

Abstract
Objective
Lupus nephritis (LN) is an immune complex-mediated glomerulonephritis. Proliferative LN (PLN, International Society of Nephrology and Renal Pathology Society (ISN/RPS) classes III and IV)) often leads to renal injury or failure despite traditional induction and maintenance therapy. Successful targeted therapeutic development requires insight into mediators of inflammation in PLN. Superoxide (SO) and its metabolites are mediators of the innate immune response through their ability to mediate reduction-oxidation signaling. Endothelial nitric oxide synthase (eNOS) modulates inflammatory responses in endothelial cells. We hypothesized that markers of SO production would be increased in active PLN and that SO production would be dependent on the activity of select enzymes in the renal cortex.
Methods
Patients with systemic lupus erythematosus were enrolled at the time of renal biopsy for active LN of all classes. Serum collected at baseline was analyzed by HPLC with electrochemical detection for markers of SO production (durable modifications of serum protein Tyr ultimately requiring SO as a substrate). Renal cortex from MRL/MpJ-FASlpr (MRL/lpr) mice with and without functional eNOS was analyzed during active disease for superoxide (SO) production with and without inhibitors of SO producing enzymes.
Results
Serum protein modifications indicative of total SO production were significantly higher in patients with PLN. These markers were increased in association with more active, inflammatory PLN. Mice lacking functional eNOS had 80% higher levels of renal cortical SO during active disease, and inhibitors of nitric oxide synthase and NADPH oxidase reduced these levels by 60% and 77%, respectively.
Conclusion
These studies demonstrate that SO production is unique to active PLN in a NOS and NADPH oxidase-dependent fashion. These findings suggest the emulating or augmenting eNOS activity or inhibiting NADPH oxidase SO production may be targets of therapy in patients with PLN. The markers of SO production used in this study could rationally be used to select SO-modulating therapies and serve as pharmacodynamic indicators for dose titration.
Keywords: Lupus nephritis, Systemic lupus erythematosus, Nitric oxide, Endothelial nitric oxide synthase, NADPH Oxidase, Proliferative lupus nephritis, Superoxide, Oxidation-reduction, Inflammation
Introduction
Lupus nephritis (LN) is an immune complex-mediated glomerulonephritis (GN) that affects approximately half of all patients with systemic lupus erythematosus (SLE). The International Society of Nephrology/Renal Pathology Society (ISN/RPS) classification system is now the standard for characterizing glomerular lesions in LN (1). However, despite knowledge of the type of lesion present, response to standard therapy is highly variable (2). Failures of the current therapeutic approaches may stem from their inability to adequately target innate immune responses distal to immune complex deposition. An understanding of mechanisms driving these downstream innate responses is essential to develop new therapies. A key mediator of these innate immune responses is the increased production of reactive oxygen species (ROS) in response to inflammatory stimuli.
Our laboratory and others demonstrated that inducible nitric oxide synthase expression was increased in PLN (3, 4) and that inducible nitric oxide synthase- and myeloperoxidase-mediated modifications of proteins (nitrotyrosine (NTyr)) were increased in the serum of lupus patients with LN, particularly among African-Americans (5). However, the importance of increased inducible nitric oxide synthase (iNOS)-mediated nitric oxide (NO) production in LN was questioned when genetic deletion of functional iNOS in lupus mouse models failed to reduce the onset of classic pathologic features of LN except vasculitis (6, 7). However, ROS production in this murine model of lupus nephritis was significantly reduced by inhibitors of iNOS (8), suggesting that driving pathogenic consequence of iNOS expression in LN is the production of SO rather than the production of NO. However, studies to date have not localized SO-producing enzyme activity to the kidney.
ROS such as superoxide (SO), hydrogen peroxide H2O2, and hydroxyl radicals (OH•) are key mediators of redox-mediated inflammatory responses (9). All are produced by enzymes in cells that are resident to or infiltrate into the glomerulus in renal disease. These enzymes include inducible nitric oxide synthase (iNOS) (4) and NADPH oxidase (NOX) (10). The SO produced by these enzymes can either transiently (akin to phosphorylation) or permanently modify proteins to initiate a process known as reduction-oxidation (redox) signaling. These modifications affect protein function, kinase activity, and transcription factor activation (9).
Many of the enzymes that produce reactive intermediates leading to more durable modifications to Tyr require SO as a common substrate; however, the enzyme source of SO in PLN is not well described. To suggest that inflammatory signaling in PLN is mediated by ROS, increased ROS production must be demonstrated in PLN relative to membranous and mesangial disease. To target SO-mediated redox inflammatory signaling, the enzyme source of SO in PLN must be identified. We hypothesized that durable modifications of serum protein Tyrs from SO production would be increased in more aggressive, PLN (PLN, ISN/RPS classes III and IV) in a manner that would associate with redox-mediated cytokine production. Furthermore, we hypothesized that ROS production would be catalyzed select enzymes in the cortex, suggesting therapeutic targets for reducing redox-mediating inflammatory signaling in LN.
Patients and Methods
Human Subjects and Methods
General Protocol
This study was designed to determine associations between systemic markers of SO production and PLN biopsy classes at baseline. Subjects with active LN but without high chronicity were enrolled if there was intent to advance therapy based on active LN. All subjects had cortical renal biopsies performed as part of routine care. Subjects were evaluated with a history and physical exam as well as blood and urine collection at baseline for traditional markers of SLE disease activity and nephritis activity. Serum collected at baseline was analyzed for systemic markers of ROS production and select cytokines known to be redox regulated (IL6 and MCP1). Baseline measures of RI production were reported by ISN/RPS biopsy class (1).
Participants
This study was in compliance with the Helsinki Declaration, revised Hong Kong 1989, and was approved by the Medical University of South Carolina Institutional Review Board. All subjects gave written informed consent before study procedures were performed.
Inclusion and Exclusion Criteria
Patients with active LN who were biopsied as part of clinical care were enrolled from the Medical University of South Carolina (MUSC) Rheumatology Clinic before biopsy or, if after biopsy, after red blood cell (RBC) counts in the urine returned to pre-biopsy levels. All subjects met 1997 criteria for SLE (11). All subjects had International Society of Nephrology/Renal Pathology Society (ISN/RPS) class II, III, IV, or V nephritis by biopsy. Only subjects whose primary rheumatologist intended to initiate induction therapy for LN were enrolled. Subjects were excluded if they had an active infection, known pregnancy, or serum Cr > 2.5.
