

Abstract
Bacillus subtilis has been a model for gram-positive bacteria and it has long been exploited for industrial and biotechnological applications. However, the availability of facile genetic tools for physiological analysis has generally lagged substantially behind traditional genetic models such as Escherichia coli and Saccharomyces cerevisiae. In this work, we have developed an efficient, precise and scarless method for rapid multiple genetic modifications without altering the chromosome of B. subtilis. This method employs upp gene as a counter-selectable marker, double-strand break (DSB) repair caused by exogenous endonuclease I-SceI and comK overexpression for fast preparation of competent cell. Foreign dsDNA can be simply and efficiently integrated into the chromosome by double-crossover homologous recombination. The DSB repair is a potent inducement for stimulating the second intramolecular homologous recombination, which not only enhances the frequency of resolution by one to two orders of magnitude, but also selects for the resolved product. This method has been successfully and reiteratively used in B. subtilis to deliver point mutations, to generate in-frame deletions, and to construct large-scale deletions. Experimental results proved that it allowed repeated use of the selectable marker gene for multiple modifications and could be a useful technique for B. subtilis.
Introduction
Bacillus subtilis has been applied as a model system for researches on the aspects of biochemistry, genetics and physiology of Gram-positive bacteria, and has long been used as an important cell factory for industrial applications [1]. In recent years, with the rapid development of post-genomic studies in B. subtilis such as genome reduction engineering [2], inverse metabolic engineering [3], [4] and synthetic biology [5], it is beneficial to create a simple method to enable multiple genetic modifications for exploration of the molecular mechanisms conveniently. Three essentials including simple and efficient procedure for transformation, suitable counter-selectable marker and high marker recycling efficiency play crucial roles in the markerless genetic modification system.
Transformation efficiency is one of the decisive factors for genetic manipulation. It is desirable to develop an approach for fast preparation of competent cells with high transformation efficiency. As we known, natural transformation is a programmed mechanism characterized by binding of free double-stranded (ds) DNA from the environment to the cell pole in rod-shaped bacteria that is widely used for chromosomal genetic manipulation [6]. Programmed competence coordinates the expression of proteins involved in DNA uptake and translocation with expression of some proteins of the recombination machinery. One of the early competence induced genes encodes the master regulator (ComK) that subsequently regulates the expression of the “late” genes to drive expression a set of genes in wild type cells that are necessary for building up the DNA uptake apparatus and recombination [7]–[10]. Overexpression of comK gene or deletion of rok gene (encoding the negative regulator of comK) is a valuable method for improvement of natural transformation efficiency and has been applied in B. subtilis [11]–[13]. Induction of comK combined with positive auto-stimulation of native comK is sufficient for the transcriptional activation of the late competence genes, which results in an increased percentage of competent cells in the population [14] and leads to establishment of the competent state when it enters the stationary growth phase [15].
The effectiveness of counter-selectable marker is correlated with counter-selection efficiency and the practicability of markerless mutation delivery system. A number of systems for marker recycling in B. subtilis have been reported based on the following counter-selectable marker: mazF [16]–[18], ysbC [19], blaI [20], lacI [21], hewl [22] and upp [23]. Besides, the site-specific recombinases system such as Cre/lox can also be used to delete the selectable marker for counter-selection. However, it cannot be applied in allele replacement and the “scar” sequence left at the modification locus might be unwanted in some cases [22], [24]. Applications of mazF and ysbC are difficult to be maintained in E. coli, whereas blaI and lacI require the corresponding auxotrophic host strains. The hewl gene is substantiated to be active against Bacillus species recently and can be used as a powerful marker for counter-selection [22]. The upp gene has been widely used in markerless modifications, such as gene disruption, point mutation, and large-scale deletion [23], [25]–[28]. Although it is feasible as a counter-selectable marker, the proportion of cells which undergo pop-out is expected to be of the order of 10−6 [23] and actually needs to be enhanced to facilitate the application of markerless system.
The I-SceI based mutagenesis system for unmarked genetic manipulation has been broadly employed in several microorganisms to promote the efficiency of resolution [29]–[33]. The exogenous endonuclease I-SceI, as a counter-selection tool, causes a unique double-strand break (DSB) with a 4-base 3′ hydroxyl overhang [34] at the I-SceI recognition site in the genome. This broken chromosomal ends are then subjected to RecA-mediated DSB repair, which is a potent inducement to stimulate homologous recombination within the regions flanked by the broken ends. Fortunately, genome of B. subtilis 168 is free of the 18 bp I-SceI recognition sequence, which was affirmed when no genome DNA degradation was detected after treatment with I-SceI [35].
In the present work, we introduce a simple and general strategy to create markerless genetic manipulations in B. subtilis. This procedure, based on competence fast preparation and repair activities of the cell, utilizes upp as counter-selectable marker in concert with DSB stimulation repair to construct various mutations efficiently. It simplifies experimental procedure and shortens the processing time to generate modification without altering the chromosome in any other condition. Experimental results demonstrate that point mutation, in-frame deletion and large-scale deletion can be readily made with this novel approach. It is simple, fast and of high efficiency in contrast to routine method, promoting it as an attractive and useful tool for genome editing.
Materials and Methods
Bacterial Strains, Plasmids, Primers and Growth Conditions
Table 1 lists the bacterial strains and plasmids used in this study. The information of primers is summarized (Table S1). All B. subtilis mutant strains were derived from the B. subtilis Marburg 168 (trpC2). Strains were grown at 37°C in Luria–Bertani (LB) liquid medium or M9 liquid medium [36] for genetics and physiology characterization and minimal medium (MM) [23] for counter-selection of the 5-fluorouracil (5FU) resistance colonies. Solid media was obtained by adding 1.5% agar to the liquid media. When required, antibiotics were added to the growth media at the following concentrations: 100 µg/ml ampicillin for E. coli selection; 100 µg/ml spectinomycin, 5 µg/ml kanamycin, 5 µg/ml chloramphenicol, and 1 µg/ml erythromycin for B. subtilis selection; 5FU was purchased from Sigma-Aldrich Corporation (Sigma-Aldrich, St Louis, MO, USA) and prepared as a stock solution of 100 mM in dimethyl sulfoxide (DMSO). Colonies that popped out the upp-cassette were selected on MM plate containing 10 µM 5FU.
