

Abstract
Nitric oxide (NO) is an endogenous antibacterial agent produced by immune cells in response to pathogens. Herein, the NO fluxes necessary to reduce bacterial adhesion of different bacteria (S. aureus, methicillin-resistant S. aureus, S. epidermidis, E. faecalis, E. coli, and P. aeruginosa) were investigated to ascertain the sensitivity of these bacteria to NO. S-nitrosothiol NO donor-modified xerogels were selected as a model NO-release surface due to their extended NO-release kinetics relative to other NO donor systems. The xerogels were coated with poly(vinyl chloride) (PVC) to achieve consistent surface energy between NO-releasing and control substrates. Fibrinogen was pre-adsorbed to these materials to more accurately mimic conditions encountered in blood and promote bacteria adhesion. Nitric oxide fluxes ranging from 20–50 pmol cm−2 s−1 universally inhibited the bacterial adhesion by >80% for each strain studied. Maximum bacteria killing activity (reduced viability by 85–98%) was observed at the greatest NO payload (1700 nmol cm−2).
1. Introduction
Implanted medical devices are employed increasingly in hospital settings to treat and/or monitor a multitude of health conditions. While some devices are utilized for hours or days (e.g., catheters), some reside in a patient’s body for years (e.g., pacemakers, orthopedic screws). Regardless of implant time or location, device-associated infections impact almost all medical implants.1 For example, central venous catheters (CVCs) result in >80,000 bloodstream infections annually with significant consequences (e.g., rigorous and expensive treatment).2, 3
Implant-associated infections are the result of protein adsorption, bacterial adhesion, and subsequent colonization.4–6 The colonized bacteria excrete an extracellular matrix offering protection from the immune response and external treatment (e.g., antibiotics) in the form of a biofilm. Eradication of biofilm bacteria requires significantly greater antibiotic concentrations than killing of planktonic bacteria.7 Without complete eradication, bacteria can proliferate and recolonize the material.8 With the emergence of antibiotic-resistant strains, device removal or replacement is often the only possible treatment option.9, 10 Fortunately, the prevention and killing of adhered bacteria have been shown to decrease the incidence of implant-associated infections.4
To date, many clinically approved devices do not adequately prevent bacterial adhesion and biofilm formation.11 The development of anti-bacterial materials is thus important for addressing device-associated infections. This goal has been pursued through the development of both surface coatings that passively reduce protein/bacterial adhesion and biomaterials that actively release an antimicrobial agent.1 Passive antibacterial materials (e.g., polyethyelene glycol, quaternary ammonium-modified interfaces)12–14 have a limited sphere of influence as they only affect bacteria in direct contact with the surface. Often, this sphere/antibacterial efficacy is reduced upon protein adsorption.1 In contrast, materials that actively release antibacterial agents can impact bacteria both in contact with and in the vicinity of a surface. Antibacterial agents that have been effectively loaded into polymeric biomaterials for subsequent release include antibiotics,15, 16 antibodies,17 silver ions,18 and nitric oxide (NO).19, 20
Nitric oxide, a reactive, diatomic radical produced by immune cells by the body response to pathogens,21, 22 has received considerable attention as an antibacterial agent due to its localized action and broad spectrum activity. Materials capable of storing and releasing NO have been the focus of intense research for the goal of reducing bacterial adhesion and implant-associated infections. 23, 24 Surface NO release has been shown to influence both bacterial adhesion and biofilm formation.25, 26 Nablo and Schoenfisch reported that NO fluxes ≥30 pmol cm−2 s−1 inhibited Pseudomonas aeruginosa adhesion by 85%.20 Charville et al. subsequently examined the reduction of bacterial adhesion due to NO flux in the presence of adsorbed fibrinogen (Fg) to better mimic in vivo conditions.19 While the adsorbed fibrinogen enhanced bacterial adhesion, a NO flux-dependent reduction of bacterial adhesion was still observed for both Gram-positive and negative bacteria.
The potential to both reduce adhesion and kill adhered bacteria sets NO apart from other antibacterial agents. Hetrick and Schoenfisch previously reported the killing efficacy of NO for P. aeruginosa as a function of NO payload.27 Herein, we further these studies by investigating the NO fluxes necessary to inhibit bacterial adhesion and reduce the viability of adhered bacteria as a function of bacteria species and class. Such studies are critical for understanding the clinical potential of NO-releasing interfaces.
2. Materials and Methods
3-Mercaptopropyltrimethoxysilane (MPTMS) was purchased from Gelest (Morrisville, PA). Methyltrimethoxysilane (MTMOS), poly(vinyl chloride) (PVC), and diethylenetriamine pentaacetic acid (DTPA) were purchased from Fluka (Buchs, Switzerland). Human fibrinogen (Fg; >95% clottable, >95% purity) was obtained from Enzyme Research Laboratories (Southbend, IN). Fibrinogen was stored at −80 °C, thawed at 37 °C for 10 min, and maintained at ambient temperature. Staphylococcus aureus (ATCC #29213), methicillin-resistant Staphylococcus aureus (MRSA; ATCC #33591), Staphylococcus epidermidis (ATCC #35983), Enterococcus faecalis (ATCC #29212), Pseudomonas aeruginosa (ATCC #19143), and Escherichia coli (ATCC #53323) were obtained from American Type Culture Collection (Manassas, VA). Nitric oxide was purchased from Praxair (Danbury, CT). Nitric oxide calibration gas (26.39 ppm; balance nitrogen), nitrogen, and argon were purchased from Airgas National Welders (Raleigh, NC).
2.1. Xerogel synthesis
Mercaptosilane-modified xerogels (40% (v/v) MPTMS/MTMOS) were prepared on glass substrates as described previously by mixing 800 μL ethanol (EtOH), 480 μL MTMOS, 320 μL MPTMS, and 25 μL hydrochloric acid (HCl; 0.5 M).28 The solution was vortexed for 1 h, and allowed to age for 4 h at room temperature. Glass microscope slides (9 × 25 mm2) were sonicated in EtOH for 30 min, then dried under a stream of nitrogen. A 30 μL aliquot of the xerogel mixture was cast onto each glass slide and dried overnight under ambient conditions, then cured in a 70 °C oven for 2 d. The resulting xerogel-modified substrates were stored at room temperature until further use.