Clinical Evaluation and sample collection
All subjects were examined prospectively in the Clinical and Translational Research Center (CTRC) or the Rheumatology Clinic at MUSC. SLE subjects were evaluated with a history, physical examination, phlebotomy, and urine collection during or within one month of the biopsy visit. All blood collections for the visit corresponding to a renal biopsy for active nephritis were performed less than or equal to 48 hours after increasing the daily equivalent prednisone dose to more than 10 mg. The baseline blood and urine collection occurred on average one day prior to the date of the biopsy.
Sample processing
All laboratory evaluations used to determine renal disease activity were performed in our Clinical Laboratory Improvement Amendments (CLIA)-approved clinical laboratory at MUSC. These laboratory measures included serum levels of C3, C4, creatinine (Cr), and anti-dsDNA antibodies and clean catch urine for protein, creatinine and microscopic analysis. All samples for analysis were promptly processed in the CTRC. Serum samples for systemic RI measures were centrifuged after clot formation and immediately frozen in aliquots at −80 °C for batch analysis.
Renal biopsy grading
A single, experienced renal pathologist (SES) reviewed the biopsies and graded them to determine the International Society of Nephrology/Renal Pathology Society (ISN/RPS) class(es), active, and chronic lesions (12), and NIH activity and chronicity indices (13). Because the clinical approach to these patients is the same, those with class III or IV disease or those with combined proliferative and membranous disease were classified as having proliferative disease.
Measurement oxidative modifications of serum protein tyrosine
As a stable, surrogate measure of SO-substrate-mediated oxidative modifications to serum proteins, modified Tyr moieties were determined semiquantitatively by high performance liquid chromatography with electrochemical detection (HPLC-EC) as described by Hensley et al. (14). Modifications to this protocol included analysis of chlorotyrosine (ClTyr, product of HOCl, produced from myeloperoxidase using SO substrate), meta-Tyr, and ortho-Tyr (mTyr and oTyr, produced by hydroxyl radical reaction with phenylalanine) through changes in solvent gradients. Serum proteins were precipitated with cold ethanol (10x volume twice) and lyophilized prior to digestion with 1mg/ml pronase (Pronase from Streptomyces griseus, cat. no. 0165921, Roche Diagnostics Corporation, Indianapolis, IN). The sample was lyophilized, resuspended in 80μl Mili-Q water, adjusted to a final concentration of 8 mg/ml, and injected into a C18 column (Shiseido CAPCELL PAK C18 UG80 S-5μm 4.6 × 250 mm) with a gradient as follows: 5% methanol, 3mg/L sodium dodecyl sulfate (SDS), 50mM sodium phosphate (pH 3.2) at 0.8 ml/minute followed by 50% methanol, 30 mg/L of SDS (pH 3.2) beginning 15 minutes after all analytes eluted. Amino acids were detected using the ESA CoulArray model 5600 electrochemical detector (250, 400, 485, 725, 760, 975, and 1000 mVolts) and UV detection at 280nm. Waveforms were collected using ESA CoulArrayWin v1.12 & v3.05 software and analyzed using in house curve fitting software in MathWorks (MATLAB R2009a). Standard curves of each analyte were created with each injection run. Pronase only blanks were used to calculate background Tyr concentrations of pronase Tyr from autodigestion. The NTyr peak was further confirmed by reduction of NTyr in the sample with 1mM sodium dithionite to form aminotyrosine (15). Each sample was analyzed in duplicate with an accompanying sample spiked with known quantities of standard. Injections were rejected and repeated when the standard deviation of duplicate measures was greater than 10% of the mean or if sample Tyr concentrations were below 25μg. Random injection of known standard quantities was performed throughout each run to detect any drift in retention time or changes in detector response. Tyr modifications were reported as concentrations of each analyte normalized to Tyr. Reported were the sum of all oxidative modifications and isolated NTyr modification.
Data collection and validation
Clinical data were first transcribed into a paper source document and then entered into a Microsoft Access database. Data were quality checked for biologic outliers, and random data checks were performed. De-identified data were exported to an Excel spreadsheet for analysis using SAS 9.3.
Statistical analysis
Descriptive statistics were calculated for all demographic and clinical characteristics. These were compared in two patient groups: those with PLN (class III or IV or these classes combined with class V) and those with non-proliferative lupus nephritis (class II or V or combined classes II and V). Chi-square tests, Fisher’s exact tests, and Wilcoxon rank sum tests were used as appropriate. A total oxidized Tyr variable representing several reactions with a common SO substrate was created by summing all modified Tyr measures reported relative to sample Tyr levels (oTyr/Tyr + mTyr/Tyr + NTyr/Tyr + ClTyr/Tyr) collected −1 ± 9 days from the biopsy date. As the data did not fit a normal distribution, this variable was compared in the two LN groups using a non-parametric Wilcoxon rank sum test. Individual oxidized Tyr markers, NIH activity and chronicity indices, and urine cytokine levels were correlated using Spearman correlations.
Murine Protocol
Mice and sample collection
The Ralph H. Johnson VA Institutional Animal Care and Use Committee approved all procedures. MRL/MpJ-FASlpr (MRL/lpr) mice purchased from Jackson Laboratory (Bar Harbor, ME) were housed under specific pathogen–free conditions. Mice were serologically tested for common murine pathogens. At 13–20 weeks of age (after the onset of proteinuria ≥100 mg/dl by dipstick), mice were anesthetized with age-matched littermate pairs for harvest of renal tissue. Renal cortical tissue was removed from kidneys, snap frozen in liquid nitrogen and stored at −80°C for analysis.
Generation of the endothelial nitric oxide synthase (eNOS) knockout (NOS3−/− MRL/lpr mice
B6.129P2-Nos3<tm1Unc>/J mice purchased from Jackson Laboratories were bred onto the MRL/MpJFas/lpr background. These NOS3−/− mice were backcrossed 9 times to MRL/lpr mice. Speed congenics techniques were used as previously described to ensure backcross of MRL/lpr susceptibility loci to the NOS3−/− mice (16). Fifteen genetic susceptibility loci and the NOS3−/− genotype were confirmed by PCR (17, 18). MRL/lpr NOS3 +/− mice were bred to generate wild type MRL/lpr (NOS3+/+) and MRL/lpr NOS3−/− (NOS3−/−) littermates for the subsequent studies. To maintain the MRL/lpr background phenotype, NOS3 −/− male mice were periodically backcrossed with the MRL/lpr female mice from Jackson Laboratories. NOS3−/− mice from these experiments had significantly lower crescentic and necrotic glomerular disease compared to NOS3+/+ mice as described by our laboratory (19).
