml) for 0, 5 or 10 min. Experiments were performed in triplicate and results show ratios of active (phosphorylated) to total Akt or ERK1/2. Error bars represent ± standard error of means of three independent experiments. *Significant (P < 0.05) decrease in phosphorylation.
DOCK2 regulates CXCL13‐mediated JNK activation
To understand the role of DOCK2 in CXCL13‐mediated PC3 cell growth, we examined its effect on the JNK signal transduction pathway. While RWPE‐1 cells expressed DOCK2, CXCR5 expression is low in these cell lines; thus, CXCL13 did not modulate JNK phosphorylation with or without DOCK2 siRNA knockdown (Fig. 4). In contrast, LNCaP and PC3 cells exhibited significant increases in JNK phosphorylation levels in the presence of CXCL13, compared to baseline/untreated cells. This activation was reduced by DOCK2 knockdown in PC3 cells, but not in LNCaP cells. Together, our results show that JNK activation following CXCL13:CXCR5 interactions requires DOCK2 expression in PC3 cells, but can be achieved using an alternative signalling cascade in LNCaP cells, which lack DOCK2.
Figure 4.
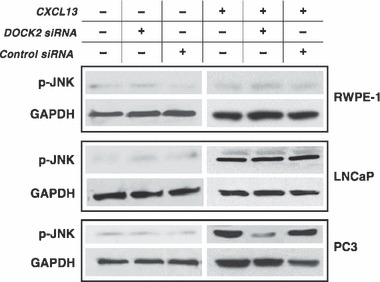
CXCL13 induces JNK activation through DOCK2 in PC3 cells. RWPE‐1, LNCaP and PC3 cells were treated with DOCK2 siRNA and corresponding controls (2 μm) in the presence or absence of CXCL13 (100 ng/ml). Lysates were collected 5 min following CXCL13 stimulation and samples were resolved on SDS–PAGE. Membranes were blotted for phospho‐JNK (46 kDa). GAPDH serves as loading control.
CXCL13 contributes to PCa cell proliferation through regulation of JNK
To determine the biological significance of DOCK2‐ and CXCL13‐mediated JNK activation, MTT cell proliferation assays were performed with or without DOCK2 silencing or JNK inhibitor. CXCL13 stimulated LNCaP and PC3 cell proliferation. In contrast, CXCL13 treatment had no effect on RWPE‐1 proliferation (Fig. 5). CXCL13‐mediated PCa cell proliferation was inhibited by anti‐CXCR5 antibody, which further confirmed the role of the CXCL13:CXCR5 axis in this process. A similar decrease in proliferation was noted with LNCaP and PC3 cells treated with JNK inhibitor. However, CXCL13‐dependent proliferation of PC3 cells was uniquely susceptible to DOCK2 siRNA. To confirm that the decreases in cell proliferation following CXCR5 blockade and DOCK2 or JNK inhibition were not a result of cell death by apoptosis, we measured caspases 3/7 activities during the same treatment conditions. Wortmannin caused PCa cell line apoptosis, as indicated by the increase in caspase3/7 activity in all three cell lines regardless of CXCL13 stimulation (Fig. 6). In contrast, lower cell proliferation observed with JNK inhibition of LNCaP cells and DOCK2 siRNA or JNK inhibition of PC3 cells was not the result of cell death, as caspase 3/7 activity remained low under these conditions after 24, 48, or 72 h culture. These data suggest that CXCL13‐mediated LNCaP and PC3 cell proliferation is CXCR5‐ and JNK‐dependent. Our findings also support the notion that CXCL13‐induced proliferation of the hormone refractory PCa cell line, PC3, requires DOCK2.
Figure 5.
