

Abstract
We report here a synthetically simple yet highly tunable and diverse visible light mediated thiol-vinyl gelation system for fabricating cell-instructive hydrogels. Gelation was achieved via a mixed-mode step-and-chain-growth photopolymerization using functionalized 4-arm poly(ethylene glycol) as backbone macromer, eosin-Y as photosensitizer, and di-thiol containing molecule as dual purpose co-initiator/cross-linker. N-vinylpyrrolidone (NVP) was used to accelerate gelation kinetics and to adjust the stiffness of the hydrogels. Visible light (wavelength: 400–700nm) was used to initiate rapid gelation (gel points: ~20 seconds) that reached completion within a few minutes. The major differences between current thiol-vinyl gelation and prior visible light mediated photopolymerization are that: (1) the co-initiator triethanolamine (TEOA) used in the previous systems was replaced with multifunctional thiols and (2) mixed-mode polymerized gels contain less network heterogeneity. The gelation kinetics and gel properties at the same PEG macromer concentration could be tuned by changing the identity of vinyl groups and di-thiol cross-linkers, as well as concentration of cross-linker and NVP. Specifically, acrylate-modified PEG afforded the fastest gelation rate, followed by acrylamide and methacrylate-functionalized PEG. Increasing NVP concentration also accelerated gelation and led to a higher network cross-linking density. Further, increasing di-thiol peptide concentration in the gel formulation increased hydrogel swelling and decreased gel stiffness. Due to the formation of thiol-ether-ester bonds following thiol-acrylate reaction, the gels degraded hydrolytically following a pseudo first order degradation kinetics. Degradation rate was controlled by adjusting thiol or NVP content in the polymer precursor solution. The cytocompatibility and utility of this hydrogel system were evaluated using in situ encapsulation of human mesenchymal stem cells (hMSC). Encapsulated hMSCs remained alive (>90%) throughout the duration of the study and the cells were differentiated down osteogenic lineage with varying degrees by controlling the rate and mode of gel degradation.
Keywords: Visible light, photopolymerization, thiol-ene, hydrogels, mesenchymal stem cells
1. Introduction
Poly(ethylene glycol) (PEG) based hydrogels are a class of hydrophilic polymers suitable for a variety of tissue engineering applications, including controlled drug delivery and directed stem cell differentiation [1–4]. The termini of PEG are hydroxyl groups that can be easily functionalized to render the otherwise soluble PEG cross-linkable [5]. Vinyl-based PEG macromers are particularly useful because of their simplicity in synthesis and purification. Various vinyl-functionalized PEG macromers have been developed for preparing cytocompatible hydrogels, including (meth)acrylate [6–9], (meth)acrylamide [10, 11], vinylsulfone [12, 13], maleimide [14], norbornene [15, 16], etc. The reactivity of the terminal vinyl group determines the mechanisms of hydrogel cross-linking. For example, PEG-di(meth)acrylate (PEGDA or PEGDM) can be homopolymerized via radical-mediated chain-growth photopolymerization [17–19]. On the other hand, multi-functional PEG-acrylate, PEG-vinylsulfone, or PEG-maleimide ‘clicks’ with thiol-bearing linkers via a step-growth nucleophilic reaction (i.e., Michael-type addition reaction) as long as the average functionality of the vinyl and thiol-bearing macromers is greater than two (f > 2) [20]. Hydrogels prepared from these step-wise and orthogonal reactions are increasingly employed as synthetic extracellular matrix (ECM) or tissue scaffold due to their improved network properties and diversity [12–14]. Step-growth hydrogels can also be prepared by radical-mediated photopolymerization. For example, Anseth and colleagues prepared multi-arm PEG-norbornene that reacts orthogonally with bi-functional thiol linker via a radical mediated thiol-ene reaction [15]. Other orthogonal click chemistries are also increasingly explored for forming PEG-based hydrogels [21–23].
While the inert or ‘blank slate’ property of PEG hydrogels contribute greatly to their biocompatibility, the utility of ‘plain’ PEG hydrogels are limited in cell culture or for tissue regeneration. Oftentimes PEG hydrogels are rendered biomimetic via functionalizing the network with bioactive motifs. For example, a fibronectin-derived RGDS is routinely immobilized in the PEG hydrogels to support cell attachment, survival, and differentiation [19, 24]. The methods for immobilizing bioactive molecules within the inert PEG microenvironment have also been extensively studied. For example, Hern and Hubbell pioneered the conjugation of oligopeptides in PEG hydrogels by homopolymerization of PEGDA with acrylated peptides [19]. This method, however, requires the functionalization of peptide primary amine groups that may be a part of the binding sites for cell surface receptors. Alternatively, the conjugation of bioactive motifs to PEG-based hydrogels can be achieved via a mixed-mode thiol-acrylate or a step-growth thiol-ene reaction mechanism [25, 26]. In these cases, the peptides to be immobilized can be synthesized with additional cysteine residues added in the peptide sequence, thereby bypassing the additional peptide modification and purification steps. In mixed-mode photopolymerization, additional chain growth reactions occur when the ratio of acrylate to thiol functionalities exceeds unity. Peptide substrates are also used as part of hydrogel cross-links to mimic structural proteins in the extracellular matrix [12, 13]. Incorporation of cell-cleavable peptide substrates in hydrogel network permits cell-mediated matrix remodeling for cellular activities such as migration and proliferation. These cell-instructive hydrogels can be formed by step-growth Michael-type addition reaction or thiol-ene photoclick reaction [12, 13, 15]. Chain-growth polymerized linear PEG macromers can also be rendered proteolytically responsive. However, additional conjugation steps are required to prepare cross-linkable PEG-peptide-PEG macromers [27–30].
Among the crosslinking methods, photopolymerization offers experimenters great degrees of control (e.g., spatial-temporally defined gelation kinetics). Both ultraviolet (UV) and visible light sources are commonly used to initiate photopolymerization for forming cell-laden hydrogels as long as an appropriate photoinitiator system is employed [31, 32]. UV light (250 nm < λ < 400 nm) mediated polymerization is typically initiated by a cleavage type (type 1) photoinitiator (e.g., Irgacure 2959) [3, 16, 24, 32, 33]. Upon photon absorption, each photoinitiator decomposes into two radical species. The radicals either propagate through vinyl groups on functionalized PEG macromers (e.g., PEGDA) via a chain-growth polymerization mechanism or abstract protons from thiol-containing molecules to form step-growth cross-links with appropriate vinyl functionalities. Alternatively, visible light (400 nm < λ < 700 nm) can be used with a type 2 initiator (e.g., eosin-Y, rose bengal, etc.) to initiate photopolymerization [34, 35]. The most commonly used visible light mediated cross-linking of hydrogels involves the excitation of dye molecules into a triplet state for extracting hydrogen atoms from amine-bearing co-initiators (e.g., triethanolamine, TEOA) [36–39]. The deprotonated radicals then initiate vinyl bond cross-linking via a chain-growth polymerization mechanism. Apart from the difference in initiation mechanisms, visible light mediated polymerizations often require co-monomers (e.g., N-vinylpyrrolidone or NVP) to accelerate gelation [10].