Table 1. Bacterial strains and plasmids used in this study.
| Strain or plasmid | Relevant description(s)a | Reference or source |
| Strains | ||
| E. coli DH5α | Standard cloning strain | Invitrogen |
| B. subtilis 168 | Wild-type strain, trpC2 | Laboratory stock |
| B. subtilis BUK | B. subtilis 168 derivate, Δupp::Para-comK | This study |
| B. subtilis BUK-1C | B. subtilis 168 derivate, Δupp::Para-comK, ΔccpN::upp-cassette | This study |
| B. subtilis BUK-1 | B. subtilis 168 derivate, Δupp::Para-comK, ccpN (Ala 130 to Ser) | This study |
| B. subtilis BUK-2C | B. subtilis 168 derivate, Δupp::Para-comK, ΔccpN::upp-cassette | This study |
| B. subtilis BUK-2 | B. subtilis 168 derivate, Δupp::Para-comK, ΔccpN | This study |
| B. subtilis BUK-3C | B. subtilis 168 derivate, Δupp::Para-comK, ΔileS::upp-cassette-ileS* | This study |
| B. subtilis BUK-3 | B. subtilis 168 derivate, Δupp::Para-comK, ileS (Ala 467 to Thr) | This study |
| B. subtilis BUKΔ20.5 kb | B. subtilis 168 derivate, ΔydcL-yddN | This study |
| B. subtilis BUKΔ75.9 kb | B. subtilis 168 derivate, ΔmutL-pksR | This study |
| Plasmidsb | ||
| pAX01 | B. subtilis integration vector, PxylA, AmpR, EmR | BGSCa |
| pDK | B. subtilis integration vector, KmR | BGSCa |
| pUC18 | E. coli cloning vector, oriT, AmpR | Laboratory stock |
| pTKRED | E. coli cloning vector, I-SceI, SpcR | Laboratory stock |
| pE194 | Bacillus/Staphylococcus cloning vector, EmR | Laboratory stock |
| pSS | Modular vector carrying upp-cassette, AmpR, CmR | This study |
| pST | B. subtilis integration vector, Para-comK, AmpR | This study |
| pEB | B. subtilis-E. coli shuttle vector, AmpR, EmR | This study |
| pEBS | B. subtilis-E. coli shuttle vector, PxylA-I-SceI, AmpR, EmR | This study |
| pEBS-cop1 | pEBS carrying a point mutation in cop-1 site, EmR | This study |
| pSS-ileS | B. subtilis integration vector, CmR | This study |
Bacillus Genetic Stock Center.
Antibiotic resistance genes in plasmids were abbreviated as follows: AmpR, ampicillin resistance; CmR, chloramphenicol resistance; EmR, erythromycin resistance; SpcR, spectinomycin resistance; KmR, kanamycin resistance.
Construction of the Modular Cassette
The upp gene, coding uracil phosphoribosyl-transferase, was fused to a chloramphenicol resistance gene (cat) and the recognition site of I-SceI endonuclease to create an “upp-cassette” for marker recycling. It was assembled from different templates by the following steps. The cat gene was PCR-generated from vector pC194 using primer pair pSS-P1/pSS-P2, with a multiple clone site (MCS) introduced by the forward primer pSS-P1. In parallel, the upp gene with its 5′ regulatory region and 3′ transcription terminator was amplified from B. subtilis 168 using primer pair pSS-P3/pSS-P4, with an 18 bp oligonucleotide of I-SceI endonuclease recognition site introduced by the reverse primer pSS-P4. Then both PCR fragments were seamlessly joined by splice overlap extension PCR (SOE-PCR) using primer pair pSS-P1/pSS-P4 to generate the upp-cassette, where the two genes were oriented in the same direction. Subsequently, the cassette was digested with AatII and SphI and cloned into the same sites of pUC18 to yield modular vector pSS (Figure 1A). The 2 kb selection-eviction upp-cassette, was amplified from pSS using appropriate primers (Table S1).
Figure 1. Plasmids for genetic modification in B. subtilis.
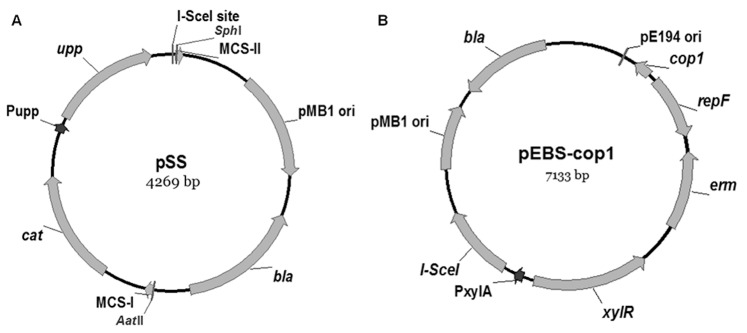
(A) A modular cassette was cloned into pUC18 to generate pSS, the modular cassette contained chloramphenicol resistance gene (cat), UPRTase encoding gene (upp) and the recognition site of I-SceI endonuclease at 3′ end of upp gene. (B) The xylose-inducible I-SceI cassette was cloned into pEB, with a temperature-sensitive replicon (repF) and erythromycin resistance gene (erm).
Construction of pEBS-cop1
To stimulate intramolecular recombination by DSB repair in B. subtilis, we constructed a temperature-sensitive shuttle vector pEBS-cop1 (Figure 1B). A region including a temperature-sensitive replication origin and the erythromycin resistance gene (erm) was amplified from pE194 using primer pair pEBS-P1/pEBS-P2 and cloned between HindIII and PstI sites of pUC18, forming an E. coli-B. subtilis shuttle vector pEB. The I-SceI gene was amplified from pTKRED using primer pair pEBS-P3/pEBS-P4, and inserted into BamHI and NcoI sites of pAX01 to generate pAX01-SceI. Subsequently, a xylose-inducible I-SceI expression cassette was PCR-generated using pEBS-P5/pEBS-P6 as primers and pAX01-SceI as template, and inserted between KpnI and PstI sites of pEB to yield pEBS with a low copy number of about 10 at 30°C [37].
To acquire a high copy number, site-directed mutagenesis was carried out to mutate the promoter of cop gene in pEBS by using Phusion TM site-directed mutagenesis kit (New England BioLabs, Ipswich, MA) according to the manufacturer’s instructions. The pEBS-cop1 was obtained which contained a C•G→G•C nucleotide transversion in the cop promoter region, with a high copy number of about 200 at 30°C [37]. It could be stably maintained under the selection pressure in B. subtilis cells at 30°C, but the copy number dramatically decreased when cells were grown at temperatures above 37°C and no growth occurred at 50°C in the presence of selective antibiotic.