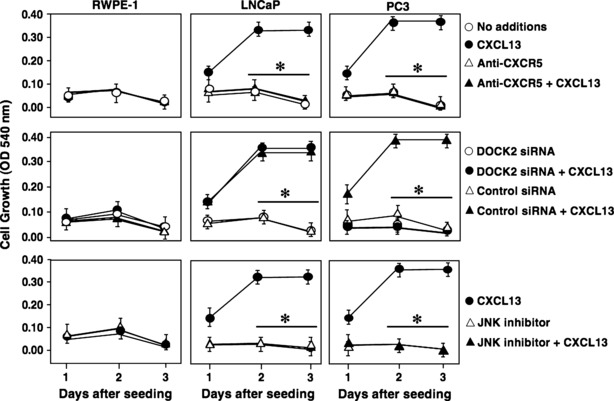
CXCL13 regulates PCa cell proliferation through JNK and DOCK2. RWPE‐1, LNCaP, and PC3 cells were grown in reduced serum conditions (2% FBS) in presence or absence of CXCL13 (100 ng/ml), isotype control antibody or anti‐CXCR5 antibody (1 μg/ml), DOCK2siRNA or control siRNA (2 μm), and/or JNK inhibitor (10 μm). MTT assay was performed every 24 h for 3 days to assess cell proliferation. Error bars represent ± standard error of means of three independent experiments. *Significant (P < 0.05) changes relative to CXCL13‐treated cells.
Figure 6.
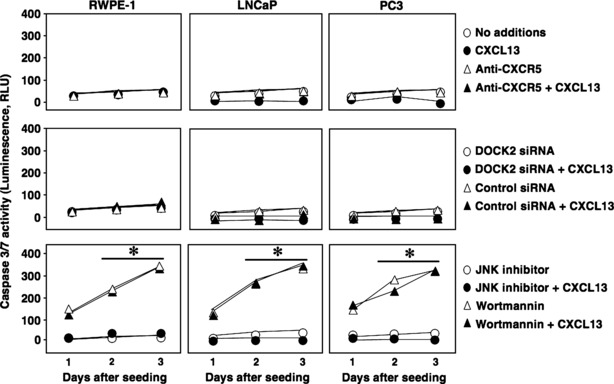
JNK inhibition and DOCK2 knockdown lead to reduction in PCa cell proliferation not due to cell death by apoptosis. RWPE‐1, LNCaP and PC3 cells were grown in reduced serum conditions (2% FBS) in presence or absence of CXCL13 (100 ng/ml), isotype control antibody or anti‐CXCR5 antibody (1 μg/ml), DOCK2 siRNA or control siRNA (2 μm), JNK inhibitor (10 μm), or wortmannin (1 μm). Caspase activity was measured using Caspase3/7 Glo assay every 24 h for 3 days. *Significant (P < 0.05) changes relative to no additions.
Discussion
Chemokine receptor stimulation has been reported to engage a broad range of physiological responses including chemotaxis and cell proliferation (23). Under normal conditions, these functions are important for maintaining homoeostasis, but deregulation of chemokine receptors leads to exacerbation and progression of some diseases, for example, cancer (24). Chemokines signal through binding to G protein‐coupled receptors (GPCRs) to activate PI3K that subsequently phosphorylates Akt, which is fully active following phosphorylation of both threonine 308 in its kinase catalytic domain and serine 473 residues in its regulatory domain (25, 26). Often, PCa cells carry somatic mutations in the phosphatase and tensin homologue (PTEN), which indirectly lead to sustained activation of Akt (27). This aberrant Akt activation causes phosphorylation of over fifty distinct substrates and regulates many processes considered hallmarks of cancer progression (28, 29). Indeed, chemokine‐mediated Akt hyperphosphorylation has been linked to cell survival and inhibition of both apoptotic and non‐apoptotic cell death.
PCa cell lines (LNCaP and PC3) have been extensively used as models to study cellular signalling in PCa progression. Both LNCaP and PC3 cell lines lack functional lipid phosphatase PTEN (30). LNCaP cells are androgen‐responsive, whereas PC3 cells are androgen‐refractory. Acquired hormone‐refractory properties have been linked to the high skeletal metastatic potential of PC3 cells, compared to that of hormone‐responsive LNCaP cells. These and other differences allow these PCa cell models to be used to provide insights into specific cellular events involved in PCa progression (31, 32).
