

Abstract
Multiple myeloma (MM) is the second most common hematological and incurable malignancy of plasma cells with low proliferative activity in the bone marrow. MM patients initially respond to conventional therapy, however, many develop resistance and recurrences occur. We have identified RKIP as a novel gene product that is differentially overexpressed in MM cell lines and MM tissues compared to other studied tumors and normal bone marrow. This overexpression consisted, in large part, of a phosphorylated inactive form of RKIP at Ser153 (p-Ser153 RKIP). In contrast to RKIP, p-Ser153 RKIP lacks its ability to inhibit the MAPK signaling pathway. The overexpression of p-Ser153 RKIP in MM cell lines and MM tissues was further validated in a mouse model carrying a human MM xenograft, namely, LAGλ-1B. Bioinformatic analyses from databases support the presence of increased RKIP mRNA expression in MM compared to normal plasma cells. In these databases, high RKIP levels in MM are also correlated with the nonhyperdiploid status and the presence of IgH translocations, parameters that generally display more aggressive clinical features and shorter patients’ survival irrespective of the treatment. Since RKIP expression regulates both the NF-κB and MAPK survival pathways, the overexpression of “inactive” p-Ser153 RKIP in MM might contribute positively to the overall cell survival/antiapoptotic phenotype and drug resistance of MM through the constitutive activation of survival pathways and downstream the transcription of anti-apoptotic gene products. The overexpression of RKIP and p-Ser153 RKIP in MM is the first demonstration in the literature, since in most tumor tissues the expression of RKIP is very low and the expression of p-Ser153 RKIP is much lower. The relationship between the levels of active RKIP and inactive p-Ser153 RKIP in MM may be of prognostic significance, and the regulation of RKIP activity may be a target for therapeutic intervention.
Keywords: NF-κB, Raf-1 kinase inhibitor protein (RKIP), apoptosis, multiple myeloma
I. INTRODUCTION
Multiple myeloma (MM) is an incurable hematological malignancy that accounts for 10% of deaths caused by blood cancers and has a median survival of approximately five years. MM is a malignancy of plasma cells with low proliferative activity in the bone marrow. Uncontrolled growth of MM within the bone marrow can lead to bone marrow failure that eventually causes anemia and, less commonly, thrombocytopenia and leukopenia. Interactions between MM cells and nonmalignant cells in the bone marrow microenvironment lead to enhanced bone loss, often resulting in the formation of osteolytic bone lesions, generalized osteoporosis, pathological fractures, hypercalcemia, and bone pain. MM is also associated with renal dysfunction most commonly caused by deposition of monoclonal proteins in the distal tubules of the collecting system of the kidney, hypercalcemia, or myeloma cell infiltration of the kidney. Most patients also show reduced production of other types of antibodies, and may also present with or develop recurrent infections.1,2
In the past decade, there have been major advances in the treatment of MM. Thalidomide, bortezomib, and lenalidomide have emerged as highly active agents in the treatment of MM. Response rates with relapsed diseases are approximately 50% with the combination of thalidomide and steroids, and 65% with a three-drug combination of thalidomide, steroids, and cyclophosphamide. Patients who are not transplant candidates are treated with standard alkylating agents therapy, namely, melphalan, prednisone, and thalidomide (MPT); melphalan, prednisone, and bortezomib (MPB); and melphalan, prednisone, and lenalidomide (MPR).2 Although initial clinical responses to drug therapy are achieved, a significant percentage of MM patients relapse and no longer respond to single or combined treatments.3,4 Hence, the resistance of MM to current therapeutic regimens remains an unsolved problem in the management of patients with MM.
Research studies on MM have provided new insights into many signaling pathways and proteins whose activation are associated with the pathogenesis of MM, including the PI3K/AKT,5 JAK/STAT3,6 transforming growth factor (TGF)-β/β-catenin,7 Wnt,8 Notch,9,10 insulin growth factor (IGF),11 pleiotrophin,12 and IKK/NF-κB signaling pathways.6,13 These pathways and gene products also regulate several antiapoptotic gene products implicated in the resistance of MM to a variety of cytotoxic therapies.