Construction of pST
Plasmid pST was constructed for upp deletion and comK expression cassette insertion at the upp locus on B. subtilis 168 chromosome. The upp upstream region (787 bp) was amplified using primer pair pST-P1/pST-P2,then cut by AatII and NdeI and inserted into MCS-I of pSS. The upp downstream region (980 bp) was amplified using primer pair pST-P3/pST-P4 and cloned between SalI and KpnI sites in MCS-II. After that, comK gene was amplified from B. subtilis 168 using primer pair pST-P5/pST-P6, and a fragment including Para promoter and araR gene was also amplified using primer pair pST-P7/pST-P8. ComK expression cassette was generated under the control of Para promoter by SOE-PCR and inserted between BglII and SalI sites to yield pST (Figure 2A).
Figure 2. Construction of host strain B. subtilis BUK.
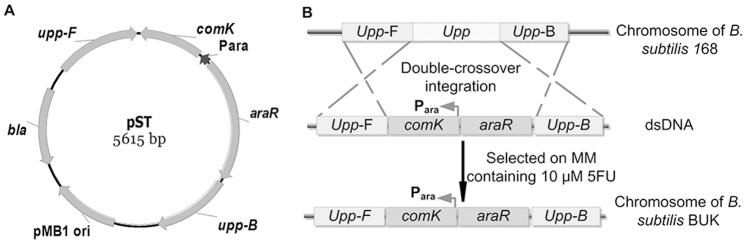
(A) Two homologous arms of upp gene (Upp-F and Upp-B) were cloned into pUC18, and the arabinose-inducible comK expression cassette was fused downstream of the front homologous arm to construct vector pST. (B) The sequence between two homologous arms of upp gene (Upp-F and Upp-B), was replaced by comK cassette via a double-crossover recombination. Crossed lines indicated double-crossover recombinant events. Upp-F and Upp-B, upstream and downstream fragment of upp gene, respectively; comK, the master regulator ComK encoding gene for B. subtilis competence development; Para, arabinose-inducible promoter of ara operon; araR, arabinose operon transcriptional repressor of B. subtilis.
Generation of B. subtilis BUK for Fast Competent Cells Preparation
pST DNA was integrated into B. subtilis 168 for constructing the UPRTase deficient host by double-crossover mechanism as previously described [38]. 5FUR recombinants were isolated on MM plate supplemented with 10 µM 5FU, which differed from 20–30 µM 5FU reported by Fabret et al. [19]. Above 10 µM 5FU concentration yielded tiny colonies and did not improve the proportion of upp-cassette pop-out. To rule out the single-crossover integration of circular pST combined with spontaneous mutation of upp, the physical structure of positive transformants were verified by PCR and DNA sequencing. One colony carrying comK cassette at the upp locus resulting from the double-crossover integration of linearized pST was designated as B. subtilis BUK (Figure 2).
The preparation of competent B. subtilis cells and transformation with dsDNA were performed as described previously by Zhang et al. [11] with slightly modifications. Briefly, an overnight LB culture of BUK cells was diluted 10-fold to fresh LB medium. When the cell density reached an OD600 (optical density at 600 nm) of 1.0, the culture was supplemented with arabinose at a final concentration of 0.4% (w/v), and continued to be shaken for 2 h. Then, the culture was ready to be transformed. One microlitre PCR product was mixed with 100 µl competent cells and the mixture was incubated at 37°C with shaking at 200 rpm for 90 min to allow the expression of antibiotic resistance gene, and then spread onto LB plates supplemented with appropriate antibiotic.
Quantitative RT-PCR
Fresh samples of cell cultures were harvested at the exponential phase grown under the same condition in M9 medium to isolated total RNA by using RNAprep pure Kit DP430 (Tiangen, Beijing, China) following the manufacturer’s instructions. RNA samples were then reversed transcribed into cDNA using Quant Reverse Transcriptase with random primers (Tiangen, Beijing, China). The qRT-PCR was carried out by Light Cycler® 480 II (Roche, Basel, Switzerland) with Real Master Mix (SYBR Green) according to the manufacturer’s instructions as follows. In brief, 100 ng of DNA-free total RNA was used in a total reaction volume of 50 µl with 0.25 mM of each primer (Table S1). The fold change values for each gene were normalized to internal control gene rrnA and calculated according to the comparative C T method [39].
Confirmation of Mutants by PCR and Sequencing
To confirm the desired mutations, genome DNA was isolated from cells using the TIANamp Bacteria DNA Kit (Tiangen, Beijing, China) following the manufacturer’s instructions. Amplification reactions were carried out using Phusion High-Fidelity DNA Polymerase (New England BioLabs, Ipswich, MA). Mutants were also checked by PCR and DNA sequencing with appropriate primers (Table S1).
Results
Construction of Induced Competence of upp-defective Strain BUK
It is well known that B. subtilis is a naturally competent species, easy to get chromosomal transformation with fully homologous DNA [6]. On the contrary, the recombination frequency for cells double-crossover chromosomal transformation with dsDNA PCR product was relatively low as reported by Fabret et al. [23]. In order to use 5FU for counter-selection and make double-crossover chromosomal transformation with dsDNA PCR product efficiently, a host strain was constructed with upp disruption and comK cassette insertion. The comK cassette was integrated at the upp locus on the chromosome seamlessly, which was designated BUK and served as the host strain for markerless mutation delivery. And the extra copy of comK enabled BUK to be easily induced into competence under arabinose induction (Figure 2B).
Induced competent cells of BUK could be rapidly prepared and had desired transformation efficiencies of ∼3–5×103 per µg of dsDNA PCR product with arabinose induction at a final concentration of 0.4% (w/v). It was much higher than that of without induction which was hard to obtain transformants. Meanwhile, the maximum of transformation efficiency was several orders of magnitude higher than that by natural transformation induced by starvation as described by Anagnostopoulos et al. [38]. Furthermore, the transformation efficiencies were of ∼1×104 per µg with linearized plasmid DNA, which was similar with the report as described previously by Zhang et al. [11].