Lucigenin assay to determine renal cortical SO production
Mouse renal cortices were homogenized in a detergent-free lysis buffer (pH 7.4) containing: 150 mM NaCl, 50 mM tris(hydroxymethyl)aminomethane (Tris), 25 mM ethylene glycol tetraacetic acid (EGTA), 25 mM ethylenediaminetetraacetic acid (EDTA), and a protease inhibitor and phosphatase inhibitor cocktail. Freshly prepared homogenates were then used to determine spontaneous SO production using lucigenin-enhanced chemiluminescence as described (20, 21). A total volume of 100 μl of homogenate was incubated for 30 minutes at 37 °C with the following: no treatment, L-NIL (10 μM), DPI (10 μM), allopurinol (50 μM), L-NAME (100 μM), SNAP (30 μM), DETA NONOate (50 μM), L-Arg (1 M), L-NIO (10 μM), rotenone (20 μM). Incubated samples (50 μl) were placed in a 24-well reading plate containing 25 μM lucigenin (Sigma) for the detection of SO in a final volume of 500 μl of Krebs solution buffered with 10 mmol/l HEPES-NaOH (pH 7.4). Readings for each sample were done using Luminoskan luminometer at 37 °C in triplicates and normalized to protein level measured by protein assay (Bio-Rad) after background subtraction. Separate assays were performed from similar experiments reported previously (19).
Statistical analysis
Raw values for lucigenin fluorescence were normalized in each experiment to control values from MRL/lpr mice with no inhibitors present. At least three experiments were performed, with three replicates each. Normalized values were compared between genotypes and inhibitor conditions using the Student t-test or Wilcoxon rank sum analysis depending on the distribution of the data. P values < 0.05 were considered significant.
Results
Study population
Enrollment
To determine if markers of RI production predicted PLN, we enrolled lupus patients before or immediately after renal biopsy from June of 1998 to February of 2011. The time of sample collection relative to renal biopsy was −1 ± 9 days. 83 patients were enrolled at the time of biopsy, and 58 subjects met entry criteria. A subset of 33 patients was selected for analysis of serum modified Tyr based on the availability of serum for analysis.
Characteristics of patients with and without PLN
A total of eight subjects with class II or V and 25 subjects with class III or IV were enrolled. The patients reflected the demographics of our clinic population (Table 1), as they were mostly young, African-American women. A majority had active, PLN (classes III and IV), and most had segmental glomerular lesions. As expected, those with PLN had lower complement and higher dsDNA antibody levels.
Table 1.
Demographic, biopsy, and laboratory characteristics of patients with proliferative (Class III or IV) or non-proliferative LN.
| Variable | Class II or V (N=8) | Class III or IV (N=25) | P-value |
|---|---|---|---|
| Sex (% female) | 75.0 | 80.0 | 0.76 |
| Race (% black) | 100.0 | 72.0 | 0.09 |
| Age: median (IQR) | 36 (26–44) | 27 (23–36) | 0.27 |
| ISN/RPS Active lesions (%) | 0 | 96 | <0.0001 |
| ISN/RPS Chronic lesions (%) | 12 | 52 | 0.10 |
| ISN/RPS Global lesions (%) | 0 | 8 | 1.0 |
| ISN/RPS Segmental lesions (%) | 38 | 92 | 0.004 |
| NIH Chronicity Score: median (IQR) | 4.5 (0.3–7.8) | 2.0 (1.0–3.5) | 0.47 |
| NIH Activity Score: median (IQR) | 1.0 (1.0–1.8) | 7.0 (5.0–9.0) | 0.0003 |
| Interstitial Fibrosis (%) | 80 | 55 | 0.59 |
| Interstitial Inflammation (%) | 20 | 70 | 0.12 |
| Urine Protein/Creatinine: median (IQR) | 1.3 (0.8–2.9) | 1.9 (0.7–4.6) | 0.66 |
| Serum dsDNA Antibodies (% positive) | 50 | 92 | 0.02 |
| Serum C3: median (IQR) | 77.6 (64.4–95.1) | 48.6 (34.3–70.4) | 0.03 |
| Serum C4: median (IQR) | 14.4 (11.2–26.8) | 9.9 (9.9–15.1) | 0.08 |
| Serum Creatinine: median (IQR) | 1.1 (0.8–2.6) | 0.9 (0.8–1.9) | 0.49 |
| Prednisone (mg/day): median (IQR) | 20 (4–35) | 30 (5.5–60) | 0.58 |
Markers of oxidative stress were higher in proliferative than membranous or mesangial lupus nephritis
During the baseline visit, serum was obtained and analyzed for oxidative modifications of Tyr as a surrogate for SO production. These values were normalized to unmodified Tyr in the same sample and reported as this ratio x 1000. These specific markers were chosen because they are stable for the life of the protein (akin to a hemoglobin A1C), and all require SO as a substrate. Levels of these markers were determined for patients within the different classes of LN. There was a significant difference in the total of oxidatively modified Tyrs between those with and without PLN (p=0.02). Those with PLN had a median ratio of 1.5 (IQR: 0.9 to 6.0), while those with non-proliferative disease had a median value of 0.8 (IQR: 0.6 to 1.2) (Figure 1A). NTyr was not significantly different between groups but trended toward being greater in those with PLN (Figure 1B). Of note, only a portion of patients with PLN had values elevated above those without PLN. Therefore, a post hoc analysis was performed to determine association with markers of SO production and alternative measures of glomerular proliferative disease. Those with ISN/RPS Active lesions had greater levels of serum total oxidative modifications of Tyr (1.5 (IQR: 1.0 to 8.3) vs. 0.8 (IQR: 0.4 to 1.1), p = 0.003, Figure 2). Finally, NIH Activity Index, not NIH Chronicity Index scores, correlated with total oxidized serum Tyr (Figure 3A, r = 0.47, p 0.005). Of not NTyr was the only isolated oxidative modification that correlated with NIH Activity Index (Figure 3B, r = 0.34, p = 0.037). In a post hoc analysis, the effect of prednisone dose at the time of biopsy was explored. Prednisone dose was no different between those with proliferative or non-proliferative lesions (Table I). Prednisone dose at biopsy did not correlate with NIH Activity Index (r = −0.01) or the sum of oxidized Tyr modifications (r = −0.30, p = 0.22). In a linear regression analysis, presnisone dose and oxidized Tyr modifications were used as input variables with NIH Activity Index as the ouput variable. The model r was similar to that of the correlation analysis (r = 0.43, p = 0.2). All of these observations suggest that SO production was increased in some patients with more active variants of PLN.