The objective of our current study is to delineate potential underlying mechanisms that regulate the resistance of MM to apoptosis induced by drugs, and thus develop new means to override resistance through manipulation of the identified resistance factors. The rationale of these studies is based on our recent reports that have identified the Raf-1 kinase inhibitor protein (RKIP) as a gene product that can regulate tumor cell resistance to both chemotherapy and immunotherapy.14,15 RKIP is a member of the phosphatidylethanolamine-binding protein (PEBP) family that functions as a potent inhibitor of different kinases in the Raf/MAPK (Raf-1 kinase) and NF-kB (TAK1, NIK) activation pathways, thereby antagonizing both cell survival and the expression of antiapoptotic gene products.14,16–18 RKIP also binds to the centrosomal and kinetochore regions of metaphase chromosomes, in which it may be involved in regulating the partitioning of chromosomes and the progression through mitosis so that its loss can result in chromosomal abnormalities.19 RKIP expression was shown to be regulated at the transcription level by Snail,20 while RKIP activity was found to be under the control of a post-translational modification, which involves a PKC-mediated phosphorylation at Ser 153.21 The phosphorylation was observed in response to growth factor stimuli and resulted in the derepression of the MAPK pathway. These results indicate that PKC regulates MAP kinase activation under physiological conditions through the phosphorylation of RKIP.
RKIP expression is significantly reduced in different cancer types, and its levels seem to regulate the tumor metastatic potential and resistance to apoptotic stimuli.22–26 Thus, we hypothesized that low RKIP expression in MM might be directly associated with the pathogenesis of MM and its resistance to therapy. To test this hypothesis, we investigated (i) the expressions of RKIP and phosphorylated RKIP (p-Ser153 RKIP) in MM cell lines and bone marrow–derived tissues from MM patients by immunohistochemistry, (ii) the expression of RKIP and p-Ser153 RKIP in vivo in mice bearing a human MM tumor xenograft, and (iii) the RKIP transcript levels in publicly available data sets containing gene expression profiles of MM and normal B cells. We further correlated our findings with other clinical parameters and patients’ outcomes.
II. OVEREXPRESSION OF RKIP IN MM
II.A. Overexpression of RKIP and p-Ser153 RKIP in MM Cell Lines and Primary MM Tumors
Given the reported role of RKIP as a positive regulator of apoptosis in a plethora of tumors, we hypothesized that MM resistance to cytotoxic drugs might be partially attributed to low RKIP levels. We tested this hypothesis by examining the expression levels, using immunohistochemistry, of RKIP in a number of MM cell lines and bone marrow–derived MM tissues (BMMCs) from MM patients. The RKIP expression in MM cell lines was compared with that detected in other tumor cell lines such as the prostate cancer cell line PC-3 and the Non-Hodgkin’s B-cell lymphoma (B-NHL) line Ramos. Interestingly, all of the tested MM cell lines, MM1.S, U266, IM9, and RPMI 8826, expressed significantly higher levels of RKIP compared to other tumor cell lines studied (Fig. 1A). These findings were further validated in MM patients–derived BMMCs whereby the majority of the MM samples were characterized by increased expression of RKIP compared to that detected in normal bone marrow (Fig. 1B). These unexpected findings suggested that MM is distinguished from the majority of other reported tumors in terms of levels of RKIP expression.22–26 Furthermore, the high level of RKIP expression in MM did not coincide with the resistant phenotype of MM. Hence, we postulated that the high RKIP expression in MM may be in its inactivated phosphorylated form.
FIGURE 1.

Overexpression of RKIP in MM cell lines and primary MM tumors. (A) Immunohistochemical (IHC) analysis of RKIP expression in the MM cell lines RPMI 8226, MM1.S and U266, as well as in Ramos and PC-3 cell lines. Following inhibition of endogenous peroxidase activity and nonspecific binding, the slides were incubated overnight at room temperature with an anti-hRKIP (1/500) antibody. The slides were further incubated with a biotinylated antirabbit secondary antibody [Universal LSAB kit (DAKO corporation, Carpinteria, California)] and then with a streptavidin-HRP conjugate as previously described.37 The color was detected using a 3, 30-diaminobenzidine tetrahydrochloride solution (DAB) according to manufacturer’s instructions (DAKO corporation, Carpinteria, California) (B) Overexpression of RKIP in representative primary MM tumors and normal bone marrow as assessed by IHC. In all cases, rabbit IgG was used as antibody specificity control. In order to reduce the variability, all samples were processed at the same time in a single experiment using a single batch of antibody diluted in PBS supplemented with normal goat serum.