We have recently reported a visible light mediated thiol-ene photopolymerization scheme to form step-growth PEG hydrogels [40, 41]. Mechanistically, an excited eosin-Y abstracts hydrogen from cysteine-bearing peptide linker to produce thiyl radicals that ‘click’ with norbornene-functionalized PEG macromers. Uniquely, this cross-linking scheme does not rely on other co-initiator (e.g., TEOA) or co-monomer (e.g., NVP), thus greatly simplifies material preparation [40]. In addition to the PEG-norbornene macromer that we previously used, we hypothesized that other vinyl macromers can also be used in this visible light mediated thiol-vinyl reaction to initiate hydrogel crosslinking. The objective of this study was to demonstrate that visible light mediated photopolymerization could be used to prepare vinyl-based PEG hydrogels without the use of cytotoxic TEOA. To this end, four PEG-based vinyl macromers were used for evaluating the potential of this new cross-linking method, including PEG-tetra-acrylate (PEG4A), PEG-tetra-acrylamide (PEG4AA), PEG-tetra-methacrylate (PEG4MA), and PEG-tetra-allylether (PEG4AE). The use of different macromers affects the cross-linking efficiency and hydrolytic stability of the resulting hydrogels. Although TEOA was not required in this visible light mediated thiol-vinyl cross-linking scheme, the addition of NVP was necessary for rapid gelation. Differing from previous visible light mediated photopolymerization of PEGDA hydrogels where TEOA was used solely as a co-initiator, the current thiol-vinyl photo-crosslinking scheme uses multifunctional thiol-containing molecules that act as co-initiator and cell-responsive linker. Furthermore, the concentration of thiols (< 8 mM) required to initiate gelation was only a fraction of the concentration of TEOA (>200 mM) typically used in prior visible light mediated gelation schemes. It should be noted that the only function of TEOA in chain-growth gelation schemes is to provide radical species. An exceptionally high concentration (e.g., 225mM [30, 42, 43]) of TEOA was often used to generate sufficient radicals to initiate gelation. The use of high TEOA concentration may cause undesired cytotoxic effect to some sensitive cell types [40]. Because of the use of dual-purpose thiol-bearing co-initiator/cross-linker, this new gelation scheme also offers many benefits for preparing cell-instructive biomimetic matrices. Here, we used human mesenchymal stem cells (hMSCs) as an example to demonstrate the cytocompatibility and utilities of this new class of hydrogels. hMSCs were encapsulated, cultured, and differentiated within cell instructive hydrogels constructed using PEG4A, unmodified protease-sensitive peptide, and NVP. The inclusion of NVP did not affect cell viability but altered cell fate determination, including spreading and osteogenic differentiation.
2. Materials & methods
2.1 Materials
4-arm PEG-hydroxyl and 4-arm PEG-amine (both 20kDa) were obtained from JenKem Technology USA. Eosin-Y disodium salt, TEOA, and NVP were purchased from Thermo Fisher. Fmoc-amino acids and peptide synthesis reagents were purchased from Chempep or Anaspec. HPLC grade acetonitrile and water were acquired from Thermo Fisher and VWR international, respectively. DMEM and DPBS were purchased from Thermo Fisher while other reagents for cell culture were obtained from Life Technologies. AlamarBlue® cell viability/metabolic indicator was procured from AbD Serotec. All other chemicals were acquired from Sigma-Aldrich unless otherwise noted.
2.2 Synthesis of PEG Macromers
PEG-tetra-acrylate (PEG4A) and PEG-tetra-acrylamide (PEG4AA) were synthesized using 4-arm PEG-hydroxyl (PEG-OH) or 4-arm PEG-amine (PEG-NH2) [10]. Prior to reaction, PEG-OH or PEG-NH2 was dried azeotropically in anhydrous toluene. Then, acryloyl chloride (8 eq. of OH or NH2 group on PEG) and triethylamine (TEA, 8 eq.) dissolved in anhydrous toluene were added slowly to the PEG/toluene solution through an addition funnel. The reaction was allowed to proceed overnight at room temperature in the dark. Next, the solution was filtered through neutral aluminum oxide to remove TEA salt and stirred for 2 h with sodium carbonate. The heterogeneous solution was filtered through a thin layer of Hyflo, followed by precipitation in cold ethyl ether.
PEG-tetra-methacrylate (PEG4MA) was synthesized by a microwave-assisted reaction [44]. Briefly, 4-arm PEG-OH was added to 8 eq. of methacrylic anhydride in a round bottom flask. The flask was placed in the microwave-assisted reactor (CEM Discover SPS) and the reaction proceeded at a microwave power of 300W and 60°C for 5 min. The product was precipitated in cold ethyl ether and collected via filtration.
PEG-tetra-allylether (PEG4AE) was synthesized as described previously [45]. First, sodium hydride (NaH, 1 eq.) was added slowly into a flask containing dried 4-arm PEG-OH dissolved in anhydrous tetrahydrofuran (THF). After the release of hydrogen gas, allyl bromide (6 eq. of OH group) was added into the flask slowly and the reaction was kept at 40°C for overnight. The product was filtered and precipitated in cold ether.
All PEG macromers were characterized by 1H NMR (AVANCE Bruker 500) and stored at −20°C until use. The degrees of functionalization for all products were at least 95%.
2.3 Peptide synthesis and purification
All peptides were synthesized by a microwave-assisted peptide synthesizer (CEM Discover) and purified to at least 90% pure by reverse phase HPLC (PerkinElmer Flaxer) as described previously [46].
2.4 Rheometry
Real-time photorheometry was used to determine gel points (i.e., cross-over time). Briefly, macromer solution was placed in the light cure cell in Bohlin CVO 100 digital rheometer. The sample was irradiated with visible light (halogen cold light lamp from AmScope, Inc., 70,000 lm/m2 or Lux) through a flexible light guide. Time-sweep measurements were operated at 10% strain, 1 Hz frequency, 0.1 N normal force, and 100μm gap size. The storage (G′) and loss (G″) moduli were recorded in the linear viscoelastic region and gel points were identified by the time at which G′ surpassed G″. Strain-sweep (0.1% to 5%) oscillatory rheometry was performed, on a Bohlin CVO 100 digital rheometer, to measure gel moduli using a parallel plate geometry (8mm) with a gap size of 750μm.
2.5 Visible light-mediated thiol-vinyl gelation
Mixed-mode thiol-vinyl hydrogels were formed by visible light mediated polymerization between PEG macromer (PEG4A, PEG4AA, PEG4MA, or PEG-4AE) and di-thiol containing cross-linker (DTT or bis-cysteine containing peptides), which were dissolved in pH 7.4 phosphate buffered solution (PBS). Eosin-Y (0.1 mM) and N-vinylpyrrolidone (NVP, 0.1 or 1 vol%) were added to the precursor solution, which was injected in a cylindrical mold and exposed under visible light for 5 min at 70,000 Lux.
2.6 Gel swelling and degradation
For gel swelling studies, each gel was prepared from 40μL of precursor solution following the gelation procedure described above. After gelation, samples were incubated in pH 7.4 PBS at 37°C on an orbital shaker for 24 h. Gels were then removed from the buffer and weighed to obtain swollen weighs (Wswollen), followed by drying in vacuuo for 24 h to obtain dried polymer weights (Wdry). Hydrogel mass swelling ratio (q) was defined as Wswollen/Wdry. Hydrogel degradation was monitored by changes in hydrogel shear moduli as time. Hydrogel shear modulus is positively correlated to the gel crosslinking density, which decreases exponentially as time for hydrolytic bulk gel degradation [20, 46]. Gels for degradation studies were incubated in PBS (pH7.4) at 37°C.
2.7 Cell culture and encapsulation
Human mesenchymal stem cells were isolated from unprocessed human bone marrow (Lonza). hMSCs (passages 2–3), on TCPS or encapsulated in hydrogels, were maintained in low glucose DMEM supplemented with 10% fetal bovine serum (FBS, Gibco), 1x antibiotic-antimycotic (Invitrogen), and 1 ng/mL basic fibroblast growth factor (bFGF, Peprotech) [47, 48]. For osteogenic differentiation, cell-laden hydrogels were placed in high glucose DMEM containing 10% FBS, 1x antibiotic-antimycotic, 100 nM dexamethasone, 50 μM ascorbic acid, and 20 mM β-glycerophosphate. Cells on TCPS or encapsulated in hydrogels were maintained in a humidified incubator with 5% CO2 at 37°C and the culture media were refreshed every 2–3 days.