Figure 1. Serum markers of superoxide production are increased in PLN.
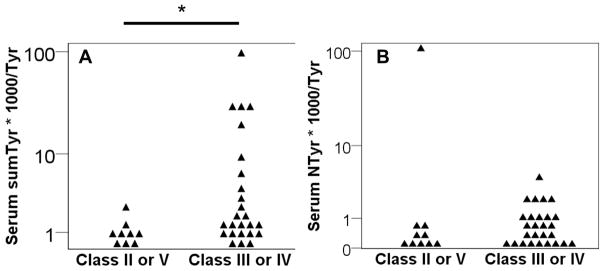
Serum 33 from patients with LN at the time of biopsy was analyzed by high performance liquid chromatography with electrochemical detection. A) The sum of all modifications as a marker of common SO substrate production (sum of ClTyr, NTyr, oTyr, and mTyr; labeled sumTyr*1000/Tyr) is reported as the log of the ratio to Tyr multiplied by 1000. B) Serum NTyr*1000/Tyr is reported separately * p = 0.02
Figure 2. Serum markers of superoxide production are increased in patients with ISN/RPS Active lesions.

SumTyr*1000/Tyr was calculated for all 33 subjects depicted in Figure 1. ISN/RPS Active classification of renal biopsies was performed. SumTyr*1000/Tyr was reported according to the presence (yes) or absence (no) of Active classification. * p = 0.003.
Figure 3. Total oxidized serum protein Tyr correlates with renal biopsy activity score.
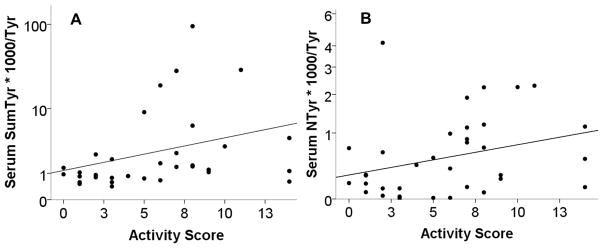
The sum of A) all serum modified Tyr or B) NTyr modification (minus one outlier to show scale) from Figure 1 was correlated to the NIH activity score from the renal biopsy performed at the time of the serum collection. Results are reported as the log of the modified serum Tyr against the biopsy activity score (r = 0.47, p = 0.005 and r = 0.34, p = 0.037 respectively).
Superoxide production in PLN renal cortical tissue was modulated by eNOS and catalyzed by nitric oxide synthase (NOS) and NADPH oxidase (NOX)
Research on targeting redox-mediated inflammatory signaling cannot progress without knowledge of the enzyme source of SO in PLN. The following experiments were designed to determine in mice the enzyme source and regulation of SO production in PLN in a fashion not possible in humans with PLN. Kidneys from MRL/lpr mice with and without a functional NOS3 gene (eNOS) were harvested during active disease. Cortical tissue homogenate was analyzed using lucigenin as a marker SO production. NOS3−/− mouse cortical tissue had significantly increased SO production when compared to levels in NOS3+/+ mice (p < 0.03, Figure 4), suggesting that eNOS modulates SO production in PLN. To determine enzyme sources of SO, inhibitors of known SO-producing enzymes were added to the homogenate. Only inhibitors of NOS and, by exclusion, NOX significantly (Figure 4, p < 0.001) reduced SO production. All other inhibitors failed to significantly reduce SO production as reported (22). For the first time, these combined data suggest that NOX is an enzyme source of SO in PLN that is modulated by low levels of eNOS-derived NO.
Figure 4. Renal cortical SO production is modulated by eNOS and increased by NOS and NOX in MRL/lpr mice with active LN.

Renal cortical tissue homogenate from MRL/lpr mice with and without a functional eNOS gene (NOS3+/+ and NOS3−/−) was analyzed for SO production by the lucigenin assay. Inhibitors of NOS and NOX significantly reduced SO production in NOS3−/− mice. Results were normalized to levels seen in wild-type (NOS3+/+) mice with no inhibitors (n = 9 replicates) and reported as the mean ± standard error. * p < 0.03 vs NOS3+/+ with same treatment. ** p < 0.001 vs. NOS3−/− control
Discussion
To our knowledge, this is the first description of increased markers of SO production specific to active PLN before induction therapy. In addition, it is the first study to identify NOX and uncoupled NOS as major enzyme sources of ROS specifically in the cortical tissue in proliferative LN. Finally, this is the first demonstration that eNOS is a key modulator of SO production in PLN. Knowledge of these enzymes sources could potentially lead to therapies designed to target the effects of redox signaling in the more clinically significant proliferative form of LN.
The findings of this study are consistent with the work of Moroni et al., in which they demonstrated increased markers of SO production in patients with LN. However, the study did not isolate this finding to patients with PLN or associate SO production with active LN as the current study has (23). Nishikawa and colleagues used modified SO dismutase (SOD) to accelerate the conversion of SO to H2O2 in the NZB/W model of lupus nephritis. In that study, SOD reduced evidence of glomerular and interstitial hypercellularity in a dose dependent fashion (24), suggesting that SO is not just a marker but is rather a mediator of disease. These findings were echoed in a study by Suwannaroj et al. (25). In that study, thiol antioxidants n-acetylcysteine (NAC) and cysteamine (CYST) were used to prevent nephritis. Both improved survival, but the ability to suppress glomerular cellularity was highly variable between these two treatments, with NAC prevailing over CYST. This differential effect suggests that natural induction of thiol antioxidant (glutathione) synthesis in vivo with NAC was a better alternative than giving an oral thiol antioxidant (CYST). Thus, therapies that naturally increase antioxidant capacity could prove to be more successful in vivo. The current study adds to the literature by identifying enzyme sources of SO production specific to PLN for therapeutic intervention.