Previous reports by Corbit et al.21 showed that the functional activities of RKIP are regulated by its phosphorylation status on Ser 153. In contrast to the unphosphorylated form, p-Ser153 RKIP lacks effective binding to Raf-1, and therefore does not inhibit the MAPK signaling pathway. In addition, p-Ser153 RKIP binds to the G protein–coupled receptor kinase-2 (GRK-2) and dissociates it from G protein–coupled receptors (GPCRs), thus preventing receptor internalization and inactivation.27 The RKIP phosphorylation at Ser153 was shown to be mediated specifically by activated classic and atypical protein kinase C (PKC) isoforms such as the PKC ζ. Thus, p-Ser153 RKIP antagonizes unphosphorylated RKIP in inhibiting survival signals and promoting apoptosis.27
Using a monospecific antibody directed against p-Ser153 RKIP, which recognizes only the phosphorylated form, we performed immunohistochemical analysis for p-Ser153 RKIP staining in the same MM cell lines and patient-derived BMMCs used for the determination of total RKIP expression. The findings revealed high p-Ser153 RKIP expressions with the same intensity range observed for total RKIP expression (Figs. 2A and 2B). In contrast, the PC-3 and Ramos cell lines as well as normal BM did not present any significant RKIP or p-Ser153 RKIP staining. It is noteworthy that the RKIP and p-Ser153 RKIP staining assessed in MM cell lines and MM-derived BMMCs was detected in both the cytoplasm and nucleus.
FIGURE 2.

MM exhibits overexpression of the inactive p-Ser153 RKIP form. (A) Expression of p-Ser153 RKIP in MM cell lines and other non-MM cell lines, as assessed by IHC. The slides were incubated overnight at room temperature with an anti-p-Ser153 RKIP antibody at 1/250 dilution in 1% BSA in PBS, and the staining was analyzed as described above. (B) IHC analysis of p-Ser153 RKIP expression in representative primary MM samples and normal bone marrow. (C) Quantitative analysis of RKIP and p-Ser153 RKIP expression in primary MM samples. The graph demonstrates the mean percentage ± SD of RKIP and p-Ser153 RKIP–positive cells counted in five random fields of each of the 20 MM samples. p = 0.001 RKIP versus p-Ser153 RKIP positive staining (Mann Whitney U-test).
A quantitative overall analysis of RKIP and p-Ser153 RKIP expression levels in MM tissues revealed a significant difference (p = 0.001) among MM samples in terms of the mean percentage of RKIP and p-Ser153 RKIP positive cells (Fig. 2C). In addition, the average percentage of p-Ser153 RKIP positive cells corresponded to ~50% of the average percentage of RKIP positive cells, indicating that for the majority of RKIP positive MM samples, a significant proportion of RKIP expression is in the p-Ser153 RKIP form.
In summary, the above findings reveal that in MM, the aberrant expression of RKIP, which should have been correlated with a good prognosis, is in its inactive phosphorylated form and may be of poor prognosis. Preliminary findings were reported at the 2008 ASH Meeting.28
II.B. Validation of High RKIP and p-Ser153 RKIP Expression in an MM Xenograft in SCID Mice
We have developed and successfully used in preclinical drug studies a murine model of human multiple myeloma derived from an MM patient, namely, LAGλ-1B.12,29,30 This stable tumor shows evidence of maintaining the same chemosensitivity and resistance to anti-MM agents as in the original patient’s derived tissue. It also displays the same human para-protein in mouse blood. The above findings for the high RKIP and p-Ser153 RKIP expression levels in MM cell lines and MM tissues were validated in the LAGλ-1B tumor xenograft. IHC analysis in biopsies of LAGλ-1B–palpable tumors 20 days postimplantation revealed high RKIP and p-Ser153 RKIP expression at almost equal levels (Fig. 3). This finding validates our in vitro findings and our hypothesis for the dominance of p-Ser153 RKIP form in MM. Furthermore, the findings suggest that this in vivo model can be used for analysis of the correlation between the expression levels of RKIP and p-Ser153 RKIP, and the response to various cytotoxic agents. It also suggests that p-Ser153 RKIP overexpression might regulate, in part, MM survival and resistance to apoptotic stimuli, and could be a potential candidate target for reversal of MM resistance to conventional drug therapy.
FIGURE 3.