For cell encapsulation, hMSCs at a final density of 5 × 106 cells/mL were suspended in sterile polymer precursor solutions (pH7.4 DPBS) containing 4 wt% PEG4A (total acrylate conc.: 8 mM) and with either 3.5 mM CGGGC or 3.5 mM KCGPQGIWGQCK peptide linker, and 1 mM CRGDS (total thiol conc.: 8 mM). All precursor solutions also contained 0.1 mM eosin-Y and either 0.1 or 1.0 vol% NVP (vinyl conc.: 9.36 or 93.6 mM). Well-mixed precursor solution (25 μl) was injected in a 1 mL cylindrical mold and exposed under a halogen lamp illuminator (AmScope) for 5 minutes at room temperature. Cured cell-laden hydrogels (~4.6 mm in diameter and 1.5 mm in thickness) were placed in cell culture media and cultured as described above.
2.8 Cell metabolic activity assay and live/dead staining
To quantify cell metabolic activity, cell-laden hydrogels were incubated in 500 μL Alamarblue® reagent (AbD Serotec, 10 % in cell culture medium) and cultured in CO2 incubator at 37 °C for 16 hours. After which, 200 μl of media were transferred to a 96-well plate for fluorescence quantification (excitation: 560 nm and emission: 590 nm) using a microplate reader (BioTek, Synergy HT). For live/dead staining, cell-laden hydrogels were incubated in buffer containing Calcein AM (0.25 μL/mL, stained live cells green. Ex: 495 nm. Em: 515 nm) and Ethidium homodimer-III (2 μL/mL, stained dead cells red. Ex: 530 nm. Em: 620 nm) for 1 hour. Stained cell-laden hydrogels were rinsed with DPBS briefly prior to imaging using a confocal microscope (Olympus Fluoview, FV1000). At least two sets of images per hydrogel sample were taken (100 μm thick, 10 μm per slice). The number of live and dead cells was counted separately using Z-stack images to determine percent cell viability, the number of live cells divided by the total cell counts.
2.9 Osteogenic differentiation and ALP assay
For osteogenic differentiation, cell-laden hydrogels were maintained in osteogenic medium as described above. At predetermined time points, alkaline phosphatase (ALP) activities of the differentiated cells were quantified following a protocol described previously. Briefly, cell metabolic activity was first determined using 10% AlamarBlue® reagent (4 h incubation) and used for ALP activity normalization. After which, cell-laden gels were rinsed with DPBS and transferred individually to 1.6 mL conical tubes containing 100 μL of RIPA buffer (Pierce). The gels were homogenized and centrifuged at 14,000 rpm for 5 minutes. The supernatants were collected and transferred to a 96-well plate. Alkaline phosphatase substrate (PNPP) solutions (100 μL per well) were added and absorbance at 405 nm was measured with a microplate reader. Measurements were taken at one-minute intervals for a total of ten minutes. The slope of the absorbance linear regression curve was used to represent the relative ALP activity. Normalized ALP activity was obtained by normalizing the slope to the corresponding cell metabolic activity and further normalized to the values obtained from day-one samples.
2.10 Statistics
Statistical analysis was conducted using a two-way analysis of variance (ANOVA) followed by a Bonferroni’s post-hoc test. Single, double, and triple asterisks represent p < 0.05, 0.001, and 0.0001, respectively. A p value < 0.05 was considered statistically significant. All experiments were conducted independently for three times and the results were presented as mean ± SD.
3. Results & Discussion
3.1 Visible light mediated thiol-vinyl gelation
Visible light curable hydrogels are commonly fabricated via a chain-growth polymerization mechanism, which yields a heterogeneous network structure and dense polyacrylate kinetic chains. To rapidly produce a stable network, at least four components are required: (1) multi-functional vinyl macromer (e.g., PEGDA), (2) photosensitizer (e.g., eosin-Y), (3) amine-based co-initiator (e.g., TEOA), and (4) mono-functional monomer (e.g., NVP) [10, 48–50]. The second visible light mediated gelation mechanism is based on a step-growth photoclick reaction recently developed by our group [40, 41]. In such gelation scheme, multi-arm PEG-norbornene (e.g., PEG4NB) was used as the macromer, while di-thiol-containing molecules and eosin-Y were used as cross-linker and initiator, respectively. Using PEG4NB and dithiothreitol (DTT), purely step-growth hydrogels were formed under 2 minutes of visible light exposure. To evaluate the possibility of using other PEG-based vinyl macromers in this visible light mediated step-growth gelation scheme, we first tested PEG4A as the macromer (Figure 1A), dithiol-containing DTT (Figure 1E) as the dual-purpose co-initiator/cross-linker, and eosin-Y (Figure 1G) as the photosensitizer. Unfortunately, gelation did not occur within the first 7 minutes of visible light exposure (gel point ~430 seconds) when 4wt% of PEG4A (8 mM acrylate), DTT (8 mM thiol), and eosin-Y (0.1 mM) were used (Figure 2A; F1 in Table 1). Theoretically, thiyl radicals generated by the visible light excited eosin-Y should be able to initiate step-growth thiol-acrylate gelation (Figure 1H). Unlike norbornene moieties, however, acrylates on PEG4A exhibited slower reaction kinetics under visible light exposure. Here, thiyl radicals were generated from di-thiol molecules by the visible light sensitized eosin-Y and were the only radical source capable of initiating the polymerization. These thiyl radicals might not have high reactivity with acrylates or not propagate fast enough to cause network cross-linking. The slow reaction kinetics observed in Figure 2A suggested a need for accelerating polymerization kinetics. Since NVP (Figure 1F) is a commonly used co-monomer for accelerating gelation kinetics in conventional visible light-mediated chain-growth gelation [10], we reasoned that it would also accelerate visible light mediated thiol-acrylate gelation. Indeed, when NVP was added at 0.1 vol.% (9.36 mM; F2 in Table 1) or 1 vol.%, (93.6 mM; F3 in Table 1), the gel points were decreased from ~430 seconds to ~231 and ~92 seconds, respectively (Figure 2B, 2C, & Table 1). Increased gelation rate in the presence of NVP could be attributed to an increased vinyl group concentration and/or rapid diffusion of radical bearing NVP [10, 43].
Figure 1.

Chemical structures of (A) PEG-tetra-acrylate (PEG4A), (B) PEG-tetra-acrylamide (PEG4AA), (C) PEG-tetra-methacrylate (PEG4MA), (D) PEG-tetra-allylether (PEG4AE), (E) dithiothreitol (DTT), (F) N-vinylpyrrolidone (NVP), (G) eosin-Y (EY). In (A–D), only one arm in each of the 4-arm PEG macromer is shown. (H) Mechanism and proposed network structures of visible light mediated mixed-mode thiol-vinyl photopolymerization.
Figure 2.

Representative in situ time sweep photorheometry data showing the effect of NVP on the evolution of storage/loss (G′/G″) moduli of visible light curable thiol-acrylate hydrogels. PEG4A (4wt% or 8mM acrylate) and DTT (8mM thiol) were used in the testing in the presence of different concentrations of co-monomer NVP: (A) 0%, (B) 0.1%, and (C) 1.0%.
Table 1.