In this study, we have shown that Akt and ERK1/2 are activated in LNCaP and PC3 cells following CXCL13 treatment to presumably support cell proliferation and/or survival. However, several studies have demonstrated PI3K‐ and Akt‐independent survival and growth signalling pathways (29, 33). Among these, the Rac GTPase subfamily of Rho guanosine triphosphatases has been reported to play an important role in integration of signals from cell surface receptors (for example, chemokine receptors, integrins and more) leading to control of cytoskeletal rearrangement, proliferation and survival of cancer cells (34). Rac activation is facilitated by DOCK2, a process leading to induction of the JNK signal transduction pathway (18, 35). Unlike LNCaP cells, hormone‐refractory cell line, PC3, was found to express DOCK2. Here, we evaluated the functional relevance of DOCK2 expression by these cell lines. CXCL13‐mediated activation of PI3K/Akt and ERK1/2 was independent of DOCK2. This finding is in partial agreement with previous studies reporting that DOCK2 is not required for transmigration of T‐lymphocytes through endothelium (36). Moreover, expression of DOCK2 by RWPE‐1, a non‐motile cell line, further implies that DOCK2 can undertake various functions in cells other than promoting migration and invasion.
We also show that DOCK2 was responsible for JNK phosphorylation and contributed to PC3 cell proliferation in response to CXCL13 stimulation. Inhibition of DOCK2 and JNK caused marked reduction in CXCL13‐dependent PC3 cell proliferation that was not associated with cell death. In contrast, CXCL13 stimulation of LNCaP cells also resulted in JNK activation, a mechanism that promoted cell proliferation independent of DOCK2, which was not expressed by these cells. CXCR5 couples to Gαq/11 in LNCaP cells, while CXCR5 is coupled to Gαi2 subunits in PC3 cells (Fig. S1). Both of these G‐proteins are able to activate the PI3K/Akt pathway by transactivating receptor tyrosine kinases, and the ERK1/2 pathway through induction of Ras causing ERK1/2 to translocate to the nucleus and initiate transcription (37, 38). Therefore, it is possible that LNCaP cells utilize the Gαq/11 protein coupled to CXCR5 to induce activation of phospholipase C‐β (PLC). This leads to subsequent cleavage of membrane phosphatidylinositol (4,5) P2 [PIP2] to the second messengers’ inositol (1,4,5) P3 [IP3] and diacylglycerol [DAG]. IP3 initiates release of calcium from intracellular stores, and DAG, in conjunction with calcium and phospholipids, activates protein kinase C (PKC) to stimulate JNK activity (Fig. 7). This would represent a divergent mechanism by which hormone‐sensitive and ‐refractory cells proliferate; however, additional studies will be required to test this postulate.
Figure 7.
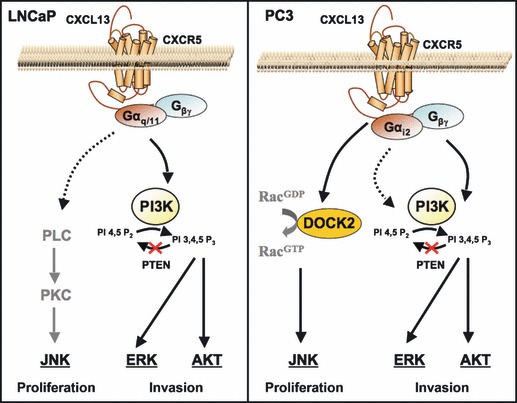
Signalling cascades modulated by CXCL13 in PCa cell lines. CXCL13 signalling through its cognate receptor, CXCR5, elicits Akt and ERK1/2 activation. In LNCaP cells, CXCL13 also regulates JNK activation presumably via Gαq/11 coupled to CXCR5, which potentially mediates phospholipase C (PLC) and protein kinase C (PKC) activation. In PC3 cells, however, JNK activation is mediated through DOCK2, a differentially expressed protein that acts as a guanine nucleotide exchange factor to activate Rac isoforms and contributes to cell proliferation.