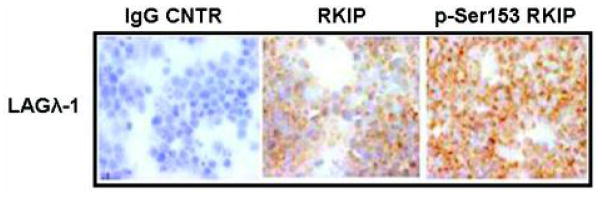
In vivo validation of high RKIP and p-Ser153 RKIP expression. Expression profiles of RKIP and p-Ser153 RKIP in MM biopsies derived from an LAGλ-1B MM human xenograft in SCID mice. The expression of both RKIP forms was assessed by IHC using the specific antibodies. All animal studies were conducted according to the University of California at Los Angeles (UCLA) Animal Research Committee protocols. Tumors were measured weekly using calipers, and their volumes calculated with the following formula: 4/3π × (width/2)2 × (length/2), portraying the 3D volume of an ellipsoid.
II.C. Computational Analysis
A special focus in the field of MM prognostication during the last few years has provided novel approaches to analyze the pharmacogenomic databases and their correlations with prognosis. The negative prognostic impact of various genetic aberrations, including specific IgH translocations and chromosome 13 deletions and, mainly, the prognostic importance of the expression profiles of individual genes or groups of genes, have been proved to be valuable tools in guiding anti-MM therapeutic decisions and determining the clinical outcome.31,32 For that reason, computational analysis of RKIP mRNA levels in different groups of MM patients was performed as described in Figure 4.
FIGURE 4.
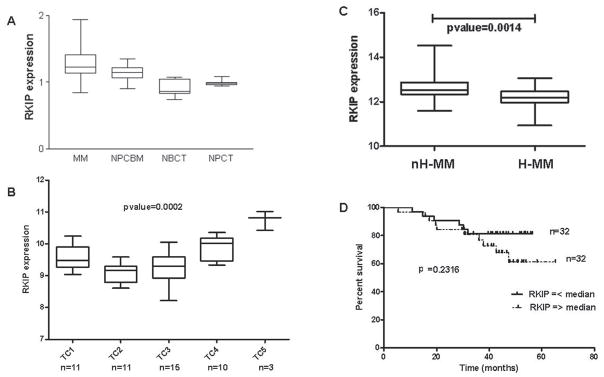
RKIP gene expression levels in MM data sets. ’The expression of RKIP was examined in independent, publicly available data sets of MM gene expression using HG-U133 series microarrays (Affymetrix, Santa Clara, CA, USA). The selection of the patients was based on the presence of recurrent IgH chromosomal translocations, hyperdiploidy, and cyclin D expression. The analyses were performed in: (a) 50 MM samples that were stratified according to the TC classification: TC1 = tumors with t(11;14) or t(6;14) translocations, overexpression of CCND1 or CCND3, and a nonhyperdiploid status; TC2 = tumors with low to moderate levels of the CCND1 gene in the absence of any primary IGH translocation but associated with a hyperdiploid status; TC3 = tumors that do not fall into any of the other groups and most of which express CCND2; TC4 = tumors showing high CCND2 levels and the presence of the t(4;14) translocation; and TC5 = tumors expressing the highest levels of CCND2 in association with either the t(14;16) or t(14;20) translocation (HG-U133A),34 and (b) 67 MM samples classified into a hyperdiploid and a nonhyperdiploid subgroup characterized by loss of chromosomes 1p, 8p, 13, and 16q and by amplification of chromosome 1q. Clinical outcome of MM patients whose neoplastic tissues were analyzed was also available (HG-U133 Plus 2.0).35 A data set obtained using Affymetrix HuGene FL microarrays was also used.33 This data set reports the gene expression data obtained from 74 MM samples and 31 cell preparations from healthy donors. Normalized expression values of RKIP were extracted from the NCBI Gene Expression Omnibus database (GEO series: GSE291234 and GSE445235) or from one author’s Web site (http://lambertlab.uams.edu).33 For HG-U133A microarrays, the mean RMA log2 intensity values of the three Affymetrix probe sets specific for RKIP (PEBP1: 210825_s_at; 211941_s_at and 205353_s_at in HGU11-A and HG-U133 Plus2 platforms) were considered. (A) Zhan database33: comparison of mean RKIP transcript levels among MM plasma cells, normal plasma cells derived from bone marrow (NPCBM), normal B cell from tonsils (NBCT), and normal plasma cells from tonsils (NPCT). p < 0.01 RKIP expression in MM plasma cells versus each of the other three groups (Mann Whitney U-test). (B) Agnelli data set34: MM patients were classified into the five TC subgroups described above. RKIP expression in the five subgroups is different with p < 001, as assessed by Kruskal-Wallis statistics. Samples harboring IgH translocations TC1, TC4, and TC8 display significantly higher RKIP expression when compared to TC2 and TC3 samples without IgH translocations (p < 0.01 for TC2 versus TC4 and TC2 versus TC5), (p < 0.05 for TC2 versus TC1, TC3 versus TC4, and TC3 versus TC5) as assessed by Dunn’s multiple comparison post test. The number of informative patients (n) is reported. (C) Carrasco data set35: nH-MM (nonhyperdiploid) samples display increased expression of RKIP when compared to H-MM samples (p < 001, Mann Whitney U-test). (D) Kaplan Meier overall survival curves for 64 patients indicating no significant difference in survival (p = 0.2316) in patients divided according to values above or below median of RKIP gene expression values.