Gel points for gel formulations used in in situ photorheometry
| Formulation | Macromer | fAa | Co-initiator/Cross-linkerb | fBa | NVP (%) | Gel point (sec) |
|---|---|---|---|---|---|---|
| F1 | PEG4A | 4 | DTT | 2 | 0 | 430 ± 38 |
| F2 | PEG4A | 4 | DTT | 2 | 0.1 | 231 ± 12 |
| F3 | PEG4A | 4 | DTT | 2 | 1 | 92 ± 11 |
|
| ||||||
| F4 | PEG4A | 4 | Cysteine | 1 | 1 | 250 ± 14 |
| F5 | PEG4A | 4 | CGGGC | 2 | 1 | 26 ± 5 |
| F6 | PEG4A | 4 | KCGPQGIWGQCK | 2 | 0.1 | 86 ± 3 |
| F7 | PEG4A | 4 | KCGPQGIWGQCK | 2 | 1 | 19 ± 2 |
|
| ||||||
| F8 | PEG4AA | 4 | DTT | 2 | 1 | 275 ± 21 |
|
| ||||||
| F9 | PEG4MA | 4 | DTT | 2 | 1 | 305 ± 36 |
|
| ||||||
| F10 | PEG4AE | 4 | DTT | 2 | 1 | > 600 |
|
| ||||||
| F11c | PEG4A | 4 | - | - | 0.1 | 78 ± 4 |
| F12c | PEG4A | 4 | - | - | 1 | 13 ± 1 |
fA and fB are the functionality of the macromers.
Thiol concentration = 8 mM.
Chain-growth photopolymerization using 8mM TEOA as co-initiator.
A major difference between acrylate and norbornene moieties is that acrylate can undergo homopolymerization with other acrylates in photopolymerization reactions while norbornene cannot, unless in the presence of a catalyst [51]. Hence, the use of acrylate-based macromers (e.g., PEG4A) with thiol-containing molecules leads to a mixed-mode photopolymerization. In such system, addition of a thiyl radical to an acrylate produces a step-growth thiol-ether-ester bond, while the propagation of carbon radicals through vinyl group results in chain-growth poly(acrylate) kinetic chains (Figure 1H). The kinetic constants of these two competing reactions are depicted by kP,C=C and kP,CT, respectively. At fixed PEG macromer and thiol concentrations, increasing NVP content in the prepolymer solution increases vinyl-to-thiol ratio (Table 2), thus shifting the network cross-linking from a pure step-growth to a mixed mode step-and-chain growth photopolymerization. Salinas and Anseth have demonstrated that, when a bi-cysteine-containing peptide was used, the chain-transfer constant (i.e., kP,C=C/kP,CT) was only slightly higher than one, indicating a high efficiency of radical-mediated thiol-acrylate conjugation and hence high percentage of peptide incorporation in the gel network (>97%) [25]. While NVP is a co-monomer that did not contribute to network cross-linking, adding NVP did increase the final gel stiffness. This is likely due to the formation of more hydrophobic and potentially more rigid poly(NVP) kinetic chains (Figure 1H) [10, 43]. Increasing NVP concentration also increased the likelihood of homopolymerization of multiple PEG-acrylate arms within the same cross-link. Although this mixed mode visible light gelation system still uses four components (i.e., PEG4A, DTT, NVP, and EY), it replaces the more cytotoxic amine-based co-initiator with multi-functional thiols that not only serve as gel cross-linker but also increase the flexibility of material design (see sections below).
Table 2.
Functional group concentrations and vinyl-to-thiol ratios
| Tetra-vinyl | NVP | Di-thiol | Ratio of total [vinyl] to [thiol] | ||
|---|---|---|---|---|---|
|
| |||||
| wt% | [acrylate] (mM) | vol.% | [vinyl] (mM) | [thiol] (mM) | |
| 4 | 8 | 0 | 0 | 8 | 1 |
| 4 | 8 | 0.1 | 9.38 | 4 | 4.35 |
| 4 | 8 | 0.1 | 9.38 | 6 | 2.90 |
| 4 | 8 | 0.1 | 9.38 | 8 | 2.17 |
| 4 | 8 | 1 | 93.8 | 4 | 25.45 |
| 4 | 8 | 1 | 93.8 | 6 | 16.97 |
| 4 | 8 | 1 | 93.8 | 8 | 12.73 |
3.2 Effect of vinyl moiety on visible light mediated gelation
In addition to PEG4A (Figure 1A), we also evaluated other commonly used PEG-based vinyl macromers for this visible light-mediated gelation scheme (Figure 1B, 1C, 1D for PEG4AA, PEG4MA, and PEG4AE, respectively). Hydrogel cross-linking efficiency varied depending on the reactivity of the cross-linkable vinyl moieties. In particular, the use of PEG4A yielded the fastest gelation kinetics (Figure 1A; F3 in Table 1), followed by PEG4AA (Figure 1B; F8 in Table 1), PEG4MA (Figure 1C; F9 in Table 1), and PEG4AE (Figure 1D; F10 in Table 1). These results were consistent with other polymerization mechanisms reported in the literature. For example, Elbert and Hubbell demonstrated that the conjugation efficiency of thiol to acrylamide is not as efficient as to acrylate [10]. The reactivity of methacrylate is also known to be lower than acrylate. The use of PEG4AE failed to result a cross-linked gel network within 10 minutes of visible light exposure. Interestingly, the same visible light-mediated photo-click reaction was used by DeForest et al. for conjugating thiol-containing molecules to allylether-bearing peptides in a hydrogel network [21, 52]. While thiol-allyether reaction is appropriate for biomolecular conjugation, it might not be efficient enough for an aqueous-based hydrogel network cross-linking because a higher cross-linking efficiency is required to reach gel point and network formation. Considering acrylate-based PEG macromers have been widely employed in the fabrication of PEG hydrogels and the fact that PEG4A yielded the fastest gelation in this system, we chose PEG4A for the majority of the subsequent studies.
3.3 Effect of thiol functionality and chemistry on visible light mediated thiol-acrylate gelation
The use of bi-functional thiol molecules (e.g., DTT) was critical for rapid gelation. As shown in Figure 3, when a mono-thiol cysteine was used, gelation did not occur until after 250 seconds of visible light exposure (F4 in Table 1). This is because the addition of a mono-thiol to an acrylate results in an effective termination in the step-growth reaction. In this case, cysteine serves only as a co-initiator. The weak gel formed after prolonged visible light exposure was likely due to chain transfer events. On the other hand, bis-cysteine containing peptides (e.g., CGGGC in Figure 3; F5 in Table 1) contributed to network cross-linking because of ‘peptide bridges’ formed between the multi-arm PEG macromers (Figure 1H). Our results also indicated that the cross-linking efficiency between acrylate and thiol was affected by the chemistry of di-thiol containing cross-linker. For example, the use of CGGGC (F5 in Table 1) or KCGPQGIWGQCK (MMP-sensitive sequence, F7 in Table 1) respectively yielded gel points of 26 ± 5 and 19 ± 2 seconds, while the use of DTT produced a gel point of 92 ± 11 seconds (F3 in Table 1). This phenomenon is similar to the work done by Salinas and Anseth using UV-based thiol-acrylate hydrogels [25], as well as and that reported by Lutolf and Hubbell using Michael-type addition hydrogels [53, 54]. It is possible that radical-mediated reactions promoted more intramolecular disulfide bond formation in DTT and less in bis-cysteine containing peptides, thus reducing the available thiyl radicals for thiol-acrylate cross-linking. Some amino acid side groups on the peptides might also affect the gelation process via altering the deprotonation of cysteine by excited eosin-Y. As a comparison, we also obtained gel points in conventional chain-growth visible light mediated gelation using PEG4A (8 mM acrylate), TEOA (8 mM), and NVP (0.1% or 1.0%). Results listed in Table 1 (F11–F12) show that the gel points for visible light mediated chain-growth polymerizations were similar to the gel points of mixed-mode polymerizations using bis-cysteine peptides as dual-purpose cross-linker/co-initiator.
Figure 3.