2.2. S-nitrosothiol-modified xerogels
The MPTMS-containing xerogels were nitrosated to form S-nitrosothiol NO donors by incubating the substrates in 0.5 M HCl containing a 10x molar excess of nitrite (versus moles of thiol) and 100 μM DTPA for 3 h.28 Of note, xerogels were kept on ice and shielded from light during nitrosation to prevent pre-mature NO donor decomposition. Slides were washed with 100 μM DTPA and stored at −20 °C in the dark until further use. Non-nitrosated xerogels were used as control (non-NO-releasing) interfaces.
2.3. Poly(vinyl chloride) coating
Poly(vinyl chloride) (1.0 g) was dissolved in tetrahydrofuran (THF; 10 mL). Approximately 300 μL of the PVC/THF solution was spin-coated onto control and S-nitrosothiol–modified xerogels using a CHEMAT Technology KW-4A Precision Spin-Coater (Northridge, CA) set to spin at 3.0 krpm. The quality of the coating process was evaluated using light microscopy. Only those PVC-coated samples without visual fault (e.g., cracking, unusual roughness features) were used for further studies. The PVC-coated xerogels were stored at −20 °C until further use.
2.4. Bacterial adhesion
Bacteria were cultured in tryptic soy broth (TSB) at 37 °C, pelleted by centrifugation, rinsed with sterile water, resuspended in a solution of phosphate buffered saline (PBS; 10 mM, pH 7.4) with 15% (v/v) glycerol, and stored at −80 °C. Secondary cultures were grown from an aliquot of the frozen stock in TSB overnight. A 1 mL portion of this solution was inoculated into 100 mL of TSB and grown to ~108 colony-forming units (CFU) per mL as measured by optical density. The resulting culture was collected by centrifugation (4500 g, 5 min) and resuspended in PBS.
To reach the desired NO flux, the PVC-coated NO-releasing xerogels were incubated at 37 °C for 0.5–96 h. The substrates were then immersed in 4 mL of a 20 μg/mL solution of Fg in PBS (pH 7.4) for 90 min at 37 °C (gentle shaking). A concentration of 20 μg/mL Fg enabled the formation of a protein monolayer (and not protein agglomerates and/or multilayers) on the PVC.29 The PVC-coated xerogels were then removed from the Fg solution and immediately immersed in 4 mL of a bacterial suspension (108 CFU/mL) at 37 °C for 60 min.
2.5. Optical microscopy for imaging of adhered bacteria
Following incubation in the bacterial suspension, the PVC-coated xerogel substrates were removed, immersed in water to remove loosely adhered bacteria, and dried with a stream of air. Adhered bacteria were imaged via phase contrast optical microscopy at 20x magnification using a Zeiss Axiovert 200 Inverted Microscope equipped with a Zeiss Axiocam. At least three different PVC-coated xerogel substrates were imaged for each NO flux. Five randomly selected regions on each slide were used to calculate an average bacterial surface coverage. The images were digitally processed by thresholding to make opaque cells contrast sharply with the transparent xerogel substrates. The relative surface coverage was translated from the number of dark pixels in the thresholded image, and normalized relative to controls (i.e., non-NO-releasing) which were scored as 1.
2.6. Adhered bacterial viability
Upon removal from the bacterial suspension, the substrates were immersed in sterile water to remove loosely adhered bacteria, and then transferred to 4 mL of PBS for 0, 6, 12, or 24 h. The PBS was supplemented with 0.5% (v/v) TSB for S. aureus, MRSA, S. epidermidis, and E. coli strains to ensure viability for 24 h. After the appropriate incubation period, the slides were gently rinsed with sterile water to remove loosely adhered bacteria. The PVC layer was then peeled away from the xerogel substrate, placed into 1 mL of PBS, and then sonicated for 15 min at 40 kHz to remove adhered bacteria.17, 27 Removal of bacteria cells via sonication was confirmed by optical microscopy of the PVC layer. After sonication, the solution was serially diluted and plated onto tryptic soy agar (TSA) nutrient plates (incubated at 37 °C). Bacterial viability was determined by counting the colonies on each plate using an IUL Flash & Go (Neutec Group, Farmingdale, NY). This plate counting method has an inherent limit of detection of 2.5 × 103 CFU/mL.30
2.7. Contact angle measurements
Static water contact angle measurements of fibrinogen-adsorbed PVC-coated, PVC-coated, and bare control and NO-releasing xerogels were acquired with a KSV Instruments Cam 200 Optical Contact Angle Meter (Helsinki, Finland). Three droplets (~10 μL) of 18.2 MΩ water were placed onto the substrate (n ≥ 3) at room temperature with enumeration of the receding angle.
2.8. Nitric oxide release measurements
Nitric oxide release was measured continuously in PBS using a Sievers 280i Chemiluminescence Nitric Oxide Analyzer (NOA) (Boulder, CO).31 Calibration of the NOA was performed for each analysis using ambient air passed through a Sievers NO zero filter and a 26.39 ppm NO gas standard (balance N2). To ensure measurement reproducibility, NO-releasing xerogels were immersed in 30 mL of deoxygenated PBS shielded from light. Liberated NO was carried to the analyzer in a nitrogen stream (200 mL min−1). Temperature control was maintained through the use of a water bath at 37 °C.
3. Results and Discussion
3.1. Material characterization
S-Nitrosothiol NO-donor-modified xerogels28 were chosen for this study to enable investigation of a large range of NO-surface fluxes. S-Nitrosothiols provide longer NO-release durations compared to N-diazeniumdiolate NO donors when incorporated within xerogel films.20, 28, 32, 33 The desired NO flux was achieved by pre-incubating the xerogels at 37 °C at set periods. To control for deviations in surface energy of the control and nitrosated xerogels (as measured by contact angles of 70 ± 1 and 54 ± 3°, respectively) that would impact bacterial adhesion,28 the xerogels were modified with a PVC coating. The PVC coatings easily foul with protein and bacteria, and therefore allow for systematic examination of bacterial adhesion.34 Contact angles for controls and NO-releasing xerogels were identical (~89 ± 4°) following PVC-modification. Neither delamination nor degradation of the PVC or xerogel layers was observed for the PVC-coated xerogels regardless of incubation period.