Rationale for the Stimulated Intramolecular Recombination in Mutation Delivery System
For the purpose of obtaining an efficient and precise markerless mutation delivery system, we take advantage of not only efficient genetic recombination during double-crossover chromosomal transformation with dsDNA fragments but also the recombinational repair activity during vegetable growth in B. subtilis. The rationale for this system stimulated by DSB is shown in Figure 3. To introduce a point mutation into the chromosome, the upstream and downstream fragments comprising the mutation site are amplified by PCR with appropriate primers, where a 30 bp region of the upstream fragment is identical to the downstream fragment. Then, a triple fusion PCR reaction joins the upp-cassette with these two fragments, generating the molecule carrying the upp-cassette flanked by 30 bp DR. This dsDNA PCR product is introduced directly into BUK, and aliquots of 10-fold diluted culture are spread on LB plates supplemented with 5 µg/ml chloramphenicol. 5FUS CmR transformants are isolated where target gene is replaced with the upp-tagged mutated copy by a double-crossover homologous recombination event. Then, strains with the expected genotype are selected for eviction of the upp-cassette. Vector pEBS-cop1 is transformed into the 5FUS CmR strain on behalf of improvement the pop-out efficiency. Expression of I-SceI endonuclease results in the cleavage of chromosome at the 18 bp recognition site, and cells can survive only by repairing the DSB or by deleting the I-SceI recognition site via intramolecular recombination between DR sequences. Optimum procedure to obtain cells lost upp-cassette (5FUR CmS) is described in detail below. A single positive transformant containing pEBS-cop1 is incubated overnight in LB medium at 30°C and then diluted 10-fold to fresh LB medium. When the culture is grown to reach an OD600 of 1.0, it is supplemented with xylose at a final concentration of 1% (w/v), and continued to be shaken about 6 h for I-SceI endonuclease expression. Aliquots of culture are spread on MM plates containing 10 µM 5FU and incubated for 24 h at 37°C. Excision of upp-cassette by the single-crossover event stimulated by DSB repair generates 5FUR CmS cells. Colonies on 5FU MM plates are picked on LB plates containing 5 µg/ml chloramphenicol and incubated for 12 h at 37°C. Only those 5FUR CmS colonies are selected for further identification by PCR, and the presence of desired mutation in chromosome is checked by DNA sequencing. Finally, cells can be easily cured of pEBS-cop1 with heat-sensitive replication by growth at nonpermissive temperatures (50°C) in the absence of the selective antibiotic.
Figure 3. Scheme for mutation delivery procedure in B. subtilis.

A. (a) For purpose of gene point-mutation, the upstream and downstream fragments carrying the mutation site (*) were amplified by PCR with primer pairs P1/P2 and P3/P4, and upp-cassette was amplified by PCR with primer pairs P5/P6, respectively. (b) A triple fusion PCR reaction joined them with the upp-cassette. (c) PCR-fused product was used directly to transform BUK, and integration of the upp-cassette by first double-crossover event yields 5FUS CmR transformants. (d) The shuttle vector pEBS-cop1 was transformed into the 5FUS CmR strain to cleavage of the chromosome at the recognition site by I-SceI under xylose induction. (e) The excision of upp-cassette through the single-crossover event between the 30 bp DR stimulated by DSB generated a 5FUR CmS strain that carried only the desired mutation without any other modification in the chromosome. B. For gene deletion, the upstream and downstream fragments carrying the same DR sequence (*) were amplified by PCR with primer pairs P7/P9 and P10/P8. The remainder of the procedure was essentially the same as above described.
For gene insertion, in-frame deletion, and genome reduction, the 30 bp DR sequence, comprised in the upstream and downstream fragments, is designed as an overlapping region for triple fusion PCR both in the forward primer of the downstream fragment and the backward primer of the upp-cassette (Figure 3B), respectively. The remainder of the procedure is essentially the same as above.
Point-mutagenesis of ccpN Gene
The transcriptional regulator CcpN mediates CcpA-independent carbon catabolite repression of two gluconeogenic genes pckA and gapB [40] and controls central carbon fluxes in B. subtilis. In order to examine the effectiveness of this strategy for site-directed genetic modification at the native locus, we generated a G-to-T point mutation at position +130 in ccpN gene. Upp-cassette was PCR-amplified using pSS as template and oligonucleotide pair ccpN-Mut-P3/ccpN-Mut-P4 as primers. 1,379 bp upstream and 1,339 bp downstream fragments of ccpN gene were amplified by PCR using two oligonucleotide pairs ccpN-Mut-P1/ccpN-Mut-P2 and ccpN-Mut-P5/ccpN-Mut-P6 as primers, respectively. Both upstream and downstream fragments of ccpN gene carried 30 bp DR sequence containing the point mutation G130T. According to the procedure described above, CmR 5FUS strain was selected after integration of the triple fusion PCR fragment into the chromosome and testified by PCR with primer pair ccpN-Mut-P7/ccpN-Mut-P8 (Figure 4A). Cultures from independently isolated CmR 5FUS colonies were subjected to 5FU counter-selection, under the xylose induction for I-SceI expression, and the loss of upp-cassette was detected by PCR. After the marker-recycling event, the intramolecular homologous recombination between two DR sequences resulted in the desired point mutation. In all the cases, ccpN mutant carried solely the expected mutation Ala130Ser (GCC to TCC).
Figure 4. Confirmation of ccpN point mutation and ccpN in-frame deletion by PCR.
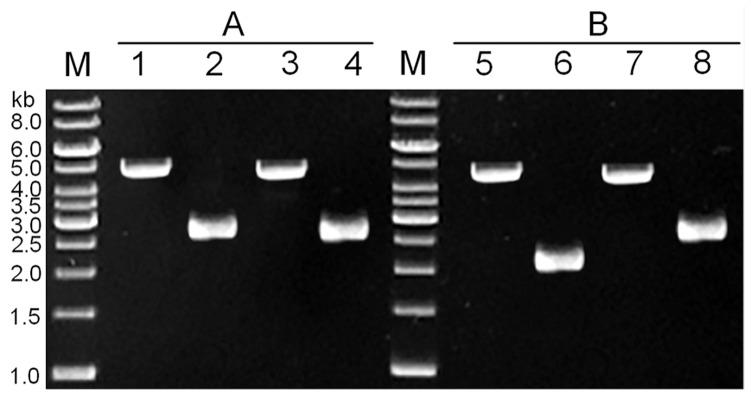
The PCR product was analyzed by agarose gel electrophoresis. A 1(Fermentas) was used as a molecular weight marker (lane M). A. Confirmation of the ccpN point mutation. Fragments were amplified using ccpN-Mut-P7/ccpN-Mut-P8 as primers. Each lane showed amplified DNA generated from a DNA template: lane 1, BUK-1C (ΔccpN::upp-cassette); lane 2, BUK-1 (ccpN-mut-Ala 130 to Ser); lane 3, dsDNA PCR fragment (positive control); lane 4, BUK (ccpN-wild type) (negative control). B. Confirmation of the ccpN in-frame deletion. Fragments were amplified using ccpN-Del-P1/ccpN-Del-P6 as primers. Each lane showed amplified DNA generated from a DNA template: lane 5, BUK-2C (ΔccpN::upp-cassette); lane 6, BUK-2 (ΔccpN); lane 7, dsDNA PCR fragment (positive control); lane 8, BUK (ccpN-wild type) (negative control).