RKIP overexpression was found to be a general event in MM cells and primary MM tumors compared to normal bone marrow and other studied tumors. The overexpression of RKIP in MM compared to normal plasma cells samples at the transcriptional level was confirmed in a publicly available gene expression data set33 (p < 0.0001) (Fig. 4A). In the perspective to identify in MM tumors the existence of a possible association between RKIP overexpression and commonly used prognostic parameters for determining MM outcome, we explored two independent gene expression data sets that include a collection of molecularly characterized and outcome-annotated clinical MM specimens.34,35 In the Agnelli et al.34 data set, RKIP expression was stratified among different TC groups classified according to the presence of IgH chromosomal translocations and hyperdiploid status (H-MM) (Fig. 4B). RKIP overexpression was found significantly associated with non–H-MM (groups TC1, TC4, and TC5). These latter MM subgroups are also composed of cases harboring IgH translocations, generally displaying more aggressive clinical features and shorter survival, irrespective of the treatment modality.36 The three main IgH translocations found in non–H-MM patients analyzed in the data set were the t(11;14), t(4;14), and t(14;16). The positive correlation of high RKIP levels with nH-MM status was further identified in the data set of Carrasco et al.35 data set, where MM patients were classified in subgroups of H-MM (which harbors numerous chromosomal trisomies and a low prevalence of IgH translocations) and non–H-MM (Fig. 4C). Similar results were also obtained by separately analyzing the three RKIP probesets (210825_s_at, 211941_s_at, and 205353_s_at) or their normalized log2 mean expression values (data not shown).
The patients belonging to the nH-MM subgroup in the data set of Carrasco et al.35 were shown to have a tendency toward a less favorable outcome. When these patients were stratified according to RKIP expression (above and below median values of expression in the data set) and correlated with the clinical outcome, no significant association with survival was observed (Fig. 4D). This finding suggests that the biological significance of RKIP in MM could not be determined only by its transcript and protein levels; but as shown in this study, could also be dictated by post-translational modifications such as the phosphorylation at Ser153, which represses RKIP activity as an antisurvival factor.
III. REMARKS
In contrast to other reported studies on the expression of RKIP in various cancer types, our findings demonstrate high levels of RKIP in MM cell lines and patients samples. The over-expression mainly concerns a p-Ser153 RKIP form. The overexpression of RKIP and p-Ser153 RKIP in MM is the first demonstration in the literature among many tumors studied. There have been no published reports (except studies performed in lung tumors, see p. 171 in this issue) to date on the analysis of p-Ser153 RKIP expression in any tumor and its possible role in the interpretation of the data. In contrast to MM, most tumors express low levels of total RKIP, and they are expected to have equal or lower levels of p-Ser153 RKIP. Given the role of RKIP as a negative regulator of survival signals induced by the activation of MAPK and NF-κB pathways, the post-translational modification of RKIP (the phosphorylation at Ser153 residue) is important to assess. Thus, p-Ser153 RKIP might contribute positively to the overall cell survival and drug resistance of MM through the constitutive activation of survival pathways and downstream the transcription of antiapoptotic genes. Furthermore, our bioinformatic analyses in reported databases support the presence of increased mRNA levels of RKIP in MM compared to normal B cells and correlate the levels with different prognostic factors (such as IgH translocations) that are responsible for more aggressive clinical features and shorter patients’ survival. However, we propose that further studies of larger cohorts are necessary to confirm our preliminary findings and to establish the relevance of the RKIP/p-Ser153 RKIP ratios as a prognostic biomarker that might be used in individualized anti-MM treatment approaches.
Acknowledgments
We acknowledge all the members of our laboratory and collaborators for their valuable contributions. We also acknowledge the Jonsson Comprehensive Cancer Center at UCLA for its continuous support and the Bodossaki Foundation for a postdoctoral fellowship support to SB. The authors also acknowledge the assistance of Kerry Choy in the preparation of this article.