SO and its metabolites can mediate changes in signaling, transcription, and enzyme activity via post translational modifications of proteins. Irreversible modifications of the aromatic amino acid Tyr can occur in the presence of OH•, HOCl, or ONOO−. These highly reactive oxidant species modify target molecules without any particular specificity to enzyme. Thus, the target species are selected by proximity to the production of the oxidizing species and susceptibility to oxidation. Less potent species such as H2O2 and NO diffuse across membranes and can induce reversible thiol oxidation or nitrosylation similar to kinase-mediated phosphorylation of serine, threonine, and Tyr but with less specificity (9). These cysteine modifications can lead to changes in enzyme activity and transcription as described below.
Mechanisms of disease from increased SO production in LN have not been well defined. However, general mechanisms proximal and distal to immune complex deposition have been described. For instance, oxidation of native dsDNA can create neo-antigens with increased binding to dsDNA antibodies (26). A similar phenomenon occurs when DNA is modified by peroxynitrite (27). However, our laboratory has demonstrated that NOS-mediated free radical production in murine LN can be reduced with inhibitors of NOS (8) in a fashion that does not affect glomerular immune complex deposition (28). Therefore, focus on pathologic redox-mediated signaling distal to immune complex deposition may be warranted. The function of endothelial cells (EC) is critical as a bridge between immune complex deposition and egress of inflammatory cells from the circulation into tissue.
EC, when activated by inflammatory stimuli, play a central role in inflammatory cell activation, chemotaxis, rolling, adhesion, and signaling for migration of inflammatory cells into tissue (29). This process is induced by redox-mediated transcriptional regulation of multiple inflammatory genes. The activity of transcription factors AP1 (cJun), NFκB, HIF1α, and p53 are all redox-regulated, some via the effect of redox modifications to Ref-1 binding (30). H2O2 leads to NFκB activation via its activation of the tyrosine kinases Lck or Src, leading to phosphorylation of IκB and translocation of NFκB to the nucleus (31). Two inflammatory cytokines, IL6 and MCP1 (CCL2), important to the pathogenesis of lupus nephritis (32–37), have in common redox-regulated NFκB, AP-1, and c-Jun response elements (38). Expression of both MCP1 and IL6 by activated EC are regulated by ROS production and can be inhibited by ROS scavengers (39) and inhibitors of NADPH oxidase (39) Whether redox-regulation of transcription of IL6 and MCP1 is important in LN is not directly tested by this study.
The tissue source of SO production in PLN is of great importance in translation of the findings to therapeutic development. Our laboratory has reported that systemic SO production is partially dependent on inducible NOS, as inhibitors of NOS reduce urine 8-isoprostane levels by half (8). Because competitive NOS inhibitors reduce SO production from uncoupled NOS (40), the source of SO in that study was likely uncoupled iNOS. Uncoupling occurs in the presence of oxidized tetrahydrobiopterin (41), a protein essential to homodimerization of NOS. The source of SO that leads to this oxidation in LN is not clear, however, this study strongly suggests that NOX is an important catalyst of SO production in PLN. The association of activity of glomerular lesions with NTyr suggests that NOS (likely inducible NOS or iNOS (3, 4)) is increased in active lesions and that both SO and NO are being produced in these lesions by NOS.
The homologs NOX1, NOX2, and NOX4 are expressed in ECs, with NOX1 and NOX4 being highly expressed in the kidney (42). These subunits are essential for NADPH oxidase catalytic activity, resulting in the formation of SO (NOX1/2) or SO and H2O2 (NOX4). Expression of NOX is prominent in the vessels, glomeruli, podocytes, and tubular cells. Regulation of SO production by NOX is PLN is not well understood. However, stimulation of Fc receptors FcγRI IIa, and IIIb in phagocytic cells induces ROS production in a fashion that may further activate Fc receptor signaling in nearby cells (43). In EC, Fc receptors also signal for ROS production in a NOX-dependent fashion (44, 45). How and whether this mechanism is essential for NOX-mediated ROS production in LN is not addressed by this study. From this study, the source of NOX-mediated ROS (mesangial cells, proximal tubular cells, podocytes, endothelial cells, or infiltrating inflammatory cells) cannot be determined and warrants further investigation.
eNOS was an important modulator of SO production in this study. Others have demonstrated in EC that low levels of NO suppress NOX-mediated SO production, in part due to S-nitrosylation of the organizer subunit p47phox (46). In mesangial cells, NOX1 transcription is also regulated by NO (47). Thus, reduced effects of eNOS-derived NO production on NOX is a potential source of increased SO production in PLN. This is consistent with the biology of human PLN, as reduced eNOS expression has been described in PLN (4). Global reduction in eNOS-derived NO production leading to endothelial dysfunction has also been described in SLE. This is evident in the observation that brachial artery flow mediated dilation (dependent on endothelial eNOS activity) is reduced in SLE patients (48). However, this study’s findings on the effect of eNOS deficiency on SO production in PLN is important for designing effective therapies to target redox signaling. Strengths of this study include its focus on SO production in human PLN and identification of NADPH oxidase as a source of SO production in murine PLN that could be targeted to improve outcomes. The impact of this study on understanding the role of SO in PLN is increased by the use of durable markers of SO-mediated protein oxidation. These markers do not fluctuate in vivo as rapidly as do urine isoprostanes, for instance (49). Use of these markers could identify individuals in whom redox signaling may play a role in the transition from humoral autoimmunity to inflammatory end organ disease. One could envision using them as pharmacodynamic indicators of treatment designed to reduce pathologic SO production. Limitations of this study include the associative nature of the human studies, making it difficult to determine the temporal relationship between SO production and inflammation. The findings indicate that there is only a subset of patients in whom markers of SO production reflect glomerular activity. However, knowledge of that subset could allow a personalized medicine approach to those in whom redox signaling may be occurring, using the markers to select therapy and as pharmacodynamic indicators of its effect. Translating the findings of the murine studies back to human disease cannot be done with complete confidence, as human disease is more heterogeneous. Thus, reduced eNOS function may be an important pathogenic pathway in only a subset of humans with PLN.