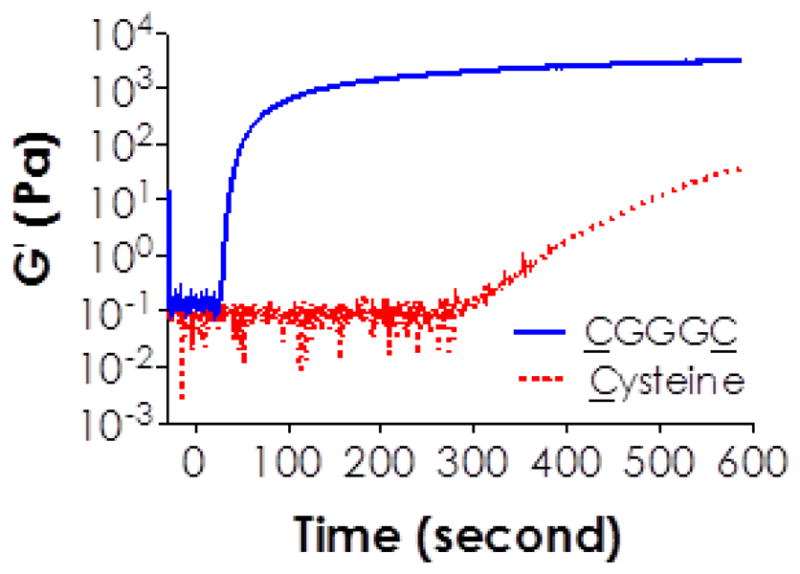
Effect of thiol-functionality (i.e., mono-functional Cysteine and bi-functional peptide CGGGC) on visible light curable thiol-acrylate hydrogelation (4wt% PEG4A or 8mM acrylate; 8 mM total thiol; and 1% NVP).
3.4 Effect of bi-functional thiol linker concentration on visible light mediated thiol-acrylate gelation
We next evaluated the influence of thiol concentration on the property of visible light cured thiol-acrylate hydrogels. At the same macromer content (i.e., 4 wt% PEG4A with 8 mM acrylate), increasing thiol concentration from 4 mM to 8 mM (i.e., 2 to 4 mM CGGGC) increased equilibrium gel swelling ratios (Figure 4), suggesting a decreased network cross-linking density. Even in the presence of 1.0% NVP, where the total vinyl concentration was ~12- to 25-fold higher than thiol concentration (Table 2), the changes in thiol concentrations still affected gel swelling. This was because one thiol group reacts with only one acrylate moiety to form a thiol-ether bond. Therefore, a higher concentration of thiol in the pre-polymer solution yielded a network with higher amounts of orthogonal thiol-ether linkages. As the thiol content was decreased, polymerization kinetics was shifted toward chain-growth polymerization that yielded hydrophobic and dense polyacrylate kinetic chains and hence produced a stiffer and less swollen hydrogel network. Therefore, adjusting the concentration of di-thiol-containing linker provides an efficient means of manipulating hydrogel properties for particular biomedical applications.
Figure 4.
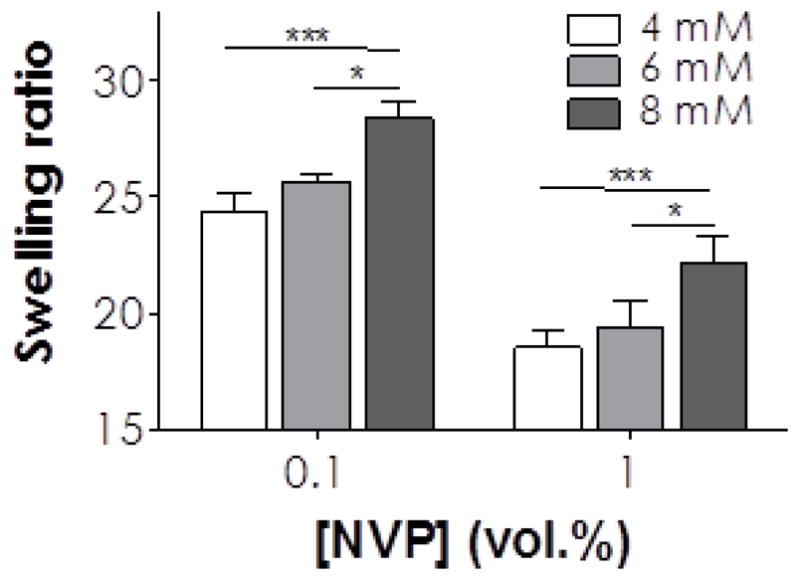
Effect of bis-cysteine peptide (CGGGC) and NVP concentrations on equilibrium swelling ratios (measured at day 1 post-gelation) of visible light cured thiol-acrylate hydrogels using 4wt% PEG4A as the macromer. Figure legends indicate total thiol concentrations on the CGGGC peptide incorporated in the polymer precursor solutions. Single and triple asterisks represent p < 0.05 and 0.0001, respectively.
3.5 Effect of macromer composition on thiol-acrylate hydrogel hydrolytic stability
The visible light cured mixed-mode polymerized hydrogels presented here could be synthesized as non-degradable gels if acrylamide-based macromers (e.g., PEG4AA) and non-cleavable di-thiol linkers were used (Figure 5A). For example, the shear moduli (G′) of hydrogels prepared from PEG4AA (4wt%) and a non-cleavable peptide CGGGC remained relatively unchanged as time (Figure 5B) regardless of NVP concentration incorporated. Since G′ correlates directly to the network cross-linking density, unchanged gel stiffness indicated the hydrolytic stability of these hydrogels. In this gel formation, two types of hydrolytically stable cross-links were formed following visible light mediated thiol-acrylamide reactions, namely poly(acrylamide) kinetic chains and thiol-ether-acrylamide bond. As expected, increasing NVP concentration resulted in stiffer hydrogels. This effect, however, reached a maximum at 4% NVP, (Figure 5B) as further increased NVP concentration from 4% to 8% did not increase gel stiffness. Another phenomenon worth noting is that the stiffness of PEG4AA hydrogels appeared to be weaker than PEG4A gels under the same gelation conditions because of the lower reactivity of acrylamide compared to acrylate as discussed earlier (Table 1). While not investigated in this contribution, proteolytic degradability could be imparted into the network by using a protease sensitive peptide (e.g., MMP-sensitive linker). This will allow researchers to investigate the effect of proteolysis on cellular fate in 3D without the compounding factor of hydrolytic gel degradation.
Figure 5.

(A) Chemical structure of a hydrolytically stable thiol-ether-acrylamide linkage formed between acrylamides (on PEG4AA) and cysteines (on CGGGC peptide). (B) Relatively stable shear moduli as a function of time and NVP concentration for hydrogels prepared from 4wt% PEG4AA and CGGGC (8mM acrylamide & 8mM SH).
We next explored the hydrolytic degradation of hydrogels formed by PEG4A and a bis-cysteine peptide cross-linker, CGGGC. Mechanistically, hydrogels prepared by PEG4A and thiol-containing linker would degrade hydrolytically following a bulk degradation mechanism due to the formation of hydrolytically labile thiol-ether-ester bonds throughout the highly swollen network (Figure 6A). For this degradation study, we fabricated PEG4A (4 wt%) hydrogels cross-linked by different peptide cross-linkers and measured the shear moduli (G′) of the hydrogels as a function of degradation time. Figure 6B and 6C show the degradation profiles in gels cross-linked by different concentration of CGGGC peptide (4, 6, and 8 mM thiol) in the presence of 0.1% or 1.0% NVP. Figure 6D and 6E represent hydrolytic degradation in gels cross-linked by MMP-sensitive peptide linker (KCGPQG↓IWGQCK) and a control peptide less susceptible to protease cleavage (KCGPQGPAGQCK) [55]. Results showed that all hydrogels formed by PEG4A and di-thiol containing linker degraded as time regardless of their initial stiffness. Figures 6B–6E also show that the degradation of these hydrogels could be easily predicted by a pseudo first-order degradation equation [20]:
| (1) |
where G′eq and G′t are the shear moduli at equilibrium swelling state (before significant degradation occurs) and at any time during degradation, respectively. kapp is the apparent degradation rate constant.