ABBREVIATIONS
- BMMCs
bone marrow mononuclear cells
- IgH
immunoglobulin heavy (chain)
- IKK
I kappa B kinase
- JAK
janus kinases
- MAPK
mitogen-activated protein kinases
- NF-κB
nuclear factor-κB
- NIK
NF-kappa B–inducing kinase
- PI3K/Akt
phosphatidylinositol 3-kinase/Akt
- PKC
protein kinase C
- STAT
signal transducers and activator of transcription
- TAK1
transforming growth factor beta–activated kinase 1
References
- 1.Li ZW, Chen H, Campbell RA, Bonavida B, Berenson JR. NF-kappaB in the pathogenesis and treatment of multiple myeloma. Curr Opin Hematol. 2008;15(4):391–9. doi: 10.1097/MOH.0b013e328302c7f4. [DOI] [PubMed] [Google Scholar]
- 2.Kyle RA, Rajkumar SV. Multiple myeloma. Blood. 2008;111(6):2962–72. doi: 10.1182/blood-2007-10-078022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kastritis E, Palumbo A, Dimopoulos MA. Treatment of relapsed/refractory multiple myeloma. Semin Hematol. 2009;46(2):143–57. doi: 10.1053/j.seminhematol.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Berenson JR. Bone disease in myeloma. Curr Treat Options Oncol. 2001;2(3):271–83. doi: 10.1007/s11864-001-0041-5. [DOI] [PubMed] [Google Scholar]
- 5.Younes H, Leleu X, Hatjiharissi E, Moreau AS, Hideshima T, Richardson P, Anderson KC, Ghobrial IM. Targeting the phosphatidylinositol 3-kinase pathway in multiple myeloma. Clin Cancer Res. 2007;13(13):3771–5. doi: 10.1158/1078-0432.CCR-06-2921. [DOI] [PubMed] [Google Scholar]
- 6.Ishikawa H, Tsuyama N, Kawano MM. Interleukin-6-induced proliferation of human myeloma cells associated with CD45 molecules. Int J Hematol. 2003;78(2):95–105. doi: 10.1007/BF02983376. [DOI] [PubMed] [Google Scholar]
- 7.Dong M, Blobe GC. Role of transforming growth factor-beta in hematologic malignancies. Blood. 2006;107(12):4589–96. doi: 10.1182/blood-2005-10-4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pearse RN. Wnt antagonism in multiple myeloma: a potential cause of uncoupled bone remodeling. Clin Cancer Res. 2006;12(20 Pt 2):6274s–8s. doi: 10.1158/1078-0432.CCR-06-0648. [DOI] [PubMed] [Google Scholar]
- 9.Jundt F, Probsting KS, Anagnostopoulos I, Muehlinghaus G, Chatterjee M, Mathas S, Bargou RC, Manz R, Stein H, Dorken B. Jagged1-induced Notch signaling drives proliferation of multiple myeloma cells. Blood. 2004;103(9):3511–5. doi: 10.1182/blood-2003-07-2254. [DOI] [PubMed] [Google Scholar]
- 10.Nefedova Y, Cheng P, Alsina M, Dalton WS, Gabrilovich DI. Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood. 2004;103(9):3503–10. doi: 10.1182/blood-2003-07-2340. [DOI] [PubMed] [Google Scholar]
- 11.Yasui H, Hideshima T, Richardson PG, Anderson KC. Novel therapeutic strategies targeting growth factor signalling cascades in multiple myeloma. Br J Haematol. 2006;132(4):385–97. doi: 10.1111/j.1365-2141.2005.05860.x. [DOI] [PubMed] [Google Scholar]
- 12.Chen H, Gordon MS, Campbell RA, Li M, Wang CS, Lee HJ, Sanchez E, Manyak SJ, Gui D, Shalitin D, Said J, Chang Y, Deuel TF, Baritaki S, Bonavida B, Berenson JR. Pleiotrophin is highly expressed by myeloma cells and promotes myeloma tumor growth. Blood. 2007;110(1):287–95. doi: 10.1182/blood-2006-08-042374. [DOI] [PubMed] [Google Scholar]
- 13.Feinman R, Siegel DS, Berenson J. Regulation of NF-kB in multiple myeloma: therapeutic implications. Clin Adv Hematol Oncol. 2004;2(3):162–6. [PubMed] [Google Scholar]