This is the first study to date to demonstrate that SO production is increased in proliferative (ISN/RPS classes III and IV) compared to non-proliferative (classes II and V) LN. This finding is strengthened by the identification of NADPH oxidase and NOS as enzyme sources of SO production and eNOS as a modulator of that production. These findings are clinically important because they suggest class-specific targets of therapy for patients with PLN, who are more likely to progress to renal failure (12, 13). Future studies could thus focus on methods of regulating the cause and consequences of NOS uncoupling and NOX activity to improve outcomes in proliferative LN and the use of oxidatively modified Tyr as a pharmacodynamic indicator of treatment success.
Summary.
Lupus nephritis (LN) is an immune complex-mediated glomerulonephritis. Superoxide and its metabolites are mediators of the innate immune response produced in the glomerulus during renal disease that mediate reduction-oxidation-mediated cellular signaling for inflammation. We hypothesized that serum markers of superoxide production would be increased in the proliferative form of LN (PLN) and that superoxide production would be dependent on the activity of select enzymes in the renal cortex. Patients with systemic lupus erythematosus were enrolled at the time of renal biopsy for active LN. Serum collected at baseline was analyzed for durable serum markers of superoxide production. Renal cortex from MRL/MpJ-FASlpr (MRL/lpr) mice with and without functional endothelial nitric oxide synthase (eNOS) was analyzed during active disease for superoxide production with and without inhibitors of superoxide producing enzymes. Serum markers of superoxide production were significantly higher in patients with PLN. Mice lacking functional eNOS had 80% higher levels of renal cortical superoxide during active disease, and inhibitors of NADPH oxidase and nitric oxide synthase (NOS) reduced these levels by 76% and 61% respectively. These studies offer the rationale for targeted therapies designed to emulate or stimulate eNOS activity or inhibit NADPH oxidase-mediated superoxide production in PLN.
Acknowledgments
This work was supported by the Arthritis Foundation, Atlanta, GA, the University Research Committee at the Medical University of SC, the Medical University of SC General Clinical Research Center [NIH grant number MO1RR001070], the Medical University of SC Clinical and Translational Science Award [grant number UL1TR000062, formerly U54RR026107], the Division of Rheumatology and Immunology Multidisciplinary Clinical Research Center [grant number P60AR062755], and National Institutes of Health [grant numbers K08AR002193, AI047469, AR045476, and AR04745], the Ralph H. Johnson VAMC Medical Research Service, and the Department of Veterans Affairs Career Development, Research Enhancement Awards.
Special thanks go to the patients who participated in this study. This project would not have been possible without coordination from Lori Ueberroth, Stephanie Slan, Tia Parker and technical support from Thomas Fleury, Jon Donohue, and Ann Hofbauer. Sally E Self, MD deserves special recognition for performing the classification of renal biopsies.
Footnotes
Conflict of Interest Statement
The authors declare no conflict of interest. None of the potential conflicts of interest (commercial or nonprofit) are relevant to this work. No commercial or noncommercial products
Contributor Information
Jim C. Oates, Department of Medicine, Division of Rheumatology, Medical University of South Carolina, Charleston, SC and Medical Service, Ralph H. Johnson VA Medical Center, Charleston, SC.
Ahmad K. Mashmoushi, Department of Medicine, Division of Rheumatology, Medical University of South Carolina, Charleston, SC.
Stephanie R. Shaftman, Department of Biostatistics, Bioinformatics & Epidemiology, Medical University of South Carolina, Charleston, SC.
Gary S. Gilkeson, Department of Medicine, Division of Rheumatology, Medical University of South Carolina, Charleston, SC and Medical Service, Ralph H. Johnson VA Medical Center, Charleston, SC.
References
- 1.Weening JJ, D’Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, Balow JE, Bruijn JA, Cook T, Ferrario F, Fogo AB, Ginzler EM, Hebert L, Hill G, Hill P, Jennette JC, Kong NC, Lesavre P, Lockshin M, Looi LM, Makino H, Moura LA, Nagata M. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int. 2004;65:521–30. doi: 10.1111/j.1523-1755.2004.00443.x. [DOI] [PubMed] [Google Scholar]
- 2.Dooley MA, Hogan S, Jennette C, Falk R. Cyclophosphamide therapy for lupus nephritis: poor renal survival in black Americans. Glomerular Disease Collaborative Network. Kidney Int. 1997;51:1188–95. doi: 10.1038/ki.1997.162. [DOI] [PubMed] [Google Scholar]
- 3.Oates JC, Christensen EF, Reilly CM, Self SE, Gilkeson GS. Prospective measure of serum 3-nitrotyrosine levels in systemic lupus erythematosus: Correlation with disease activity. Proceedings of the Association of American Physicians. 1999;111:611–21. doi: 10.1046/j.1525-1381.1999.99110.x. [DOI] [PubMed] [Google Scholar]
- 4.Furusu A, Miyazaki M, Abe K, Tsukasaki S, Shioshita K, Sasaki O, Miyazaki K, Ozono Y, Koji T, Harada T, Sakai H, Kohno S. Expression of endothelial and inducible nitric oxide synthase in human glomerulonephritis. Kidney Int. 1998;53:1760–8. doi: 10.1046/j.1523-1755.1998.00907.x. [DOI] [PubMed] [Google Scholar]