In this case of point-mutagenesis of ccpN, a direct correlation between the concentration of xylose and the pop-out efficiency was observed (Table 2). In contrast to the efficiency of 2.8×10−6 without xylose induction, the proportion of 5FUR cells stimulated by DSB was between 3×10−5 and 8×10−4 as the ratio of 5FUR colonies to the viable cells. Moreover, the upp-cassette pop-out efficiency in the presence of 1% xylose was about 21-fold higher than that in the presence of 0.5% xylose, and was about 300-fold higher than that of without xylose induction.
Table 2. The efficiencies of first double-crossover recombination and counter-selectiona.
| Mutantb | DR (bp) | The first double-crossoverc | xylose induction concentrationd | |||
| 0% | 0.5% | 1.0% | 2.0% | |||
| ccpN* | 30 | (4.8±0.2)×103 | (2.8±0.1)×10−6 | (3.9±0.4)×10−5 | (8.4±0.1)×10−4 | (3.1±0.3)×10−4 |
| ΔccpN | 30 | (3.6±0.3)×103 | (2.7±0.4)×10−6 | (4.0±0.3)×10−5 | (7.8±0.2)×10−4 | (4.3±0.3)×10−4 |
| ileS* | 1691 | (4.2±0.2)×103 | (7.4±0.4)×10−6 | (4.3±0.2)×10−5 | (8.8±0.4)×10−4 | (4.7±0.5)×10−4 |
Data are means ± SD from three independent experiments for genetic manipulations.
ccpN*, point mutation of ccpN gene (G130T); ileS*, point mutation of ileS gene (G1399A); ΔccpN, in-frame deletion of ccpN gene.
The first double-crossover recombination efficiency was calculated as the number of CmR colonies/µg of dsDNA PCR product.
Counter-selection efficiency was calculated as Nr/Nt. Nr, number of 5FUR CmS colonies in the culture treated by different concentration of xylose; Nt, number of total colonies in culture treated by different concentration of xylose.
Point-mutagenesis of Essential Gene ileS
Essential genes are those indispensable for the survival of an organism under certain conditions. Generally, essential gene is hard to be manipulated because its disruption causes cell death [41]. Here, we try an attempt to deliver a G-to-A point mutation at position +1399 in an essential gene ileS coding the isoleucyl-tRNA synthetase. The major experimental process was similar with point-mutagenesis of ccpN except for the following minor revision: either upstream or downstream fragment comprised a complete mutant essential gene that kept the cells survival in the first double-crossover recombination event.
The mutagenesis of ileS gene was conducted by restriction-ligation method rather than SOE-PCR, since SOE-PCR of several large fragments is not easy to be operated and unwished mutations may be introduced. A 1,733 bp upstream fragment and a 3,093 bp downstream fragment of the ileS gene were amplified by PCR using two oligonucleotide pairs ileS-Mut-P1/ileS-Mut-P2 and ileS-Mut-P5/ileS-Mut-P6 as primers, respectively. The downstream fragment comprised a complete mutant essential gene ileS. Then, the upstream fragment was cut by AatII and XhoI and inserted into MCS-I of pSS. And the downstream fragment was subsequently cloned between SalI and BamHI sites in MCS-II of pSS to yield pSS-ileS. The remainder of the procedure is essentially the same as above mentioned in ccpN point-mutation. The transformation efficiency was ∼4.2×103 per µg of linearized pSS-ileS DNA. After cleavage by I-SceI endonuclease, the pop-out efficiencies of ileS point mutation were between ∼4×10−5 and ∼9×10−4 (Table 2). In addition, the cell growth traits were similar between the parental strain BUK and mutant strain B. subtilis BUK-3 in M9 medium, suggesting that ileS point-mutation basically did not affected its physiological characteristics.
In-Frame Deletion of ccpN Gene
Here, we generated an in-frame deletion of ccpN gene with this system. A 1,110 bp upstream and a 1,140 bp downstream fragments flanked by ccpN gene were PCR-generated using two primer pairs ccpN-Del-P1/ccpN-Del-P2 and ccpN-Del-P5/ccpN-Del-P6, respectively. Upp-cassette was PCR-amplified using pSS as template and oligonucleotide pair ccpN-Del-P3/ccpN-Del-P4 as primers. Triple fusion PCR product was used to transform BUK. Integration of upp-cassette into chromosome inactivated the ccpN gene, yielding 5FUS CmR colonies. Cultures from independently isolated colonies were subjected to 5FU counter-selection, and the proportion of pop-out events was determined by PCR. Mutation process and deletion junctions were testified by PCR with primer pair ccpN-Del-P1/ccpN-Del-P6 and DNA sequencing (Figure 4B). After eviction of upp-cassette, the remaining ORF encoded the expected 98-amino-acid peptide that contained 51 N-terminal and 47 C-terminal amino acids of ccpN. The proportion of 5FUR cells was 2.7×10−6 without xylose addition. However, the upp-cassette pop-out efficiency was still low and needed to be improved. By DSB repair stimulation, the proportion of 5FUR cells raised to between 4×10−5 and 8×10−4 and that was high enough for fast selection of the desired mutant. The observed highest upp-cassette pop-out efficiency was 7.8×10−4 at a final xylose concentration of 1% (w/v) (Table 2).
The validity of this method was further tested via investigating the effects of ccpN point-mutation and inactivation. In comparison with that of the reference strain BUK, both transcriptional levels of pckA and gapB were up-regulated in BUK-1 and BUK-2. CcpN point-mutation and inactivation seriously affected their physiological characteristics and caused growth defect. Compared with BUK strain, the specific growth rate of BUK-1 decreased from 0.63±0.03 h−1 to 0.48±0.01 h−1 and the specific growth rate of BUK-2 drastically decreased to 0.28±0.02 h−1.