Figure 6.

(A) Schematic showing the structure and hydrolysis mechanism of a thiol-ether-ester bond formed by visible light mediated thiol-acrylate reaction. (B–C) The influence of thiol concentration on gel degradation as measured by time-dependent gel shear moduli. Hydrogels were prepared from 4wt% PEG4A and CGGGC with: (B) 0.1% or (C) 1.0% NVP. Figure legends indicate total thiol concentrations on the CGGGC peptide incorporated in the polymer precursor solutions. (D–E) The influence of peptide cross-linker on gel degradation as measured by time-dependent gel shear moduli. Hydrogels were prepared from 4wt% PEG4A and MMP-sensitive linker KCGPQGIWGQCK or a protease stable peptide linker KCGPQGPAGQCK with: (D) 0.1% or (E) 1.0% NVP.
Eq. (1) describes the degradation kinetics of hydrogels prepared with 0.1% NVP very well (R2 = 0.95~0.99, Tables 3 & 4), indicating that the degradation of these gels was mainly governed by the hydrolysis rate of thiol-ether-ester bond. Furthermore, the apparent degradation rate constant (kapp) was higher in hydrogels containing higher concentrations of bis-cysteine peptides. This phenomenon could be attributed to the formation of more thiol-ether-ester bonds in hydrogels containing higher amounts of peptide. Since the hydrolytic degradation rate of bulk-degrading hydrogels was dictated by the concentration of hydrolytically labile units, higher thiol-ether-ester concentration would cause faster gel degradation.
Table 3.
Hydrolytic degradation rate constants of visible light cured thiol-acrylate hydrogels with different thiol and NVP concentrations (with CGGGC as the peptide linker).
| NVP (%) | [acrylate] (mM) | [SH] (mM) | kapp (day−1) | R2 |
|---|---|---|---|---|
| 0.1 | 8 | 4 | 0.032 ± 0.002 | 0.95 |
| 8 | 6 | 0.060 ± 0.003 | 0.97 | |
| 8 | 8 | 0.168 ± 0.005 | 0.99 | |
|
| ||||
| 1.0 | 8 | 4 | 0.019 ± 0.001 | 0.89 |
| 8 | 6 | 0.026 ± 0.002 | 0.91 | |
| 8 | 8 | 0.049 ± 0.003 | 0.94 | |
Table 4.
Hydrolytic degradation rate constants of visible light cured thiol-acrylate hydrogels with different peptide linker and different NVP concentration.
| Peptide linker | NVP (%) | kapp (day−1) | R2 |
|---|---|---|---|
| KCGPQG↓IWGQCK (MMP sensitive linker) | 0.1 | 0.167 ± 0.009 | 0.98 |
| 1.0 | 0.035 ± 0.002 | 0.91 | |
|
| |||
| KCGPQGPAGQCK (MMP insensitive linker) | 0.1 | 0.131 ± 0.006 | 0.98 |
| 1.0 | 0.025 ± 0.001 | 0.88 | |
We further examined the degradation of visible light cured thiol-acrylate hydrogels cross-linked with 1.0% NVP and found that these degradation profiles fit less perfectly using exponential curve fitting (R2 = 0.88~0.94, Tables 3 & 4). One possible explanation is that the hydrolytic degradation of thiol-acrylate hydrogels in the presence of high NVP concentration was affected by the dense and hydrophobic poly(NVP) chains. Among other factors, local water concentration could also be affected in the presence of dense poly(NVP) chains. Water concentration was assumed to be a constant during hydrolysis of ester bonds in a pseudo-first order degradation kinetics. Further, the empirically obtained kapp values for gels cross-linked with 0.1% NVP were 1.68 to 3.42 fold higher than that in 1.0% NVP containing gels. These results suggest that the presence of high concentration of poly(NVP) chains not only partially affects the predictability of hydrolytic gel degradation (i.e., less perfect curve fitting) but also slows down the degradation rate (i.e., lower kapp). Although not explored in this contribution, one could easily tune the gel degradation rate by mixing hydrolytic-labile PEG-acrylate and hydrolytic-stable PEG-acrylamide in different ratios while keeping the concentration of bis-cysteine containing peptide constant.
3.6 Cytocompatibility of visible light cured thiol-acrylate hydrogels for hMSCs
Next, we evaluated the cytocompatibility of our visible light-cured thiol-acrylate hydrogels. We photoencapsulated hMSCs in PEG4A hydrogels formed with different cross-linkers (non-protease sensitive CGGGC or MMP-sensitive KCGPQG↓IWGQCK) and NVP concentrations (0.1% or 1.0%). In growth conditions, the encapsulated hMSCs retained high viability for the duration of the study (Figure 7). Live/dead staining images and metabolic activity assay revealed high cell viability (>90%) one day following photoencapsulation (Figure 7A, 7C). It is worth noting that hMSCs encapsulated in PEG4A/CGGGC hydrogels cross-linked with 0.1% NVP maintained high metabolic activity for the duration of 21 days (Figure 7B). The use of conventional PEGDA/TEOA hydrogels, however, caused significant hMSC death as shown in our previous report (0.75% TEOA, and 0.1% NVP) [40]. This suggests that the visible light cured thiol-vinyl hydrogels are highly cytocompatible, at least for the cell type studied here. Although the use of higher NVP concentration (e.g., 1.0%) increased gel stiffness, it did not significantly affect cell viability compared to a softer gel cross-linked with 0.1% NVP one-day post encapsulation. After culturing in growth medium for 21 days, however, hMSCs encapsulated in hydrogels containing 1.0% NVP (i.e., stiffer gels) showed higher percentage of dead cells and slightly lower cellular metabolic activity (Figure 7C). The slight decrease in cell viability in 1.0% NVP containing PEG4A hydrogels after culturing for 21 days might be a result of cytotoxicity induced by cross-linked poly(NVP). However, this was less likely since hydrogels containing poly(NVP) have been shown to support cell attachment and proliferation compared to gels without poly(NVP) [56–58]. The most likely explanation for the decreased cell viability in 1.0% NVP containing hydrogel was a lower degree of hydrogel degradation (Fig. 6D, 6E). We have recently shown that hMSC encapsulated in thio-ene hydrogels with lower degradability exhibited lower viability [59]. Another notable difference between day-1 and day-21 live/dead images was a lower cell counts in day-21 images, especially for hydrogels contained 0.1% NVP. This was likely due to faster hydrolytic degradation (Figure 6D, 6E) and higher swelling in hydrogels with lower cross-linking density (i.e., gel swelling/expansion increases the distance between cells).
Figure 7.

(A) Viability of hMSCs encapsulated in visible light cured PEG4A-peptide hydrogels with different formulations as indicated. Sequence of MMPLinker: KCGPQGIWGQCK. (B) Metabolic activity of hMSCs encapsulated in PEG-peptide hydrogels with different formulations as indicated. (C) Live/Dead staining of hMSCs encapsulated in PEG-peptide hydrogels with different formulations as indicated. All hydrogels contained 1mM of integrin ligand CRGDS peptide (Scale: 100μm).