Earlier observations designated JNK in cell death pathways, whereas other studies established the growth‐supporting role of JNK (39, 40). JNK inhibition has been shown to suppress population growth of various cell types, including T cells, KB‐3 human carcinoma cells, HepG2 cells, Chang liver cells, HT29 cells, Jurkat cells, and melanoma cells, relying on induction of G2/M cell cycle arrest (41, 42, 43). Indeed, treatment of prostate carcinoma‐bearing mice with antisense oligonucleotides of JNK leads to inhibition of tumour growth (44). Our results allude to a positive role of DOCK2 and JNK in PCa cell population growth, a process that has not been described previously. However, DOCK2/JNK‐dependent regulation of PC3 cell cycle in response to CXCL13 requires further examination. Taken together, our results suggest a novel role of DOCK2 in hormone‐refractory PCa cell population growth (Fig. 7). While blockade of CXCL13:CXCR5 interactions using antibodies and pharmacological reagents holds great promise, our results suggest that differential DOCK2 expression by advanced or hormone‐refractory PCa should be considered when determining mechanisms of chemokine‐mediated JNK activation and tumour growth.
Supporting information
Fig. S1 CXCR‐associated G proteins in prostate cancer cell lines. CXCR5 was immuno‐precipiated (IP) from LNCaP and PC3 cell line lysates to pull down associated proteins from total cell lysates. The CXCR5‐IP cell lysates were resolved by SDS‐PAGE and the expression of Gai1, Gai2, Gai3, Gas, Gaq/11, Ga12 and Ga13 were examined by immunoblot.
Supporting info item
References
- 1. Walczak JR, Carducci MA (2007) Prostate cancer: a practical approach to current management of recurrent disease. Mayo Clin. Proc. 82, 243–249. [DOI] [PubMed] [Google Scholar]
- 2. Msaouel P, Pissimissis N, Halapas A, Koutsilieris M (2008) Mechanisms of bone metastasis in prostate cancer: clinical implications. Best Pract. Res. Clin. Endocrinol. Metab. 22, 341–355. [DOI] [PubMed] [Google Scholar]
- 3. Klocker H, Culig Z, Kaspar F, Hobisch A, Eberle J, Reissigl A et al. (1994) Androgen signal transduction and prostatic carcinoma. World J. Urol. 12, 99–103. [DOI] [PubMed] [Google Scholar]
- 4. Edwards J, Bartlett JM (2005) The androgen receptor and signal‐transduction pathways in hormone‐refractory prostate cancer. Part 2: Androgen‐receptor cofactors and bypass pathways. BJU Int. 95, 1327–1335. [DOI] [PubMed] [Google Scholar]
- 5. Kakinuma T, Hwang ST (2006) Chemokines, chemokine receptors, and cancer metastasis. J. Leukoc. Biol. 79, 639–651. [DOI] [PubMed] [Google Scholar]
- 6. Koizumi K, Hojo S, Akashi T, Yasumoto K, Saiki I (2007) Chemokine receptors in cancer metastasis and cancer cell‐derived chemokines in host immune response. Cancer Sci. 98, 1652–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rodriguez‐Frade JM, Martinez AC, Mellado M (2005) Chemokine signaling defines novel targets for therapeutic intervention. Mini Rev. Med. Chem. 5, 781–789. [DOI] [PubMed] [Google Scholar]
- 8. Vandercappellen J, Van Damme J, Struyf S (2008) The role of CXC chemokines and their receptors in cancer. Cancer Lett. 267, 226–244. [DOI] [PubMed] [Google Scholar]
- 9. Singh S, Singh R, Singh UP, Rai SN, Novakovic KR, Chung LWK et al. (2009) Clinical and biological significance of CXCR5 expressed by prostate cancer specimens and cell lines. Int. J. Cancer 125, 2288–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Singh S, Singh R, Sharma PK, Singh UP, Rai SN, Chung LWK et al. (2009) Serum CXCL13 positively correlates with prostatic disease, prostate‐specific antigen and mediates prostate cancer cell invasion, integrin clustering and cell adhesion. Cancer Lett. 283, 29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lisignoli G, Toneguzzi S, Piacentini A, Cattini L, Lenti A, Tschon M et al. (2003) Human osteoblasts express functional CXC chemokine receptors 3 and 5: activation by their ligands, CXCL10 and CXCL13, significantly induces alkaline phosphatase and beta‐N‐acetylhexosaminidase release. J. Cell. Physiol. 194, 71–79. [DOI] [PubMed] [Google Scholar]