Deletion of Large-scale Chromosomal Regions
For B. subtilis, a large number of potentially dispensable chromosomal loci can be inferred from the complete nucleotide sequence of the chromosome [42]. Fragments such as prophage (-like) regions and the largest operon pks (polyketide synthase operon, 1781306–1857233 SubtiList coordinates) are not evolved into regions encoding very indispensable functions, meanwhile, genome minimization affects neither cell viability nor the key physiological [43]. With these considerations in mind, both the 20.5 kb prophage (-like) region (528148–548697 SubtiList coordinates) and the 75.9 kb pks operon were chosen as the targets to be deleted, so as to test the availability of marker-free system for genome reduction engineering.
We used a new strategy to obtain markerless large-scale deletion mutants (Figure 5). First, we amplified fragment A and fragment B flanking the region to be deleted, fragment C locating at 4–5 kb downstream from the end of fragment A, and fragment D locating at 4–5 kb upstream of fragment B (the length of these fragments is about 1,000 bp, respectively). Fragments A and B were combined to get fragment A-B by SOE-PCR. Upp-cassette, fragment A-B and fragment C were fused by triple fusion PCR. Next, 1 µl purified PCR product was used to transform BUK. Transformants were selected on LB plate containing 5 µg/mL chloramphenicol and the positive colony was designated as BUK-I. Another triple fusion PCR product, which contained fragment D, kanamycin resistance gene amplified from pDK and fragment B containing the I-SceI recognition site, was transformed into BUK-I and selected on LB plate containing 5 µg/mL kanamycin. The objective transformant was named BUK-II. For construction of large-scale deletion, pEBS-cop1 was delivered into BUK-II and cultivated in LB liquid medium at 30°C, with adding 1% xylose at the middle-log growth phase to induce the expression of I-SceI endonuclease. Culture was spread on MM plates containing 10 µM 5FU to obtain colonies in which the upp-cassette was excised by intramolecular homologous recombination between two B homologous regions. The chloramphenicol- and kanamycin-sensitive colonies were selected from 5FU MM plate and subjected to further identification by PCR.
Figure 5. Markerless deletion of large chromosomal regions stimulated by DSB.

A. (a) Two dsDNA fragments were generated by fusion PCR and integrated into genome by double-crossover events. (b) The endonuclease I-SceI was expressed from pEBS-cop1 under the induction of xylose, and the genome of B. subtlis BUK-II was subjected to I-SceI cleavage. (c) Deletion mutants were isolated on MM plate containing 10 µM 5FU and confirmed by PCR. A, B, C, D represent DNA segments selected for homologous recombination. B. Verification of the 20.5 kb and 75.9 kb deletions in BUK by PCR. Fragments were amplified using the primer pairs 20.5 kb-DEL-P15/20.5 kb-DEL-P16 (a), 20.5 kb-DEL-P17/20.5 kb-DEL-P18 (b), 75.9 kb-DEL-P15/75.9 kb-DEL-P16 (c), 75.9 kb-DEL-P17/75.9 kb-DEL-P18 (d). 1 kb DNA Ladder (Fermentas) was used as a molecular weight marker (lane M). (a) Amplification of the ydcR gene (534334–535389 SubtiList coordinates) in the 20.5 kb DNA region of B. subtilis; (b) Amplification of the 20.5 kb DNA fragment of B. subtilis; (c–d) Confirmation of the 75.9 kb deletion of BUK was the same as that of 20.5 kb deletion. Amplification of the pksG gene (1789763–1791405 SubtiList coordinates) in the 75.9 kb DNA region of B. subtilis (c) and amplification of the 75.9 kb DNA fragment of B. subtilis (d). Each lane showed amplified DNA generated from a DNA template: lane 1, BUKΔ20.5 kb; lane 2, BUK-II (20.5); lane 3, BUK; lane 4, BUKΔ75.9 kb; lane 5, BUK-II (76.9); lane 6, BUK.
After cleavage by I-SceI endonuclease, the pop-out efficiencies of 20.5 kb and 75.9 kb large-scale deletions were between ∼5×10−6 and ∼3×10−5, which was improved about one to two orders of magnitude compared to that of ∼3×10−7 under no xylose addition (Table 3). In addition, for validation the pop-out frequency with one-ended double-strand break, strain BUK-I carrying pEBS-cop1 was used to generate large-scale deletion. As shown in Table 3, the pop-out frequency with one-ended double-strand break was between 1.5×10−6 and 1.5×10−5 which was much lower than that of with two-ended double-strand break. Both of these large-scale fragments were individually deleted and no significantly different phenotype was found among the deleted mutants (data not shown).
Table 3. Effect of the number of I-SceI sites on the counter-selection for large-scale deletiona.
| Mutantb | DR (bp) | The first double-crossoverc | Numbers of DSB | xylose induction concentrationd | |||
| 0% | 0.5% | 1.0% | 2.0% | ||||
| 20.5 kb-DEL | 1000 | (4.5±0.1)×103 | one-ended | (2.8±0.4)×10−7 | (1.7±0.2)×10−6 | (4.4±0.1)×10−6 | (1.5±0.2)×10−5 |
| two-ended | (3.2±0.2)×10−7 | (5.4±0.1)×10−6 | (1.4±0.4)×10−5 | (2.9±0.3)×10−5 | |||
| 75.9 kb-DEL | 1000 | (4.3±0.2)×103 | one-ended | (2.5±0.3)×10−7 | (1.5±0.4)×10−6 | (4.6±0.1)×10−6 | (1.1±0.3)×10−5 |
| two-ended | (3.0±0.2)×10−7 | (6.0±0.3)×10−6 | (1.0±0.2)×10−5 | (2.6±0.1)×10−5 | |||
Data are means ± SD from three independent experiments for genetic manipulations.
20.5 kb-DEL, large deletion of 20.5 kb DNA sequence (528148–548697 SubtiList coordinates) of BUK; 75.9 kb-DEL, large deletion of 75.9 kb DNA sequence (1781306–1857233 SubtiList coordinates) of BUK.
The first double-crossover recombination efficiency was calculated as the number of CmR colonies/µg of dsDNA PCR product.
Counter-selection efficiency was calculated as Nr/Nt. Nr, number of 5FUR CmS colonies in the culture treated by different concentration of xylose; Nt, number of total colonies in culture treated by different concentration of xylose.