- 5.Oates JC, Shaftman SR, Self SE, Gilkeson GS. Association of serum nitrate and nitrite levels with longitudinal assessments of disease activity and damage in systemic lupus erythematosus and lupus nephritis. Arthritis Rheum. 2008;58:263–72. doi: 10.1002/art.23153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Njoku C, Self SE, Ruiz P, Hofbauer AF, Gilkeson GS, Oates JC. Inducible nitric oxide synthase inhibitor SD-3651 reduces proteinuria in MRL/lpr mice deficient in the NOS2 gene. J Investig Med. 2008;56:911–9. doi: 10.231/JIM.0b013e3181889e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilkeson GS, Mudgett JS, Seldin MF, Ruiz P, Alexander AA, Misukonis MA, Pisetsky DS, Weinberg JB. Clinical and serologic manifestations of autoimmune disease in MRL-lpr/lpr mice lacking nitric oxide synthase type 2. J Exp Med. 1997;186:365–73. doi: 10.1084/jem.186.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Njoku CJ, Patrick KS, Ruiz P, Jr, Oates JC. Inducible nitric oxide synthase inhibitors reduce urinary markers of systemic oxidant stress in murine proliferative lupus nephritis. J Investig Med. 2005;53:347–52. doi: 10.2310/6650.2005.53705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Janssen-Heininger YM, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG, van der Vliet A. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med. 2008;45:1–17. doi: 10.1016/j.freeradbiomed.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiebeler A, Park JK, Muller DN, Lindschau C, Mengel M, Merkel S, Banas B, Luft FC, Haller H. Growth arrest specific protein 6/Axl signaling in human inflammatory renal diseases. Am J Kidney Dis. 2004;43:286–95. doi: 10.1053/j.ajkd.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 11.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 12.Austin HA, 3rd, Muenz LR, Joyce KM, Antonovych TA, Kullick ME, Klippel JH, Decker JL, Balow JE. Prognostic factors in lupus nephritis. Contribution of renal histologic data. Am J Med. 1983;75:382–91. doi: 10.1016/0002-9343(83)90338-8. [DOI] [PubMed] [Google Scholar]
- 13.Austin HA, 3rd, Muenz LR, Joyce KM, Antonovych TT, Balow JE. Diffuse proliferative lupus nephritis: identification of specific pathologic features affecting renal outcome. Kidney Int. 1984;25:689–95. doi: 10.1038/ki.1984.75. [DOI] [PubMed] [Google Scholar]
- 14.Hensley K, Williamson KS, Floyd RA. Measurement of 3-nitrotyrosine and 5-nitro-gamma-tocopherol by high-performance liquid chromatography with electrochemical detection. Free Radic Biol Med. 2000;28:520–8. doi: 10.1016/s0891-5849(00)00155-6. [DOI] [PubMed] [Google Scholar]
- 15.Crowley JR, Yarasheski K, Leeuwenburgh C, Turk J, Heinecke JW. Isotope dilution mass spectrometric quantification of 3-nitrotyrosine in proteins and tissues is facilitated by reduction to 3-aminotyrosine. Anal Biochem. 1998;259:127–35. doi: 10.1006/abio.1998.2635. [DOI] [PubMed] [Google Scholar]
- 16.Vyse TJ, Todd JA. Genetic analysis of autoimmune disease. Cell. 1996;85:311–8. doi: 10.1016/s0092-8674(00)81110-1. [DOI] [PubMed] [Google Scholar]
- 17.Vidal S, Kono DH, Theofilopoulos AN. Loci predisposing to autoimmunity in MRL-Fas lpr and C57BL/6-Faslpr mice. The Journal of clinical investigation. 1998;101:696–702. doi: 10.1172/JCI1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watson ML, Rao JK, Gilkeson GS, Ruiz P, Eicher EM, Pisetsky DS, Matsuzawa A, Rochelle JM, Seldin MF. Genetic analysis of MRL-lpr mice: relationship of the Fas apoptosis gene to disease manifestations and renal disease-modifying loci. J Exp Med. 1992;176:1645–56. doi: 10.1084/jem.176.6.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilkeson GS, Mashmoushi AK, Ruiz P, Caza TN, Perl A, Oates JC. Endothelial nitric oxide synthase reduces crescentic and necrotic glomerular lesions, reactive oxygen production, and MCP1 production in murine lupus nephritis. PLoS ONE. 2013 doi: 10.1371/journal.pone.0064650. submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hagiwara M, Bledsoe G, Yang ZR, Smith RS, Jr, Chao L, Chao J. Intermedin ameliorates vascular and renal injury by inhibition of oxidative stress. Am J Physiol Renal Physiol. 2008;295:F1735–43. doi: 10.1152/ajprenal.90427.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupte SA, Levine RJ, Gupte RS, Young ME, Lionetti V, Labinskyy V, Floyd BC, Ojaimi C, Bellomo M, Wolin MS, Recchia FA. Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J Mol Cell Cardiol. 2006;41:340–9. doi: 10.1016/j.yjmcc.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 22.Gilkeson GS, Mashmoushi AK, Ruiz P, Caza TN, Perl A, Oates JC. Endothelial Nitric Oxide Synthase Reduces Crescentic and Necrotic Glomerular Lesions, Reactive Oxygen Production, and MCP1 Production in Murine Lupus Nephritis. PloS one. 2013;8:e64650. doi: 10.1371/journal.pone.0064650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moroni G, Novembrino C, Quaglini S, De Giuseppe R, Gallelli B, Uva V, Montanari V, Messa P, Bamonti F. Oxidative stress and homocysteine metabolism in patients with lupus nephritis. Lupus. 2010;19:65–72. doi: 10.1177/0961203309346906. [DOI] [PubMed] [Google Scholar]
- 24.Nishikawa M, Igarashi R, Nakazawa T, Aikawa E. Rescue of (NZB x NZW) F1 mice from oxygen-derived free radical injury by use of phosphatidylcholine-modified superoxide dismutase. Lab Anim Sci. 1999;49:560–4. [PubMed] [Google Scholar]
- 25.Suwannaroj S, Lagoo A, Keisler D, McMurray RW. Antioxidants suppress mortality in the female NZB x NZW F1 mouse model of systemic lupus erythematosus (SLE) Lupus. 2001;10:258–65. doi: 10.1191/096120301680416940. [DOI] [PubMed] [Google Scholar]
- 26.Cooke MS, Mistry N, Wood C, Herbert KE, Lunec J. Immunogenicity of DNA damaged by reactive oxygen species--implications for anti-DNA antibodies in lupus. Free Radic Biol Med. 1997;22:151–9. doi: 10.1016/s0891-5849(96)00283-3. [DOI] [PubMed] [Google Scholar]