- 14.Baritaki S, Katsman A, Chatterjee D, Yeung KC, Spandidos DA, Bonavida B. Regulation of tumor cell sensitivity to TRAIL-induced apoptosis by the metastatic suppressor Raf kinase inhibitor protein via Yin Yang 1 inhibition and death receptor 5 up-regulation. J Immunol. 2007;179(8):5441–53. doi: 10.4049/jimmunol.179.8.5441. [DOI] [PubMed] [Google Scholar]
- 15.Baritaki S, Yeung K, Palladino M, Berenson J, Bonavida B. Pivotal roles of snail inhibition and RKIP induction by the proteasome inhibitor NPI-0052 in tumor cell chemoimmunosensitization. Cancer Res. 2009;69(21):8376–85. doi: 10.1158/0008-5472.CAN-09-1069. [DOI] [PubMed] [Google Scholar]
- 16.Granovsky AE, Rosner MR. Raf kinase inhibitory protein: a signal transduction modulator and metastasis suppressor. Cell Res. 2008;18(4):452–7. doi: 10.1038/cr.2008.43. [DOI] [PubMed] [Google Scholar]
- 17.Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, Sedivy JM, Kolch W. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401(6749):173–7. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- 18.Yeung KC, Rose DW, Dhillon AS, Yaros D, Gustafsson M, Chatterjee D, McFerran B, Wyche J, Kolch W, Sedivy JM. Raf kinase inhibitor protein interacts with NF-kappaB-inducing kinase and TAK1 and inhibits NF-kappaB activation. Mol Cell Biol. 2001;21(21):7207–17. doi: 10.1128/MCB.21.21.7207-7217.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eves EM, Shapiro P, Naik K, Klein UR, Trakul N, Rosner MR. Raf kinase inhibitory protein regulates aurora B kinase and the spindle checkpoint. Mol Cell. 2006;23(4):561–74. doi: 10.1016/j.molcel.2006.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beach S, Tang H, Park S, Dhillon AS, Keller ET, Kolch W, Yeung KC. Snail is a repressor of RKIP transcription in metastatic prostate cancer cells. Oncogene. 2008;27(15):2243–8. doi: 10.1038/sj.onc.1210860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corbit KC, Trakul N, Eves EM, Diaz B, Marshall M, Rosner MR. Activation of Raf-1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J Biol Chem. 2003;278(15):13061–8. doi: 10.1074/jbc.M210015200. [DOI] [PubMed] [Google Scholar]
- 22.Fu Z, Smith PC, Zhang L, Rubin MA, Dunn RL, Yao Z, Keller ET. Effects of raf kinase inhibitor protein expression on suppression of prostate cancer metastasis. J Natl Cancer Inst. 2003;95(12):878–89. doi: 10.1093/jnci/95.12.878. [DOI] [PubMed] [Google Scholar]
- 23.Fu Z, Kitagawa Y, Shen R, Shah R, Mehra R, Rhodes D, Keller PJ, Mizokami A, Dunn R, Chinnaiyan AM, Yao Z, Keller ET. Metastasis suppressor gene Raf kinase inhibitor protein (RKIP) is a novel prognostic marker in prostate cancer. Prostate. 2006;66(3):248–56. doi: 10.1002/pros.20319. [DOI] [PubMed] [Google Scholar]
- 24.Hagan S, Al-Mulla F, Mallon E, Oien K, Ferrier R, Gusterson B, Garcia JJ, Kolch W. Reduction of Raf-1 kinase inhibitor protein expression correlates with breast cancer metastasis. Clin Cancer Res. 2005;11(20):7392–7. doi: 10.1158/1078-0432.CCR-05-0283. [DOI] [PubMed] [Google Scholar]
- 25.Al-Mulla F, Hagan S, Behbehani AI, Bitar MS, George SS, Going JJ, Garcia JJ, Scott L, Fyfe N, Murray GI, Kolch W. Raf kinase inhibitor protein expression in a survival analysis of colorectal cancer patients. J Clin Oncol. 2006;24(36):5672–9. doi: 10.1200/JCO.2006.07.5499. [DOI] [PubMed] [Google Scholar]
- 26.Schuierer MM, Bataille F, Hagan S, Kolch W, Bosserhoff AK. Reduction in Raf kinase inhibitor protein expression is associated with increased Ras-extracellular signal-regulated kinase signaling in melanoma cell lines. Cancer Res. 2004;64(15):5186–92. doi: 10.1158/0008-5472.CAN-03-3861. [DOI] [PubMed] [Google Scholar]