Discussion
In this study, we report on the development of an easy-to-implement and highly efficient cloning-independent genetic manipulation system in B. subtilis. This system exploits upp as a counter-selectable marker in concert with DSB stimulation repair caused by I-SceI endonuclease. To the best of our knowledge, it is the first report of establishment of a markerless mutation delivery system in B. subtilis stimulated by DSB repair. It bypasses the time-consuming restriction/ligation-dependent vector construction procedure and enhances the pop-out frequency by one to two orders of magnitude after I-SceI cleavage. The DSB-stimulated genetic modification method was successfully used to deliver two point mutations, to realize an in-frame deletion, and to delete two large-scale genomic regions with high efficiency.
A low-frequency of first double-crossover recombinant with dsDNA PCR product is an obstacle for genetic manipulation system. To promote the efficiency of competent cell formation and simplify the preparation process, we generated the host strain BUK by overexpressing comK gene encoding the competence transcription factor at the upp locus. After a short period of arabinose induction, this engineered strain can be transformed at high efficiency of ∼3–5×103 per µg dsDNA PCR product (Table 2). It is so far the highest genetic recombination efficiency ever reported in B. subtilis transformed by double-crossover mechanism with dsDNA PCR product containing heterologous DNA sequences in the central region. In fact, the efficiencies of recombination with PCR fragments vary depending on the intergration sites and the fitness of mutants (data not shown). In addition, the transformation efficiency was of ∼1×104 per µg of linearized plasmid DNA which was consistent with the result of Zhang et al. [11].
In this markerless mutation delivery system, the counter-selection efficiencies were ∼2.8×10−6 without xylose addition in point-mutation and in-frame deletion of ccpN. In contrast, the pop-out frequency could be enhanced by one to two orders of magnitude under xylose induction. Since the I-SceI cleavage serves not only as a selection tool, but also as a stimulator of the resolution process. From a technical point of view, introduction of insertion, in-frame deletion or point-mutation into the chromosome are equivalent processes. However, construction of large-scale deletion poses an especial issue. Since the frequency of intramolecular recombination depends on the host cell and the physical distance between homologous regions, it is difficult to increase intramolecular recombination efficiency for large deletion of genome. By using the DSB-stimulated replacement method, we deleted two large chromosomal regions of 20.5 kb and 75.9 kb in BUK easily. On the one hand, compared with the high efficiencies of point mutation and in-frame deletion, only between 5×10−6 and 3×10−5 of the positive transformants survived in large-scale deletions, but all of them were accurate deletion mutants. Since this effect depends on the concentrations of I-SceI in the cell, which can be regulated by different induction concentrations. On the other hand, the one- or two-ended DSB with short patches of homology could be repaired by error-prone single-strand annealing which unwanted mutations might accumulate or by error-free homologous recombination. Indeed, when applying the DSB-stimulated gene replacement method, only one or two DNA lesions are inflicted on the chromosome, thus there are no other hotspots for mutation [31]. According to our experiment results, no increase of mutation rate was observed in cells subjected to DSB-stimulated gene replacement.
This method has been successfully applied to introduce insertions, point mutations and deletions into B. subtilis chromosome over thirty various sites in our laboratory. It represents a substantial advance in reiterative genetic manipulation that will save time, effort and expense in functional genomics studies. All these results have proved that this method allows repeated use of the selectable marker for multiple modifications in B. subtilis and could be potential applicable in other microorganisms with underdeveloped genetic tools.
Supporting Information
Primers used in this study.
(XLS)
Acknowledgments
We thank Dr. Zhenquan Lin for the help of qRT-PCR analysis.
Funding Statement
This work was supported by National Program on Key Basic Research Project (2011CBA00804, 2012CB725203), National Natural Science Foundation of China (NSFC-21206112, NSFC-21176182), National High-tech R&D Program of China (2012AA022103, 2012AA02A702) and the Innovation Foundation of Tianjin University (1308). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Van Dijl JM, Hecker M (2013) Bacillus subtilis: from soil bacterium to super-secreting cell factory. Microb Cell Fact 12: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morimoto T, Kadoya R, Endo K, Tohata M, Sawada K, et al. (2008) Enhanced recombinant protein productivity by genome reduction in Bacillus subtilis . DNA Res 15: 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tannler S, Zamboni N, Kiraly C, Aymerich S, Sauer U (2008) Screening of Bacillus subtilis transposon mutants with altered riboflavin production. Metab Eng 10: 216–226. [DOI] [PubMed] [Google Scholar]
- 4. Tanaka K, Henry CS, Zinner JF, Jolivet E, Cohoon MP, et al. (2013) Building the repertoire of dispensable chromosome regions in Bacillus subtilis entails major refinement of cognate large-scale metabolic model. Nucleic Acids Res 41: 687–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nandagopal N, Elowitz MB (2011) Synthetic biology: integrated gene circuits. Science 333: 1244–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kidane D, Ayora S, Sweasy JB, Graumann PL, Alonso JC (2012) The cell pole: the site of cross talk between the DNA uptake and genetic recombination machinery. Crit Rev Biochem Mol Biol 47: 531–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berka RM, Hahn J, Albano M, Draskovic I, Persuh M, et al. (2002) Microarray analysis of the Bacillus subtilis K-state: genome-wide expression changes dependent on ComK. Mol Microbiol 43: 1331–1345. [DOI] [PubMed] [Google Scholar]
- 8. Ogura M, Yamaguchi H, Kobayashi K, Ogasawara N, Fujita Y, et al. (2002) Whole-genome analysis of genes regulated by the Bacillus subtilis competence transcription factor ComK. J Bacteriol 184: 2344–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dubnau D (1999) DNA uptake in bacteria. Annu Rev Microbiol 53: 217–244. [DOI] [PubMed] [Google Scholar]