Initial experiments revealed that S. aureus, MRSA, and E. coli did not readily adhere to control PVC-coated substrates (<0.10 relative surface coverage). Low absolute adhesion for these strains would make accurate measurements of the relative adhesion to NO-releasing substrates difficult due to the error inherent in the optical microscopy evaluation. To enhance bacterial adhesion and mimic in vivo biofouling more closely, PVC-coated slides were pre-incubated in fibrinogen (Fg) solutions prior to bacteria exposure. Contact angles of Fg-adsorbed PVC-coated control and NO-releasing xerogels were both 55 ± 5° (decreased from 89°), confirming Fg adsorption and the expected surface alteration. Charville et al. previously reported that NO-release does not influence Fg adsorption onto PVC interfaces.19 Soaking the Fg-adsorbed surfaces in PBS for 24 h had no effect on the contact angle indicating that the Fg layer is not weakly bound.
The NO-release properties of the bare and PVC-coated S-nitrosothiol xerogels are provided in Table 1. As expected, the PVC topcoat did not significantly alter the NO-release properties of the xerogels since the NO release is thermally triggered and NO readily diffuses through PVC.28, 34 The substrates released NO for at least 14 d at low NO fluxes (e.g., 0.16 pmol cm−2 s−1). The average NO flux employed for the bacteria studies ranged from 0.5–50 pmol cm−2 s−1, with 50 pmol cm−2 s−1 selected as the greatest flux due to unpreventable loss of NO during the pre-incubation of the xerogels in Fg prior to bacteria exposure. Prior to immersion in bacteria solutions, the greatest NO flux measured for the PVC-coated NO-releasing xerogels was 260 pmol cm−2 s−1 (Table 1). The NO flux changes by a large degree initially due to the pseudo first-order NO-release kinetics common for S-nitrosothiol-modified xerogels.28
Table 1.
Nitric oxide-release properties of bare and PVC-coated 40% S-nitrosothiol-modified xerogels. Data are mean ± standard deviation for at least three separately prepared substrates.
| NO-release properties | 40% MPTMS/MTMOS xerogel | PVC-coated 40% MPTMS/MTMOS xerogel |
|---|---|---|
| tmax (min)a | 1.1 ± 0.2 | 1.2 ± 0.2 |
| [NO]max (pmol cm−2 s−1) | 290 ± 70 | 260 ± 60 |
| [NO]1 h (pmol cm−2 s−1) | 64 ± 10 | 58 ± 8 |
| [NO]5 h (pmol cm−2 s−1) | 26 ± 5 | 22 ± 4 |
| [NO]12 h (pmol cm−2 s−1) | 9 ± 1 | 8 ± 1 |
| [NO]24 h (pmol cm−2 s−1) | 3.4 ± 0.5 | 3.2 ± 0.5 |
| [NO]48 h (pmol cm−2 s−1) | 1.2 ± 0.1 | 1.1 ± 0.1 |
| [NO]96 h (pmol cm−2 s−1) | 0.5 ± 0.1 | 0.5 ± 0.1 |
| Total NO (μmol cm−2) | 2.6 ± 0.4 | 2.5 ± 0.3 |
Time to reach maximum NO flux
3.2. Bacterial adhesion
The degree of bacterial adhesion to PVC-coated control and NO-releasing xerogels was examined using large bacteria concentrations (~108 CFU/mL). Although such concentrations are not biologically relevant for most implants, a starting concentration of ~108 CFU/mL of bacteria allowed for larger absolute surface coverage for all bacterial strains, a requirement for studying differences in bacterial adhesion via optical microscopy. Using the NO-release data provided in Table 1, S-nitrosothiol-modified xerogels were pre-incubated at 37 °C for periods between 30 min and 4 d to achieve a wide-range of average NO fluxes over the subsequent 1 h bacteria assay (0.50 ± 0.08, 1.0 ± 0.1, 2.5 ± 0.4, 5.0 ± 0.8, 10 ± 2, 20 ± 4, 35 ± 5, and 50 ± 10 pmol cm−2 s−1). The resulting NO fluxes allowed for investigation of NO’s anti-bacterial adhesion properties. Increased error was noted for the larger NO fluxes due to the pseudo first-order NO-release kinetics that rapidly change at early time points (e.g., NO flux is reduced by >50% between 1 and 5 h).
Based on prior adhesion results,19, 20, 27 we predicted a negative correlation between bacterial adhesion and NO flux. Indeed, a NO flux-dependent decrease in bacterial adhesion was observed for all six bacterial strains investigated (Figure 1) relative to controls (with 1.0 indicating identical adhesion to the control substrates). S. aureus, MRSA, E. faecalis, and E. coli showed a greater susceptibility to NO from 0–20 pmol cm−2 s−1 with little to no additional reduction in bacterial adhesion (surface coverage) at increased NO fluxes. This maximum plateau in reduced adhesion corroborates a previous report by Nablo and Schoenfisch where NO release resulted in a large initial reduction in bacterial adhesion but was followed by little additional effect beyond a certain NO flux threshold.20 In contrast, P. aeruginosa and S. epidermidis required greater NO fluxes to inhibit adhesion by 50% (20 and 35 pmol cm−2 s−1, respectively) and 80% (35 and 50 pmol cm−2 s−1, respectively). While increased NO fluxes were necessary to reduce adhesion for these particular strains, the NO release still decreased adhesion by >80%, analogous to the relative inhibition observed for other strains (Table 2).
Figure 1.

The NO flux-dependent relative adhesion of A) S. aureus, B) MRSA, C) S. epidermidis, D) E. faecalis, E) E. coli, and F) P. aeruginosa to pre-adsorbed Fg PVC-coated xerogels after 1 h of exposure. A relative adhesion of 1.0 represents the adhesion of the strain to control (i.e., non-NO-releasing) substrates. Data are mean ± standard deviation.
Table 2.