Although images shown in Figure 7 demonstrate high cell viability in these hydrogels, cell morphology was easily manipulated by altering gel compositions. For example, hMSCs encapsulated in CGGGC cross-linked hydrogels with 0.1% NVP retained their round morphology after 21 days of culture (left column in Figure 7C). On the other hand, extensive cell spreading could be observed in hydrogels cross-linked with 0.1% NVP and MMP-sensitive peptide linker, indicating that hMSCs actively remodeled their local matrix by means of protease-mediated matrix cleavage (middle column in Figure 7C). These results are consistent with prior efforts on creating cell-instructive matrices for culture and differentiation of hMSCs [47]. More interestingly, when we increased the concentration of NVP from 0.1% to 1.0% without changing other macromer compositions, we no longer observed the enhanced spreading seen in gels with 0.1% NVP, even though these gels contained the same concentration of MMP-sensitive peptide linker (right column in Figure 7C). The restriction in cell spreading might be a result of increased local gel cross-linking density that decreased proteases secretion, reduced cell traction forces [60], or both. It should be noted that we did not conduct gel degradation experiments using exogenously added proteases because: (1) Exogenously added protease will cause gel degradation in a surface erosion mechanism if enzymatic reaction is faster than enzyme diffusion in hydrogels [16]. This situation is not relevant to cell-laden hydrogels because proteases are not infiltrated from the external media. They are secreted by the cells encapsulated within the hydrogels. Furthermore, we did not observe any surface erosion occurring in hydrogels encapsulated with hMSCs; (2) If enzyme diffusion is faster than substrate cleavage, hydrogels will degrade homogenously throughout the gel. This situation is also irrelevant to cell-laden hydrogels because studies have shown that cell-mediated gel degradation is restricted very locally to the surrounding of the cells [47, 59].
3.7 Osetogenic differentiation of hMSCs encapsulated in visible light cured thiol-acrylate hydrogels
In addition to cell survival, we also assessed the potential of this gel system to support osteogenic differentiation of hMSCs. We encapsulated hMSCs in hydrogels formed by PEG4A and MMP-sensitive peptide linker with 0.1% or 1.0% NVP. Cell-laden hydrogels were cultured in standard osteogenic media for 21 days. Similar to the images shown in Figure 7C, the degree of hMSCs spreading was affected significantly by the extent of gel cross-linking. Cells encapsulated in hydrogels with higher degree of chain-growth cross-links (i.e., with 1.0% NVP) were not able to spread out (Figure 8A). Cell metabolic activity in osteogenic media was also influenced by the degree of network cross-linking (Figure 8B). Furthermore, we found that the use of NVP at high concentration hinders osteogenic differentiation (Figure 8C), likely due to restricted gel degradation with a high cross-linking density. Burdick and colleagues have recently reported the profound impact of degradation in chemically cross-linked hydrogel on hMSC differentiation [60]. In highly cross-linked hydrogels (e.g., +UV gels in the study by Burdick et al. or the thiol-acrylate gels cross-linked with 1.0% NVP as shown here), the encapsulated hMSCs failed to exert traction forces upon surrounding matrix, which resulted in reduced osteogenic differentiation.
Figure 8.

Osteogenic differentiation of hMSCs encapsulated in MMP-sensitive PEG-peptide hydrogels formed by visible light mediated mixed mode photopolymerization. (A) Cell morphologies revealed by Live/Dead staining and confocal imaging using gels crosslinked with 0.1% or 1.0% NVP (Scale: 100μm). (B) Metabolic activity of encapsulated hMSCs cultured in osteogenic media. (C) Normalized ALP activity of encapsulated hMSCs. Triple asterisks represent p < 0.0001.
It is worth noting that although thiols and acrylates can react via a nucleophilic addition reaction, the use of eosin-Y and visible light irradiation significantly accelerates crosslinking speed (i.e., complete gelation within 5 minutes). This visible light mediated gelation scheme also offers additional advantages in spatial-temporal control of gel properties without the concern of UV-mediated cellular damage. In addition, this visible light curable thiol-vinyl gel system has the following unique features that distinguish this system from the conventional visible light initiated photopolymerized hydrogels. Firstly, the gelation does not rely on cytotoxic TEOA or other amine-based molecules as co-initiators and hence provides enhanced cytocompatibility for sensitive cell types [32, 40]. Secondly, the gel network can be easily tuned from a near step-growth structure to a prone chain-growth structure, depending on the amounts of bis-cysteine containing cross-linker and/or co-monomer NVP used. Finally, these thiol-vinyl hydrogels can be designed to remain stable or be degraded via hydrolysis, proteolysis, or combination of these mechanisms. This gelation system provides a facile way of using visible light to form biomimetic matrix with highly controllable cell-matrix interaction and matrix properties.
Conclusion
In conclusion, we have developed a new form of visible light curable and cytocompatible PEG-based thiol-vinyl hydrogels with high tunability in gelation and degradation. Further, these gels supported the growth and osteogenic differentiation of hMSCs in 3D. The components used in this gel platform (e.g., PEG-acrylate, initiator eosin-Y, and bis-cysteine containing peptide cross-linkers) have been widely employed by many research groups for cell-laden hydrogel preparation. With this platform, one can obtain highly tunable degradation profiles by simply mixing various macromer components without complicated chemical synthesis and material preparation. The use of visible light to form hydrogels with diverse degradation profiles should have great potential in promoting tissue regeneration and/or controlled release applications.
Acknowledgments
This project was funded by the National Institutes of Health (R21EB013717), IUPUI department of Biomedical Engineering (Faculty start-up fund), and Indiana CTSI (Indiana Diabetes Research Center Pilot & Feasibility Grant).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cushing MC, Anseth KS. Hydrogel cell cultures. Science. 2007;316:1133–4. doi: 10.1126/science.1140171. [DOI] [PubMed] [Google Scholar]
- 2.Lin CC, Anseth KS. PEG hydrogels for the controlled release of biomolecules in regenerative medicine. Pharm Res. 2009;26:631–43. doi: 10.1007/s11095-008-9801-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mellott MB, Searcy K, Pishko MV. Release of protein from highly cross-linked hydrogels of poly(ethylene glycol) diacrylate fabricated by UV polymerization. Biomaterials. 2001;22:929–41. doi: 10.1016/s0142-9612(00)00258-1. [DOI] [PubMed] [Google Scholar]
- 4.Slaughter BV, Khurshid SS, Fisher OZ, Khademhosseini A, Peppas NA. Hydrogels in regenerative medicine. Adv Mater. 2009;21:3307–29. doi: 10.1002/adma.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J, Kao WJ. Synthesis of polyethylene glycol (PEG) derivatives and PEGylated-peptide blopolymer conjugates. Biomacromolecules. 2003;4:1055–67. doi: 10.1021/bm034069l. [DOI] [PubMed] [Google Scholar]
- 6.Sawhney AS, Pathak CP, Hubbell JA. Interfacial photopolymerization of poly(ethylene glycol)-based hydrogels upon alginate poly(l-lysine) microcapsules for enhanced biocompatiblity. Biomaterials. 1993;14:1008–16. doi: 10.1016/0142-9612(93)90194-7. [DOI] [PubMed] [Google Scholar]