- 10. Burton B, Dubnau D (2010) Membrane-associated DNA transport machines. Cold Spring Harb Perspect Biol 2: a000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang XZ, Zhang YH (2011) Simple, fast and high-efficiency transformation system for directed evolution of cellulase in Bacillus subtilis . Microb Biotechnol 4: 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hoa TT, Tortosa P, Albano M, Dubnau D (2002) Rok (YkuW) regulates genetic competence in Bacillus subtilis by directly repressing comK . Mol Microbiol 43: 15–26. [DOI] [PubMed] [Google Scholar]
- 13. Smits WK, Kuipers OP, Veening JW (2006) Phenotypic variation in bacteria: the role of feedback regulation. Nat Rev Microbiol 4: 259–271. [DOI] [PubMed] [Google Scholar]
- 14. Mironczuk AM, Kovacs AT, Kuipers OP (2008) Induction of natural competence in Bacillus cereus ATCC14579. Microb Biotechnol 1: 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hamoen LW, Venema G, Kuipers OP (2003) Controlling competence in Bacillus subtilis: shared use of regulators. Microbiology 149: 9–17. [DOI] [PubMed] [Google Scholar]
- 16. Zhang XZ, Yan X, Cui ZL, Hong Q, Li SP (2006) mazF, a novel counter-selectable marker for unmarked chromosomal manipulation in Bacillus subtilis . Nucleic Acids Res 34: e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morimoto T, Ara K, Ozaki K, Ogasawara N (2009) A new simple method to introduce marker-free deletions in the Bacillus subtilis genome. Genes Genet Syst 84: 315–318. [DOI] [PubMed] [Google Scholar]
- 18. Yu H, Yan X, Shen W, Shen Y, Zhang J, et al. (2010) Efficient and precise construction of markerless manipulations in the Bacillus subtilis genome. J Microbiol Biotechnol 20: 45–53. [PubMed] [Google Scholar]
- 19. Defoor E, Kryger MB, Martinussen J (2007) The orotate transporter encoded by oroP from Lactococcus lactis is required for orotate utilization and has utility as a food-grade selectable marker. Microbiology 153: 3645–3659. [DOI] [PubMed] [Google Scholar]
- 20. Brans A, Filee P, Chevigne A, Claessens A, Joris B (2004) New integrative method to generate Bacillus subtilis recombinant strains free of selection markers. Appl Environ Microbiol 70: 7241–7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang C, Zhang X, Yao Z, Lu Y, Lu F, et al. (2011) A new method for multiple gene inactivations in Bacillus subtilis 168, producing a strain free of selectable markers. Can J Microbiol 57: 427–436. [DOI] [PubMed] [Google Scholar]
- 22. Wang Y, Weng J, Waseem R, Yin X, Zhang R, et al. (2012) Bacillus subtilis genome editing using ssDNA with short homology regions. Nucleic Acids Res 40: e91–e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fabret C, Ehrlich SD, Noirot P (2002) A new mutation delivery system for genome-scale approaches in Bacillus subtilis . Mol Microbiol 46: 25–36. [DOI] [PubMed] [Google Scholar]
- 24. Yan X, Yu HJ, Hong Q, Li SP (2008) Cre/lox system and PCR-based genome engineering in Bacillus subtilis . Appl Environ Microbiol 74: 5556–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kristich CJ, Manias DA, Dunny GM (2005) Development of a method for markerless genetic exchange in Enterococcus faecalis and its use in construction of a srtA mutant. Appl Environ Microbiol 71: 5837–5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goh YJ, Azcarate-Peril MA, O'Flaherty S, Durmaz E, Valence F, et al. (2009) Development and application of a upp-based counterselective gene replacement system for the study of the S-layer protein SlpX of Lactobacillus acidophilus NCFM. Appl Environ Microbiol 75: 3093–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Keller KL, Bender KS, Wall JD (2009) Development of a markerless genetic exchange system for Desulfovibrio vulgaris Hildenborough and its use in generating a strain with increased transformation efficiency. Appl Environ Microbiol 75: 7682–7691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peters B, Junker A, Brauer K, Muhlthaler B, Kostner D, et al. (2013) Deletion of pyruvate decarboxylase by a new method for efficient markerless gene deletions in Gluconobacter oxydans . Appl Microbiol Biotechnol 97: 2521–2530. [DOI] [PubMed] [Google Scholar]
- 29. Flannagan RS, Linn T, Valvano MA (2008) A system for the construction of targeted unmarked gene deletions in the genus Burkholderia . Environ Microbiol 10: 1652–1660. [DOI] [PubMed] [Google Scholar]
- 30. Horzempa J, Shanks RM, Brown MJ, Russo BC, O'Dee DM, et al. (2010) Utilization of an unstable plasmid and the I-SceI endonuclease to generate routine markerless deletion mutants in Francisella tularensis . J Microbiol Methods 80: 106–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Posfai G, Kolisnychenko V, Bereczki Z, Blattner FR (1999) Markerless gene replacement in Escherichia coli stimulated by a double-strand break in the chromosome. Nucleic Acids Res 27: 4409–4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wingler LM, Cornish VW (2011) Reiterative Recombination for the in vivo assembly of libraries of multigene pathways. Proc Natl Acad Sci U S A 108: 15135–15140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Billerbeck S, Panke S (2012) A genetic replacement system for selection-based engineering of essential proteins. Microb Cell Fact 11: 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Monteilhet C, Perrin A, Thierry A, Colleaux L, Dujon B (1990) Purification and characterization of the in vitro activity of I-SceI, a novel and highly specific endonuclease encoded by a group I intron. Nucleic Acids Res 18: 1407–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Itaya M (2012) Sequential insertion of multiple I-SceI recognition sites at designed loci of the Bacillus subtilis 168 genome. Biosci Biotechnol Biochem 76: 180–182. [DOI] [PubMed] [Google Scholar]
- 36.Harwood CR, Cutting SM (1990) Molecular biological methods for Bacillus: Chichester, John Wiley & Sons, Ltd.
- 37. Villafane R, Bechhofer DH, Narayanan CS, Dubnau D (1987) Replication control genes of plasmid pE194. J Bacteriol 169: 4822–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Anagnostopoulos C, Spizizen J (1961) Requirements for Transformation in Bacillus Subtilis . J Bacteriol 81: 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 40. Servant P, Le Coq D, Aymerich S (2005) CcpN (YqzB), a novel regulator for CcpA-independent catabolite repression of Bacillus subtilis gluconeogenic genes. Mol Microbiol 55: 1435–1451. [DOI] [PubMed] [Google Scholar]
- 41. Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, et al. (2003) Essential Bacillus subtilis genes. Proc Natl Acad Sci U S A 100: 4678–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, et al. (1997) The complete genome sequence of the gram-positive bacterium Bacillus subtilis . Nature 390: 249–256. [DOI] [PubMed] [Google Scholar]
- 43. Westers H, Dorenbos R, van Dijl JM, Kabel J, Flanagan T, et al. (2003) Genome engineering reveals large dispensable regions in Bacillus subtilis . Mol Biol Evol 20: 2076–2090. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers used in this study.
(XLS)