Linear regression analysis and NO flux required to inhibit adhesion of each bacteria strain by 50 and 80% relative to control (i.e., non-NO-releasing) surfaces. Data are mean ± standard deviation.
| Bacteria strain | Gram class | Linear regression slope (relative adhesion/pmol cm−2 s−1)a | r2 | NO flux required to reduce adhesion by 50% (pmol cm−2 s−1) | NO flux required to reduce adhesion by 80% (pmol cm−2 s−1) | Absolute surface coverage to controls (%) |
|---|---|---|---|---|---|---|
| S. aureus | + | −0.043 ± 0.005 | 0.922 | 10 | 20 | 17.1 ± 2.7 |
| MRSA | + | −0.037 ± 0.005 | 0.914 | 10 | 20 | 16.8 ± 2.3 |
| S. epidermidis | + | −0.017 ± 0.002 | 0.939 | 35 | 50 | 14.7 ± 1.9 |
| E. faecalis | + | −0.043 ± 0.004 | 0.954 | 10 | 20 | 13.5 ± 2.7 |
| E. coli | − | −0.040 ± 0.004 | 0.953 | 10 | 20 | 19.5 ± 3.5 |
| P. aeruginosa | − | −0.036 ± 0.003 | 0.984 | 20 | 35 | 47.2 ± 4.2 |
Slopes calculated from NO fluxes between 0–20 pmol cm−2 s−1
Linear regressions of the relative bacteria adhesion (measured as surface coverage) as a function of NO flux were performed from 0–20 pmol cm−2 s−1 to compare the relative susceptibility of each bacterial strain to NO (Table 2). This range was chosen because all strains showed a linear (r2 = 0.91–0.98) reduction in adhesion, allowing for direct comparison between strains. This linearity only held true until ~80% reduction in bacterial adhesion. All of the strains studied except S. epidermidis were characterized as having similar linear regression slopes, ranging from −0.036 to −0.043 relative bacterial adhesion/pmol cm−2 s−1 of NO. Slight variations in slope were independent of the class of bacteria (Gram-positive or Gram-negative) and likely an attribute of the broad-spectrum activity of NO arising from multiple mechanisms of action on bacteria (e.g., nitrosative and oxidative stress).35 In our study, S. epidermidis was the only bacterium with a relatively low susceptibility to NO with respect to bacterial adhesion (slope of −0.017). Charville et al. reported similar behavior when investigating the role of Fg and NO release on bacterial adhesion,19 S. epidermidis is indeed unique in that it has Fg-binding proteins such as serine-aspartate repeat G (SdrG) present on its surface.36 While S. aureus also has Fg-binding proteins (e.g., Clumping Factor A),37 the dissociation constant (KD) of SdrG is two orders of magnitude lower than Clumping Factor A, indicating a significantly increased binding strength.38 The large affinity of SdrG to Fg may decrease the susceptibility of S. epidermidis to the anti-adhesive effects of surface-derived NO. Despite the relatively low efficacy of NO in preventing S. epidermidis adhesion, NO release at fluxes >20 pmol cm−2 s−1 further diminished the bacterial surface coverage with an 81% reduction in adhesion compared to control surfaces for a flux of 50 pmol cm−2 s−1. Greater NO fluxes may further inhibit S. epidermidis adhesion; however, the maximum average NO flux that could be achieved over the 1 h study using the S-nitrosothiol-modified xerogels was limited to 50 pmol cm−2 s−1.
3.3. Bacteria surface viability
Reduction of bacterial adhesion is a crucial initial step in the design of antibacterial interfaces that prevent colonization on implantable devices. However, the most effective antibacterial surfaces should both reduce bacterial adhesion and kill any bacteria that do adhere to minimize bacteria proliferation and biofilm formation.39 While our prior work with P. aeruginosa demonstrated reduced viability of adhered bacteria to a NO-releasing interface in a dose-dependent manner,27 this study was limited to one bacteria strain. As such, the effect of NO release on the viability of adhered bacteria was not systematically investigated with respect to bacterial strains. Based on prior literature,19, 27, 40–42 we hypothesized that the viability of adhered bacteria would be reduced in a NO dose-dependent manner regardless of strain due to NO’s broad spectrum action and modes of killing.1, 43, 44 The effect of total NO payload (i.e., cumulative NO release) over time was thus investigated against as a function of bacteria strain to evaluate the potential for surface-derived NO to kill adhered bacteria broadly. After the 1 h bacterial adhesion assay, the substrates were incubated for 0, 6, 12, or 24 h in PBS (E. faecalis and P. aeruginosa) or 0.5% TSB in PBS (S. aureus, MRSA, S. epidermidis, and E. coli) to provide bacteriostatic conditions. As not all bacteria survived in PBS for 24 h due to lack of nutrients, the 0.5% TSB was required for some strains to preserve bacterial viability. The viability was confirmed by quantifying planktonic bacteria of each strain in PBS and 0.5% TSB in PBS. The average time-dependent NO fluxes and payloads, provided in Table 3, ranged from 0.5–50 pmol cm−2 s−1 and 38–1700 nmol cm−2, respectively, over the 24 h of incubation. Since the NO release followed pseudo first-order kinetics, substrates with an initial average NO flux of 20, 35, and 50 pmol cm−2 s−1 delivered 48, 53, and 77% of their 24 h NO payload in the first 6 h, respectively (Table 3). In contrast, substrates with lower initial average NO fluxes (e.g., 0.5 and 1.0 pmol cm−2 s−1) delivered NO in a more sustained manner, with only ~25% of the 24 h NO payload released during the first 6 h.
Table 3.