- 27.Griffiths HR. Is the generation of neo-antigenic determinants by free radicals central to the development of autoimmune rheumatoid disease? Autoimmun Rev. 2008;7:544–9. doi: 10.1016/j.autrev.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 28.Weinberg JB, Granger DL, Pisetsky DS, Seldin MF, Misukonis MA, Mason SN, Pippen AM, Ruiz P, Wood ER, Gilkeson GS. The role of nitric oxide in the pathogenesis of spontaneous murine autoimmune disease: increased nitric oxide production and nitric oxide synthase expression in MRL-lpr/lpr mice, and reduction of spontaneous glomerulonephritis and arthritis by orally administered NG-monomethyl-L- arginine. J Exp Med. 1994;179:651–60. doi: 10.1084/jem.179.2.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–15. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- 30.Shah D, Wanchu A, Bhatnagar A. Interaction between oxidative stress and chemokines: possible pathogenic role in systemic lupus erythematosus and rheumatoid arthritis. Immunobiology. 2011;216:1010–7. doi: 10.1016/j.imbio.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 31.Kabe Y, Ando K, Hirao S, Yoshida M, Handa H. Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal. 2005;7:395–403. doi: 10.1089/ars.2005.7.395. [DOI] [PubMed] [Google Scholar]
- 32.Brunner HI, Bennett MR, Mina R, Suzuki M, Petri M, Kiani AN, Pendl J, Witte D, Ying J, Rovin BH, Devarajan P. Association of noninvasively measured renal protein biomarkers with histologic features of lupus nephritis. Arthritis Rheum. 2012;64:2687–97. doi: 10.1002/art.34426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Tucci M, Narain S, Barnes EV, Sobel ES, Segal MS, Richards HB. Urinary biomarkers in lupus nephritis. Autoimmun Rev. 2006;5:383–8. doi: 10.1016/j.autrev.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 34.Kiberd BA. Interleukin-6 receptor blockage ameliorates murine lupus nephritis. J Am Soc Nephrol. 1993;4:58–61. doi: 10.1681/ASN.V4158. [DOI] [PubMed] [Google Scholar]
- 35.Kulkarni O, Pawar RD, Purschke W, Eulberg D, Selve N, Buchner K, Ninichuk V, Segerer S, Vielhauer V, Klussmann S, Anders HJ. Spiegelmer inhibition of CCL2/MCP-1 ameliorates lupus nephritis in MRL-(Fas)lpr mice. J Am Soc Nephrol. 2007;18:2350–8. doi: 10.1681/ASN.2006121348. [DOI] [PubMed] [Google Scholar]
- 36.Wada T, Yokoyama H, Su SB, Mukaida N, Iwano M, Dohi K, Takahashi Y, Sasaki T, Furuichi K, Segawa C, Hisada Y, Ohta S, Takasawa K, Kobayashi K, Matsushima K. Monitoring urinary levels of monocyte chemotactic and activating factor reflects disease activity of lupus nephritis. Kidney Int. 1996;49:761–7. doi: 10.1038/ki.1996.105. [DOI] [PubMed] [Google Scholar]
- 37.Marks SD, Williams SJ, Tullus K, Sebire NJ. Glomerular expression of monocyte chemoattractant protein-1 is predictive of poor renal prognosis in pediatric lupus nephritis. Nephrol Dial Transplant. 2008;23:3521–6. doi: 10.1093/ndt/gfn270. [DOI] [PubMed] [Google Scholar]
- 38.Champion ChiP Transcription Factor Search Portal. 2012 [cited 2012 December 27]; Available from: http://www.sabiosciences.com/chipqpcrsearch.php?app=TFBS.
- 39.Volk T, Hensel M, Schuster H, Kox WJ. Secretion of MCP-1 and IL-6 by cytokine stimulated production of reactive oxygen species in endothelial cells. Mol Cell Biochem. 2000;206:105–12. doi: 10.1023/a:1007059616914. [DOI] [PubMed] [Google Scholar]
- 40.Bendall JK, Alp NJ, Warrick N, Cai S, Adlam D, Rockett K, Yokoyama M, Kawashima S, Channon KM. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ Res. 2005;97:864–71. doi: 10.1161/01.RES.0000187447.03525.72. [DOI] [PubMed] [Google Scholar]
- 41.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. The Journal of clinical investigation. 2003;111:1201–9. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 43.Pricop L, Gokhale J, Redecha P, Ng SC, Salmon JE. Reactive oxygen intermediates enhance Fc gamma receptor signaling and amplify phagocytic capacity. J Immunol. 1999;162:7041–8. [PubMed] [Google Scholar]
- 44.Sumiyoshi K, Mokuno H, Iesaki T, Shimada K, Miyazaki T, Kume A, Kiyanagi T, Kuremoto K, Watanabe Y, Tada N, Daida H. Deletion of the Fc receptors gamma chain preserves endothelial function affected by hypercholesterolaemia in mice fed on a high-fat diet. Cardiovasc Res. 2008;80:463–70. doi: 10.1093/cvr/cvn206. [DOI] [PubMed] [Google Scholar]
- 45.Stielow C, Catar RA, Muller G, Wingler K, Scheurer P, Schmidt HH, Morawietz H. Novel Nox inhibitor of oxLDL-induced reactive oxygen species formation in human endothelial cells. Biochem Biophys Res Commun. 2006;344:200–5. doi: 10.1016/j.bbrc.2006.03.114. [DOI] [PubMed] [Google Scholar]
- 46.Selemidis S, Dusting GJ, Peshavariya H, Kemp-Harper BK, Drummond GR. Nitric oxide suppresses NADPH oxidase-dependent superoxide production by S-nitrosylation in human endothelial cells. Cardiovasc Res. 2007;75:349–58. doi: 10.1016/j.cardiores.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 47.Pleskova M, Beck KF, Behrens MH, Huwiler A, Fichtlscherer B, Wingerter O, Brandes RP, Mulsch A, Pfeilschifter J. Nitric oxide down-regulates the expression of the catalytic NADPH oxidase subunit Nox1 in rat renal mesangial cells. FASEB J. 2006;20:139–41. doi: 10.1096/fj.05-3791fje. [DOI] [PubMed] [Google Scholar]
- 48.Kiss E, Soltesz P, Der H, Kocsis Z, Tarr T, Bhattoa H, Shoenfeld Y, Szegedi G. Reduced flow-mediated vasodilation as a marker for cardiovascular complications in lupus patients. J Autoimmun. 2006;27:211–7. doi: 10.1016/j.jaut.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 49.Ahmadzadehfar H, Oguogho A, Efthimiou Y, Kritz H, Sinzinger H. Passive cigarette smoking increases isoprostane formation. Life Sci. 2006;78:894–7. doi: 10.1016/j.lfs.2005.05.099. [DOI] [PubMed] [Google Scholar]