- 12. Song G, Ouyang G, Bao S (2005) The activation of Akt/PKB signaling pathway and cell survival. J. Cell Mol. Med. 9, 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Caraglia M, Marra M, Leonetti C, Meo G, D’Alessandro AM, Baldi A et al. (2007) R115777 (Zarnestra)/Zoledronic acid (Zometa) cooperation on inhibition of prostate cancer proliferation is paralleled by Erk/Akt inactivation and reduced Bcl‐2 and bad phosphorylation. J. Cell. Physiol. 211, 533–543. [DOI] [PubMed] [Google Scholar]
- 14. Kue PF, Daaka Y (2000) Essential role for G proteins in prostate cancer cell growth and signaling. J. Urol. 164, 2162–2167. [PubMed] [Google Scholar]
- 15. Vivanco I, Palaskas N, Tran C, Finn SP, Getz G, Kennedy NJ et al. (2007) Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer Cell 11, 555–569. [DOI] [PubMed] [Google Scholar]
- 16. Durand CA, Westendorf J, Tse KW, Gold MR (2006) The Rap GTPases mediate CXCL13‐ and sphingosine1‐phosphate‐induced chemotaxis, adhesion, and Pyk2 tyrosine phosphorylation in B lymphocytes. Eur. J. Immunol. 36, 2235–2249. [DOI] [PubMed] [Google Scholar]
- 17. Weston CR, Davis RJ (2002) The JNK signal transduction pathway. Curr. Opin. Genet. Dev. 12, 14–21. [DOI] [PubMed] [Google Scholar]
- 18. Sanui T, Inayoshi A, Noda M, Iwata E, Stein JV, Sasazuki T et al. (2003) DOCK2 regulates Rac activation and cytoskeletal reorganization through interaction with ELMO1. Blood 102, 2948–2950. [DOI] [PubMed] [Google Scholar]
- 19. Chung CY, Lee S, Briscoe C, Ellsworth C, Firtel RA (2000) Role of Rac in controlling the actin cytoskeleton and chemotaxis in motile cells. Proc. Natl. Acad. Sci. USA 97, 5225–5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fukui Y, Hashimoto O, Sanui T, Oono T, Koga H, Abe M et al. (2001) Haematopoietic cell‐specific CDM family protein DOCK2 is essential for lymphocyte migration. Nature 412, 826–831. [DOI] [PubMed] [Google Scholar]
- 21. Kunisaki Y, Tanaka Y, Sanui T, Inayoshi A, Noda M, Nakayama T et al. (2006) DOCK2 is required in T cell precursors for development of Valpha14 NK T cells. J. Immunol. 176, 4640–4645. [DOI] [PubMed] [Google Scholar]
- 22. Nombela‐Arrieta C, Mempel TR, Soriano SF, Mazo I, Wymann MP, Hirsch E et al. (2007) A central role for DOCK2 during interstitial lymphocyte motility and sphingosine‐1‐phosphate‐mediated egress. J. Exp. Med. 204, 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cotton M, Claing A (2009) G protein‐coupled receptors stimulation and the control of cell migration. Cell. Signal. 21, 1045–1053. [DOI] [PubMed] [Google Scholar]
- 24. Zlotnik A (2006) Chemokines and cancer. Int. J. Cancer 119, 2026–2029. [DOI] [PubMed] [Google Scholar]
- 25. Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kandel ES, Hay N (1999) The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp. Cell Res. 253, 210–229. [DOI] [PubMed] [Google Scholar]
- 27. Fang J, Ding M, Yang L, Liu LZ, Jiang BH (2007) PI3K/PTEN/AKT signaling regulates prostate tumor angiogenesis. Cell. Signal. 19, 2487–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ghosh PM, Malik S, Bedolla R, Kreisberg JI (2003) Akt in prostate cancer: possible role in androgen‐independence. Curr. Drug Metab. 4, 487–496. [DOI] [PubMed] [Google Scholar]