Nitric oxide payloads (nmol cm−2) at 6, 12, and 24 h after the 1 h adhesion assay for initial average NO fluxes from 0.5–50 pmol cm−2 s−1 NO fluxes examined. Data are mean ± standard deviation.
| Initial NO flux (pmol cm−2 s−1) | NO payload (nmol cm−2)
|
||
|---|---|---|---|
| 6 h | 12 h | 24 h | |
| 0.5 | 9.5 ± 0.8 | 19 ± 2 | 38 ± 2 |
| 1.0 | 17 ± 2 | 33 ± 3 | 66 ± 4 |
| 2.5 | 40 ± 3 | 72 ± 5 | 105 ± 9 |
| 5.0 | 90 ± 9 | 140 ± 10 | 240 ± 20 |
| 10 | 150 ± 20 | 250 ± 20 | 370 ± 20 |
| 20 | 270 ± 20 | 410 ± 30 | 550 ± 50 |
| 35 | 390 ± 50 | 560 ± 50 | 740 ± 60 |
| 50 | 1300 ± 200 | 1500 ± 200 | 1700 ± 200 |
To ensure that only bacteria adhered to the PVC-coated xerogel and not to the sides or back of the glass substrates were enumerated, the outer PVC layer was physically removed from the glass slide and sonicated. These bacteria were then plated onto TSA plates and enumerated to quantify viable bacteria.27 Optical microscopy confirmed that this sonication step removed >93% of the adhered bacteria and thus the viability quantification was an accurate measurement. Relative adhered bacterial viability was calculated as a function of time by dividing the viable bacteria concentration at x = 6, 12, or 24 h by the viable bacteria at t = 0 for a given bacteria strain and NO flux (Equation 1). A relative viability of 1 would thus indicate that the NO release
| (1) |
had zero effect on the killing of adhered bacteria. Of note, the viability of bacteria adhered to control substrates was constant (negligible death) between the 0 and 24 h time points.
To ascertain whether NO was solely preventing adhesion or also killing adhered bacteria during the 1 h adhesion period, relative adhesion and relative viability were compared for the S. aureus and E. coli strains (Figure 2). The relative surface coverage as determined by microscopy and relative adhesion of viable bacteria were generally comparable at all NO fluxes after a 1 h exposure. For example, S. aureus relative viability at substrates with an average NO flux of 50 pmol cm−2 s−1 was ~0.15 that of control substrates, in line with the ~0.10 relative adhesion observed with microscopy (Figure 2A). The positive agreement between surface coverage and viable bacteria counts immediately following the adhesion assay indicates that surface NO release initially only influences bacterial adhesion and not adhered bacterial cell death.
Figure 2.
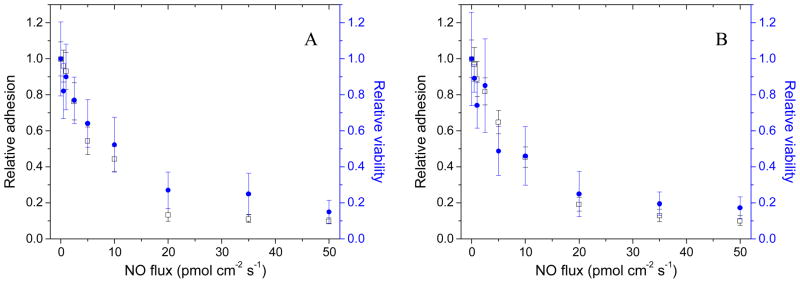
The NO flux-dependent relative adhesion (open) and viability (filled) immediately following the 1 h adhesion period for A) S. aureus and B) E. coli to pre-adsorbed Fg PVC-coated xerogels. A relative adhesion or relative viability of 1.0 represents the adhesion or viability of the strain to control (i.e., non-NO-releasing) substrates. Data are mean ± standard error of the mean.
While NO works primarily to prevent bacterial adhesion initially (during ~1 h), NO uniquely kills adherent bacteria at longer exposure times making it an unusual antibacterial agent. This time-dependent viability of bacteria adhered to the NO-releasing surfaces is provided in Figure 3 and Tables 4, 5, and 6 for 6, 12, and 24 h NO-release conditions. The reduction in adhered bacteria viability was dependent on NO payload for all strains, albeit time dependent. For example, nearly all strains (MRSA, S. epidermidis, E. faecalis, E. coli, and P. aeruginosa) exhibited reduced viability at NO-releasing substrates at 6 h. Unexpectedly, the viability of adhered S. aureus was minimal prior to the 24 h time point (Table 6). As indicated by the data, this strain of S. aureus may require a minimal NO threshold to induce killing. S. aureus is known to resist NO via metabolic activities and as such killing may require greater NO concentrations, although it is uncertain why MRSA was not affected in this manner.45
Figure 3.

Relative viability of S. aureus, MRSA, S. epidermidis, E. faecalis, E. coli, and P. aeruginosa at 3 different initial NO fluxes (1.0, 10, and 50 pmol cm−2 s−1) at incubation times of 6, 12, and 24 h.
Table 4.
Relative viability of bacteria adhered to NO-releasing surfaces after 6 h incubation in PBS or PBS with 0.5% TSB (bacteriostatic conditions). A relative viability of 1.00 represents the change in viability from t = 0 h. Data are mean ± standard error of the mean, representing a minimum of three separate experiments.
| Bacteria strain | Initial average NO flux (pmol cm−2 s−1)
|
|||||||
|---|---|---|---|---|---|---|---|---|
| 0.5 | 1.0 | 2.5 | 5.0 | 10 | 20 | 35 | 50 | |
| S. aureus | 1.05 ± 0.19a | 1.06 ± 0.19a | 0.85 ± 0.17 | 0.69 ± 0.28 | 0.61 ± 0.23 | 0.84 ± 0.31 | 0.86 ± 0.21 | 0.87 ± 0.23 |
| MRSA | 0.93 ± 0.28 | 0.93 ± 0.30 | 0.46 ± 0.17 | 0.39 ± 0.12 | 0.42 ± 0.16 | 0.46 ± 0.15 | 0.45 ± 0.18 | 0.32 ± 0.12 |
| S. epidermidis | 1.18 ± 0.30a | 0.84 ± 0.21 | 0.75 ± 0.22 | 1.01 ± 0.30a | 0.41 ± 0.18 | 0.28 ± 13 | 0.25 ± 0.10 | 0.24 ± 0.08 |
| E. faecalis | 0.96 ± 0.30 | 0.94 ± 0.20 | 0.42 ± 0.05 | 0.24 ± 0.07 | 0.22 ± 0.06 | 0.27 ± 10 | 0.33 ± 0.10 | 0.29 ± 0.09 |
| E. coli | 1.16 ± 0.10a | 1.16 ± 0.15a | 0.51 ± 0.17 | 0.21 ± 0.10 | 0.22 ± 0.08 | 0.27 ± 0.08 | 0.17 ± 0.05 | 0.16 ± 0.04 |
| P. aeruginosa | 0.92 ± 0.06 | 1.10 ± 0.15a | 0.41 ± 0.18 | 0.20 ± 0.07 | 0.18 ± 0.06 | 0.20 ± 0.08 | 0.06 ± 0.02 | 0.05 ± 0.01 |
Relative viability >1 indicates bacteria proliferation
Table 5.