- 27.Lorenz K, Lohse MJ, Quitterer U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature. 2003;426(6966):574–9. doi: 10.1038/nature02158. [DOI] [PubMed] [Google Scholar]
- 28.Baritaki S, Huerta-Yepez S, Cabera-Munoz M, Rivera-Pazos C, Waterman B, Yeung K, Chen H, Berenson J, Bonavida B. Unique Pattern of Raf-1 kinase Inhibitory protein (RKIP) expression in multiple myeloma (MM): RKIP overexpression and RKIP phosphorylation is common in MM tumor cells. ASH Meeting Abstracts. Blood. 2008;112(11):2729. [Google Scholar]
- 29.Campbell RA, Manyak SJ, Yang HH, Sjak-Shie NN, Chen H, Gui D, Popoviciu L, Wang C, Gordon M, Pang S, Bonavida B, Said J, Berenson JR. LAGlambda-1: a clinically relevant drug resistant human multiple myeloma tumor murine model that enables rapid evaluation of treatments for multiple myeloma. Int J Oncol. 2006;28(6):1409–17. [PubMed] [Google Scholar]
- 30.Campbell RA, Sanchez E, Steinberg JA, Baritaki S, Gordon M, Wang C, Shalitin D, Chen H, Pang S, Bonavida B, Said J, Berenson JR. Antimyeloma effects of arsenic trioxide are enhanced by melphalan, bortezomib and ascorbic acid. Br J Haematol. 2007;138(4):467–78. doi: 10.1111/j.1365-2141.2007.06675.x. [DOI] [PubMed] [Google Scholar]
- 31.Stewart AK, Fonseca R. Prognostic and therapeutic significance of myeloma genetics and gene expression profiling. J Clin Oncol. 2005;23(26):6339–44. doi: 10.1200/JCO.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 32.Yeung J, Chang H. Genomic aberrations and immunohistochemical markers as prognostic indicators in multiple myeloma. J Clin Pathol. 2008;61(7):832–6. doi: 10.1136/jcp.2007.049585. [DOI] [PubMed] [Google Scholar]
- 33.Zhan F, Hardin J, Kordsmeier B, Bumm K, Zheng M, Tian E, Sanderson R, Yang Y, Wilson C, Zangari M, Anaissie E, Morris C, Muwalla F, van Rhee F, Fassas A, Crowley J, Tricot G, Barlogie B, Shaughnessy J., Jr Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood. 2002;99(5):1745–57. doi: 10.1182/blood.v99.5.1745. [DOI] [PubMed] [Google Scholar]
- 34.Agnelli L, Fabris S, Bicciato S, Basso D, Baldini L, Morabito F, Verdelli D, Todoerti K, Lambertenghi-Deliliers G, Lombardi L, Neri A. Upregulation of translational machinery and distinct genetic subgroups characterise hyperdiploidy in multiple myeloma. Br J Haematol. 2007;136(4):565–73. doi: 10.1111/j.1365-2141.2006.06467.x. [DOI] [PubMed] [Google Scholar]
- 35.Carrasco DR, Tonon G, Huang Y, Zhang Y, Sinha R, Feng B, Stewart JP, Zhan F, Khatry D, Protopopova M, Protopopov A, Sukhdeo K, Hanamura I, Stephens O, Barlogie B, Anderson KC, Chin L, Shaughnessy JD, Jr, Brennan C, Depinho RA. High-resolution genomic profiles define distinct clinicopathogenetic subgroups of multiple myeloma patients. Cancer Cell. 2006;9(4):313–25. doi: 10.1016/j.ccr.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 36.Fonseca R, Bergsagel PL, Drach J, Shaughnessy J, Gutierrez N, Stewart AK, Morgan G, Van Ness B, Chesi M, Minvielle S, Neri A, Barlogie B, Kuehl WM, Liebisch P, Davies F, Chen-Kiang S, Durie BG, Carrasco R, Sezer O, Reiman T, Pilarski L, Avet-Loiseau H. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23(12):2210–21. doi: 10.1038/leu.2009.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huerta-Yepez S, Vega M, Escoto-Chavez SE, Murdock B, Sakai T, Baritaki S, Bonavida B. Nitric oxide sensitizes tumor cells to TRAIL-induced apoptosis via inhibition of the DR5 transcription repressor Yin Yang 1. Nitric Oxide. 2009;20(1):39–52. doi: 10.1016/j.niox.2008.08.001. [DOI] [PubMed] [Google Scholar]