- 29. Blanco‐Aparicio C, Renner O, Leal JF, Carnero A (2007) PTEN, more than the AKT pathway. Carcinogenesis 28, 1379–1386. [DOI] [PubMed] [Google Scholar]
- 30. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI et al. (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947. [DOI] [PubMed] [Google Scholar]
- 31. Maitland NJ, Macintosh CA, Hall J, Sharrard M, Quinn G, Lang S (2001) In vitro models to study cellular differentiation and function in human prostate cancers. Radiat. Res. 155, 133–142. [DOI] [PubMed] [Google Scholar]
- 32. Mitchell S, Abel P, Ware M, Stamp G, Lalani E (2000) Phenotypic and genotypic characterization of commonly used human prostatic cell lines. BJU Int. 85, 932–944. [DOI] [PubMed] [Google Scholar]
- 33. Li P, Lee H, Guo S, Unterman TG, Jenster G, Bai W et al. (2003) AKT‐independent protection of prostate cancer cells from apoptosis mediated through complex formation between the androgen receptor and FKHR. Mol. Cell. Biol. 23, 104–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ridley AJ (2001) Rho family proteins: coordinating cell responses. Trends Cell Biol. 11, 471–477. [DOI] [PubMed] [Google Scholar]
- 35. Nishihara H, Maeda M, Oda A, Tsuda M, Sawa H, Nagashima K et al. (2002) DOCK2 associates with CrkL and regulates Rac1 in human leukemia cell lines. Blood 100, 3968–3974. [DOI] [PubMed] [Google Scholar]
- 36. Shulman Z, Pasvolsky R, Woolf E, Grabovsky V, Feigelson SW, Erez N et al. (2006) DOCK2 regulates chemokine‐triggered lateral lymphocyte motility but not transendothelial migration. Blood 108, 2150–2158. [DOI] [PubMed] [Google Scholar]
- 37. Delcourt N, Bockaert J, Marin P (2007) GPCR‐jacking: from a new route in RTK signalling to a new concept in GPCR activation. Trends Pharmacol. Sci. 28, 602–607. [DOI] [PubMed] [Google Scholar]
- 38. Piiper A, Zeuzem S (2004) Receptor tyrosine kinases are signaling intermediates of G protein‐coupled receptors. Curr. Pharm. Des. 10, 3539–3545. [DOI] [PubMed] [Google Scholar]
- 39. Smith A, Ramos‐Morales F, Ashworth A, Collins M (1997) A role for JNK/SAPK in proliferation, but not apoptosis, of IL‐3‐dependent cells. Curr. Biol. 7, 893–896. [DOI] [PubMed] [Google Scholar]
- 40. Yang YM, Bost F, Charbono W, Dean N, McKay R, Rhim JS et al. (2003) C‐Jun NH(2)‐terminal kinase mediates proliferation and tumor growth of human prostate carcinoma. Clin. Cancer Res. 9, 391–401. [PubMed] [Google Scholar]
- 41. Ennis BW, Fultz KE, Smith KA, Westwick JK, Zhu D, Boluro‐Ajayi M et al. (2005) Inhibition of tumor growth, angiogenesis, and tumor cell proliferation by a small molecule inhibitor of c‐Jun N‐terminal kinase. J. Pharmacol. Exp. Ther. 313, 325–332. [DOI] [PubMed] [Google Scholar]
- 42. Mingo‐Sion AM, Marietta PM, Koller E, Wolf DM, Van Den Berg CL (2004) Inhibition of JNK reduces G2/M transit independent of p53, leading to endoreduplication, decreased proliferation, and apoptosis in breast cancer cells. Oncogene 23, 596–604. [DOI] [PubMed] [Google Scholar]
- 43. Sundberg LJ, Galante LM, Bill HM, Mack CP, Taylor JM (2003) An endogenous inhibitor of focal adhesion kinase blocks Rac1/JNK but not Ras/ERK‐dependent signaling in vascular smooth muscle cells. J. Biol. Chem. 278, 29783–29791. [DOI] [PubMed] [Google Scholar]
- 44. Heasley LE, Han SY (2006) JNK regulation of oncogenesis. Mol. Cells 21, 167–173. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 CXCR‐associated G proteins in prostate cancer cell lines. CXCR5 was immuno‐precipiated (IP) from LNCaP and PC3 cell line lysates to pull down associated proteins from total cell lysates. The CXCR5‐IP cell lysates were resolved by SDS‐PAGE and the expression of Gai1, Gai2, Gai3, Gas, Gaq/11, Ga12 and Ga13 were examined by immunoblot.
Supporting info item