Relative viability of bacteria adhered to NO-releasing surfaces after 12 h incubation in PBS or PBS with 0.5% TSB (bacteriostatic conditions). A relative viability of 1.00 represents the change in viability from t = 0 h. Data are mean ± standard error of the mean, representing a minimum of three separate experiments.
| Bacteria strain | Initial average NO flux (pmol cm−2 s−1)
|
|||||||
|---|---|---|---|---|---|---|---|---|
| 0.5 | 1.0 | 2.5 | 5.0 | 10 | 20 | 35 | 50 | |
| S. aureus | 0.69 ± 0.06 | 0.61 ± 0.25 | 0.65 ± 0.09 | 0.39 ± 0.09 | 0.54 ± 0.13 | 0.56 ± 0.16 | 0.60 ± 0.15 | 0.49 ± 0.06 |
| MRSA | 0.75 ± 0.18 | 0.82 ± 0.26 | 0.34 ± 0.13 | 0.34 ± 0.14 | 0.23 ± 0.08 | 0.54 ± 0.13 | 0.28 ± 0.10 | 0.21 ± 0.06 |
| S. epidermidis | 0.72 ± 0.20 | 0.75 ± 0.22 | 0.40 ± 0.15 | 0.57 ± 0.17 | 0.34 ± 0.11 | 0.24 ± 0.09 | 0.20 ± 0.08 | 0.20 ± 0.08 |
| E. faecalis | 0.91 ± 0.04 | 0.61 ± 0.10 | 0.34 ± 0.11 | 0.20 ± 0.08 | 0.21 ± 0.07 | 0.21 ± 0.06 | 0.16 ± 0.05 | 0.11 ± 0.03 |
| E. coli | 0.95 ± 0.22 | 0.73 ± 0.22 | 0.38 ± 0.13 | 0.16 ± 0.04 | 0.14 ± 0.05 | 0.07 ± 0.02 | 0.10 ± 0.03 | 0.08 ± 0.02 |
| P. aeruginosa | 0.57 ± 0.02 | 0.46 ± 0.10 | 0.33 ± 0.05 | 0.25 ± 0.12 | 0.16 ± 0.06 | 0.08 ± 0.02 | 0.05 ± 0.02 | 0.04 ± 0.01 |
Table 6.
Relative viability of bacteria adhered to NO-releasing surfaces after 24 h incubation in PBS or PBS with 0.5% TSB (bacteriostatic conditions). A relative viability of 1.00 represents the change in viability from t = 0 h. Data are mean ± standard error of the mean, representing a minimum of three separate experiments.
| Bacteria strain | Initial average NO flux (pmol cm−2 s−1)
|
|||||||
|---|---|---|---|---|---|---|---|---|
| 0.5 | 1.0 | 2.5 | 5.0 | 10 | 20 | 35 | 50 | |
| S. aureus | 0.67 ± 0.17 | 0.66 ± 0.19 | 0.69 ± 0.23 | 0.43 ± 0.16 | 0.25 ± 0.12 | 0.38 ± 0.12 | 0.22 ± 0.07 | 0.15 ± 0.06 |
| MRSA | 0.66 ± 0.13 | 0.72 ± 0.18 | 0.27 ± 0.11 | 0.25 ± 0.09 | 0.21 ± 0.06 | 0.18 ± 0.06 | 0.19 ± 0.07 | 0.15 ± 0.04 |
| S. epidermidis | 0.80 ± 0.12 | 0.80 ± 0.19 | 0.38 ± 0.12 | 0.45 ± 0.16 | 0.34 ± 0.13 | 0.20 ± 0.08 | 0.15 ± 0.05 | 0.14 ± 0.04 |
| E. faecalis | 0.83 ± 0.07 | 0.50 ± 0.20 | 0.34 ± 0.02 | 0.21 ± 0.03 | 0.23 ± 0.02 | 0.22 ± 0.04 | 0.13 ± 0.05 | 0.09 ± 0.02 |
| E. coli | 0.76 ± 0.11 | 0.36 ± 0.07 | 0.36 ± 0.08 | 0.14 ± 0.05 | 0.11 ± 0.04 | 0.08 ± 0.02 | 0.09 ± 0.03 | 0.05 ± 0.01 |
| P. aeruginosa | 0.66 ± 0.03 | 0.34 ± 0.04 | 0.29 ± 0.11 | 0.13 ± 0.04 | 0.14 ± 0.04 | 0.04 ± 0.01 | 0.04 ± 0.01 | 0.02 ± 0.004 |
The lowest 24 h NO payload investigated (38 nmol cm−2) eradicated 17–34% of the adhered bacteria regardless of bacteria species. Conversely, the greatest NO payload (1700 nm cm−2) caused the maximal reduction in adhered bacteria viability at 24 h exposure to substrates with an initial NO flux of 50 pmol cm−2 s−1 were 85, 85, 86, 92, 95, and 98% for S. aureus, MRSA, S. epidermidis, E. faecalis, E. coli, and P. aeruginosa, respectively (Table 6). Therefore, substrates with an initial NO flux of 50 pmol cm−2 s−1 delivering a payload of 1700 nmol cm−2 over 24 h inhibited adhesion of all strains by >80% with concomitant eradication of ≥85% of the adhered bacteria. After the 1 h adhesion period, the initial NO flux of 50 pmol cm−2 s−1 resulted in a bacterial viability of 5 × 105 – 1 × 106 CFU cm−2 for each of the six strains examined. As such, enough viable bacteria were present that could still colonize the material.8 Of note, significantly larger concentrations of bacteria were employed in this study compared to what might be encountered clinically.
The NO payloads necessary to decrease the adhered bacteria viability by 50 and 80% after 24 h are provided in Table 7. Cumulative NO release levels of 66–240 and 240–1700 nmol cm−2 were required to reduce adhered bacteria viability by 50 and 80%, respectively. P. aeruginosa and E. coli were the most vulnerable strains with respect to NO-based toxicity, a result that was previously reported for planktonic bacteria.46 Importantly, the amount of bacteria killed is not proportional to the amount of NO released.
Table 7.
The required surface-derived total NO payload to decrease adhered bacteria viability by 50 and 80% after 24 h incubation in bacteriostatic media. Data are mean ± standard deviation.
| NO payload that reduced adhered bacteria viability by 50% (nmol cm−2) | NO payload that reduced adhered bacteria viability by 80% (nmol cm−2) | |
|---|---|---|
| S. aureus | 240 ± 20 | 1700 ± 200 |
| MRSA | 105 ± 9 | 550 ± 50 |
| S. epidermidis | 105 ± 9 | 550 ± 50 |
| E. faecalis | 66 ± 4 | 740 ± 60 |
| E. coli | 66 ± 4 | 240 ± 20 |
| P. aeruginosa | 66 ± 4 | 240 ± 20 |
The rate of the total NO delivery also affected bacteria viability. While the cumulative NO payloads of xerogels with initial NO fluxes of 2.5 and 0.5 pmol cm−2 s−1 were approximately equivalent at 6 and 24 h, respectively (38–40 nmol cm−2, Table 3), E. faecalis viability at 6 h to surfaces with the greater initial NO flux (0.42 ± 0.05) was significantly lower than the viability at 24 h to surfaces with an initial NO flux of 0.5 pmol cm−2 s−1 (0.83 ± 0.07). This data suggests the bactericidal properties of NO-releasing surfaces are dependent on the NO payload, rate of delivery, and duration of NO release.
While the NO payloads used in this study were bactericidal to multiple strains, the cytotoxicity to mammalian cells represents an important consideration for future therapeutic potential. Nablo and Schoenfisch previously reported that NO-releasing PVC-coated xerogels were only slightly cytotoxic to L929 fibroblasts after 24 h incubation after an initial NO flux of ~50 pmol cm−2 s−1.47 These previous results reveal that NO fluxes capable of greatly reducing bacterial adhesion and viability typically avoid cytotoxic effects against mammalian cells.
4. Conclusion
Herein, the adhesion of S. aureus, MRSA, S. epidermidis, E. faecalis, E. coli, and P. aeruginosa at pre-adsorbed Fg interfaces was investigated to understand the influence of NO release adhered bacteria viability. An average NO flux from 20–50 pmol cm−2 s−1 over a 1 h exposure period decreased bacterial adhesion by >80% regardless of bacterial strain. With the exception of S. epidermidis, the adhesion of Gram-positive and Gram-negative bacteria were equally affected by surface-derived NO release. S. epidermidis required significantly greater NO fluxes (50 pmol cm−2 s−1) to inhibit adhesion to the level observed for other strands (>80%) relative to control xerogels. S. epidermidis’ Fg-binding membrane protein affinity to the adsorbed Fg likely influences both general adhesion and NO’s decreased anti-adhesive properties. We are currently testing this hypothesis with other S. epidermidis strains. Regardless of bacteria species, the number of viable bacteria adhered to the NO-releasing substrates decreases in a dose-dependent manner, but only after a set residence time. In other words, NO is not biocidal immediately upon surface adhesion. Future experiments must optimize the NO concentration (flux) and NO-release kinetics to maximize both antibacterial adhesion and biocidal activity, but minimize toxicity to mammalian cells. Of course, the ultimate test will require thorough in vivo evaluation with biofilm and infection models.25
Acknowledgments
The authors acknowledge research support from the National Institutes of Health (NIH Grant EB000708).
References
- 1.Hetrick EM, Schoenfisch MH. Chem Soc Rev. 2006;35:780–789. doi: 10.1039/b515219b. [DOI] [PubMed] [Google Scholar]
- 2.Mermel LA. Ann Intern Med. 2000;132:391–402. doi: 10.7326/0003-4819-132-5-200003070-00009. [DOI] [PubMed] [Google Scholar]
- 3.Walz JM, Memtsoudis SG, Heard SO. J Intensive Care Med. 2010;25:131–138. doi: 10.1177/0885066609358952. [DOI] [PubMed] [Google Scholar]
- 4.Rodrigues LR. In: Bacterial Adhesion: Chemistry, Biology and Physics. Linke D, Goldman A, editors. Vol. 715. Springer-Verlag Berlin; Berlin: 2011. pp. 351–367. [Google Scholar]
- 5.Pascual A. Clin Microbiol and Infec. 2002;8:256–264. doi: 10.1046/j.1469-0691.2002.00418.x. [DOI] [PubMed] [Google Scholar]
- 6.Emmerson M. New Horiz-Sci Pract Acute Med. 1998;6:S3–S10. [PubMed] [Google Scholar]
- 7.Katsikogianni M, Missirlis YF. Eur Cell Mater. 2004;8:37–57. doi: 10.22203/ecm.v008a05. [DOI] [PubMed] [Google Scholar]
- 8.Busscher HJ, van der Mei HC, Subbiahdoss G, Jutte PC, van den Dungen JJAM, Zaat SAJ, Schultz MJ, Grainger DW. Science Translational Medicine. 2012;4:153rv110. doi: 10.1126/scitranslmed.3004528. [DOI] [PubMed] [Google Scholar]
- 9.Ceri H, Olson ME, Stremick C, Read RR, Morck D, Buret A. J Clin Microbiol. 1999;37:1771–1776. doi: 10.1128/jcm.37.6.1771-1776.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams I, Venables WA, Lloyd D, Paul F, Critchley I. Microbiology-Uk. 1997;143:2407–2413. doi: 10.1099/00221287-143-7-2407. [DOI] [PubMed] [Google Scholar]
- 11.Estivill D, Arias A, Torres-Lana A, Carrillo-Munoz AJ, Arevalo MP. J Microbiol Methods. 2011;86:238–242. doi: 10.1016/j.mimet.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 12.Yao C, Li XS, Neoh KG, Shi ZL, Kang ET. J Membr Sci. 2008;320:259–267. [Google Scholar]
- 13.Thebault P, de Givenchy ET, Geribaldi S, Levy R, Vandenberghe Y, Guittard F. J Fluor Chem. 2010;131:592–596. [Google Scholar]
- 14.Carpenter AW, Worley BV, Slomberg DL, Schoenfisch MH. Biomacromolecules. 2012;13:3334–3342. doi: 10.1021/bm301108x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson EM, Noble ML, Garty S, Ma HY, Bryers JD, Shen TT, Ratner BD. Biomaterials. 2009;30:5675–5681. doi: 10.1016/j.biomaterials.2009.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schierholz JM, Steinhauser H, Rump AFE, Berkels R, Pulverer G. Biomaterials. 1997;18:839–844. doi: 10.1016/s0142-9612(96)00199-8. [DOI] [PubMed] [Google Scholar]
- 17.Rojas IA, Slunt JB, Grainger DW. J Control Release. 2000;63:175–189. doi: 10.1016/s0168-3659(99)00195-9. [DOI] [PubMed] [Google Scholar]
- 18.Monteiro DR, Gorup LF, Takamiya AS, Ruvollo-Filho AC, Camargo ERd, Barbosa DB. Int J Antimicro Ag. 2009;34:103–110. doi: 10.1016/j.ijantimicag.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 19.Charville GW, Hetrick EM, Geer CB, Schoenfisch MH. Biomaterials. 2008;29:4039–4044. doi: 10.1016/j.biomaterials.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nablo BJ, Schoenfisch MH. Biomacromolecules. 2004;5:2034–2041. doi: 10.1021/bm049727w. [DOI] [PubMed] [Google Scholar]
- 21.Mannick JB. Proc Am Thorac Soc. 2006;3:161–165. doi: 10.1513/pats.200505-048BG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wink DA, Mitchell JB. Free Radic Biol Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 23.Riccio DA, Schoenfisch MH. Chem Soc Rev. 2012;41:3731–3741. doi: 10.1039/c2cs15272j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carpenter AW, Schoenfisch MH. Chem Soc Rev. 2012;41:3742–3752. doi: 10.1039/c2cs15273h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nablo BJ, Prichard HL, Butler RD, Klitzman B, Schoenfisch MH. Biomaterials. 2005;26:6984–6990. doi: 10.1016/j.biomaterials.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 26.Holt J, Hertzberg B, Weinhold P, Storm W, Schoenfisch M, Dahners L. J Orthop Trauma. 2011;25:432–437. doi: 10.1097/BOT.0b013e3181f9ac8a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hetrick EM, Schoenfisch MH. Biomaterials. 2007;28:1948–1956. doi: 10.1016/j.biomaterials.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Riccio DA, Dobmeier KP, Hetrick EM, Privett BJ, Paul HS, Schoenfisch MH. Biomaterials. 2009;30:4494–4502. doi: 10.1016/j.biomaterials.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Young BR, Pitt WG, Cooper SL. J Colloid Interface Sci. 1988;125:246–260. [Google Scholar]
- 30.Breed RS, Dotterrer WD. J Bacteriol. 1916;1:321–331. doi: 10.1128/jb.1.3.321-331.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coneski PN, Schoenfisch MH. Chem Soc Rev. 2012;41:3753–3758. doi: 10.1039/c2cs15271a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marxer SM, Chen TY, Schoenfisch MH. Abstr Pap Am Chem Soc. 2001;222:U95–U95. [Google Scholar]
- 33.Marxer SM, Rothrock AR, Nablo BJ, Robbins ME, Schoenfisch MH. Chem Mat. 2003;15:4193–4199. [Google Scholar]
- 34.Nablo BJ, Schoenfisch MH. Biomacromolecules. 2004;5:2034–2041. doi: 10.1021/bm049727w. [DOI] [PubMed] [Google Scholar]
- 35.Fang FC. J Clin Invest. 1997;99:2818–2825. doi: 10.1172/JCI119473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nilsson M, Frykberg L, Flock JI, Pei L, Lindberg M, Guss B. Infect Immun. 1998;66:2666–2673. doi: 10.1128/iai.66.6.2666-2673.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDevitt D, Nanavaty T, HousePompeo K, Bell E, Turner N, McIntire L, Foster T, Hook M. Eur J Biochem. 1997;247:416–424. doi: 10.1111/j.1432-1033.1997.00416.x. [DOI] [PubMed] [Google Scholar]
- 38.Davis SL, Gurusiddappa S, McCrea KW, Perkins S, Hook M. J Biol Chem. 2001;276:27799–27805. doi: 10.1074/jbc.M103873200. [DOI] [PubMed] [Google Scholar]
- 39.Hendricks SK, Kwok C, Shen MC, Horbett TA, Ratner BD, Bryers JD. J Biomed Mater Res. 2000;50:160–170. doi: 10.1002/(sici)1097-4636(200005)50:2<160::aid-jbm10>3.0.co;2-m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nablo BJ, Chen TY, Schoenfisch MH. J Am Chem Soc. 2001;123:9712–9713. doi: 10.1021/ja0165077. [DOI] [PubMed] [Google Scholar]
- 41.Nablo BJ, Rothrock AR, Schoenfisch MH. Biomaterials. 2005;26:917–924. doi: 10.1016/j.biomaterials.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 42.Nablo BJ, Schoenfisch MH. J Biomed Mater Res Part A. 2003;67A:1276–1283. doi: 10.1002/jbm.a.20030. [DOI] [PubMed] [Google Scholar]
- 43.Carpenter AW, Schoenfisch MH. Chem Soc Rev. 2012;41:3742–3752. doi: 10.1039/c2cs15273h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jones M, Ganopolsky J, Labbé A, Wahl C, Prakash S. Appl Microbiol Biotechnol. 2010;88:401–407. doi: 10.1007/s00253-010-2733-x. [DOI] [PubMed] [Google Scholar]
- 45.Richardson AR, Libby SJ, Fang FC. Science. 2008;319:1672–1676. doi: 10.1126/science.1155207. [DOI] [PubMed] [Google Scholar]
- 46.Privett BJ, Deupree SM, Backlund CJ, Rao KS, Johnson CB, Coneski PN, Schoenfisch MH. Mol Pharm. 2010;7:2289–2296. doi: 10.1021/mp100248e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nablo BJ, Schoenfisch MH. Biomaterials. 2005;26:4405–4415. doi: 10.1016/j.biomaterials.2004.11.015. [DOI] [PubMed] [Google Scholar]