- 7.Weber LM, He J, Bradley B, Haskins K, Anseth KS. PEG-based hydrogels as an in vitro encapsulation platform for testing controlled beta-cell microenvironments. Acta Biomater. 2006;2:1–8. doi: 10.1016/j.actbio.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Lin-Gibson S, Jones RL, Washburn NR, Horkay F. Structure-property relationships of photopolymerizable poly(ethylene glycol) dimethacrylate hydrogels. Macromolecules. 2005;38:2897–902. [Google Scholar]
- 9.Browning MB, Wilems T, Hahn M, Cosgriff-Hernandez E. Compositional control of poly(ethylene glycol) hydrogel modulus independent of mesh size. J Biomed Mater Res A. 2011;98A:268–73. doi: 10.1002/jbm.a.33109. [DOI] [PubMed] [Google Scholar]
- 10.Elbert DL, Hubbell JA. Conjugate addition reactions combined with free-radical cross-linking for the design of materials for tissue engineering. Biomacromolecules. 2001;2:430–41. doi: 10.1021/bm0056299. [DOI] [PubMed] [Google Scholar]
- 11.Browning MB, Cosgriff-Hernandez E. Development of a biostable replacement for PEGDA hydrogels. Biomacromolecules. 2012;13:779–86. doi: 10.1021/bm201707z. [DOI] [PubMed] [Google Scholar]
- 12.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, et al. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. Proc Natl Acad Sci USA. 2003;100:5413–8. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lutolf MP, Hubbell JA. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 14.Phelps EA, Enemchukwu NO, Fiore VF, Sy JC, Murthy N, Sulchek TA, et al. Maleimide cross-linked bioactive PEG hydrogel exhibits improved reaction kinetics and cross-linking for cell encapsulation and in situ delivery. Adv Mater. 2012;24:64–70. doi: 10.1002/adma.201103574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. A versatile synthetic extracellular matrix mimic via thiol-norbornene photopolymerization. Adv Mater. 2009;21:5005–10. doi: 10.1002/adma.200901808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin CC, Raza A, Shih H. PEG hydrogels formed by thiol-ene photo-click chemistry and their effect on the formation and recovery of insulin-secreting cell spheroids. Biomaterials. 2011;32:9685–95. doi: 10.1016/j.biomaterials.2011.08.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ward JH, Peppas NA. Preparation of controlled release systems by free-radical UV polymerizations in the presence of a drug. J Control Release. 2001;71:183–92. doi: 10.1016/s0168-3659(01)00213-9. [DOI] [PubMed] [Google Scholar]
- 18.Sawhney AS, Pathak CP, Hubbell JA. Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(alpha-hydroxy acid) diacrylate macromers. Macromolecules. 1993;26:581–7. [Google Scholar]
- 19.Hern DL, Hubbell JA. Incorporation of adhesion peptides into nonadhesive hydrogels useful for tissue resurfacing. J Biomed Mater Res. 1998;39:266–76. doi: 10.1002/(sici)1097-4636(199802)39:2<266::aid-jbm14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 20.Metters A, Hubbell J. Network formation and degradation behavior of hydrogels formed by Michael-type addition reactions. Biomacromolecules. 2005;6:290–301. doi: 10.1021/bm049607o. [DOI] [PubMed] [Google Scholar]
- 21.DeForest CA, Polizzotti BD, Anseth KS. Sequential click reactions for synthesizing and patterning three-dimensional cell microenvironments. Nat Mater. 2009;8:659–64. doi: 10.1038/nmat2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grover GN, Lam J, Nguyen TH, Segura T, Maynard HD. Biocompatible hydrogels by oxime click chemistry. Biomacromolecules. 2012;13:3013–7. doi: 10.1021/bm301346e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alge DL, Azagarsamy MA, Donohue DF, Anseth KS. Synthetically tractable click hydrogels for three-dimensional cell culture formed using tetrazine-norbornene chemistry. Biomacromolecules. 2013;14:949–53. doi: 10.1021/bm4000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burdick JA, Anseth KS. Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering. Biomaterials. 2002;23:4315–23. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 25.Salinas CN, Anseth KS. Mixed mode thiol-acrylate photopolymerizations for the synthesis of PEG-peptide hydrogels. Macromolecules. 2008;41:6019–26. [Google Scholar]
- 26.Lin CC, Anseth KS. Glucagon-like peptide-1 functionalized PEG hydrogels promote survival and function of encapsulated pancreatic beta-cells. Biomacromolecules. 2009;10:2460–7. doi: 10.1021/bm900420f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.West JL, Hubbell JA. Polymeric biomaterials with degradation sites for proteases involved in cell migration. Macromolecules. 1999;32:241–4. [Google Scholar]
- 28.Mann BK, Gobin AS, Tsai AT, Schmedlen RH, West JL. Smooth muscle cell growth in photopolymerized hydrogels with cell adhesive and proteolytically degradable domains: synthetic ECM analogs for tissue engineering. Biomaterials. 2001;22:3045–51. doi: 10.1016/s0142-9612(01)00051-5. [DOI] [PubMed] [Google Scholar]
- 29.Sokic S, Papavasiliou G. Controlled proteolytic cleavage site presentation in biomimetic PEGDA hydrogels enhances neovascularization in vitro. Tissue Eng A. 2012;18:2477–86. doi: 10.1089/ten.tea.2012.0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turturro MV, Christenson MC, Larson JC, Young DA, Brey EM, Papavasiliou G. MMP-Sensitive PEG Diacrylate Hydrogels with Spatial Variations in Matrix Properties Stimulate Directional Vascular Sprout Formation. PLoS One. 2013:8. doi: 10.1371/journal.pone.0058897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elisseeff J, Anseth K, Sims D, McIntosh W, Randolph M, Langer R. Transdermal photopolymerization for minimally invasive implantation. Proc Natl Acad Sci USA. 1999;96:3104–7. doi: 10.1073/pnas.96.6.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bryant SJ, Nuttelman CR, Anseth KS. Cytocompatibility of UV and visible light photoinitiating systems on cultured NIH/3T3 fibroblasts in vitro. J Biomater Sci Polym Ed. 2000;11:439–57. doi: 10.1163/156856200743805. [DOI] [PubMed] [Google Scholar]
- 33.Lin CC, Anseth KS. Controlling affinity binding with peptide-functionalized poly(ethylene glycol) hydrogels. Adv Funct Mater. 2009;19:2325–31. doi: 10.1002/adfm.200900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kizilel S, Perez-Luna VH, Teymour F. Photopolymerization of poly(ethylene glycol) diacrylate on eosin-functionalized surfaces. Langmuir. 2004;20:8652–8. doi: 10.1021/la0496744. [DOI] [PubMed] [Google Scholar]
- 35.Kizilel S, Sawardecker E, Teymour F, Perez-Luna VH. Sequential formation of covalently bonded hydrogel multilayers through surface initiated photopolymerization. Biomaterials. 2006;27:1209–15. doi: 10.1016/j.biomaterials.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 36.Fouassier JP, Allonas X, Burget D. Photopolymerization reactions under visible lights: principle, mechanisms and examples of applications. Prog Org Coating. 2003;47:16–36. [Google Scholar]
- 37.Kizilel S, Perez-Luna VH, Teymour F. Mathematical model for surface-initiated photopolymerization of poly(ethylene glycol) diacrylate. Macromol Theory Simulat. 2006;15:686–700. [Google Scholar]
- 38.Turturro MV, Papavasiliou G. Generation of mechanical and biofunctional gradients in PEG diacrylate hydrogels by perfusion-based frontal photopolymerization. J Biomater Sci Polym Ed. 2012;23:917–39. doi: 10.1163/092050611X566450. [DOI] [PubMed] [Google Scholar]
- 39.Kim SH, Chu CC. Visible light induced dextran-methacrylate hydrogel formation using (-)riboflavin vitamin B2 as a photoinitiator and L-arginine as a co-initiator. Fibers Polym. 2009;10:14–20. [Google Scholar]
- 40.Shih H, Lin CC. Visible-light-mediated thiol-ene hydrogelation using eosin-Y as the only photoinitiator. Macromol Rapid Comm. 2013;34:269–73. doi: 10.1002/marc.201200605. [DOI] [PubMed] [Google Scholar]
- 41.Shih H, Fraser AK, Lin CC. Interfacial thiol-ene photoclick reactions for forming multilayer hydrogels. ACS Appl Mater Interfaces. 2013;5:1673–80. doi: 10.1021/am302690t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cruise GM, Hegre OD, Scharp DS, Hubbell JA. A sensitivity study of the key parameters in the interfacial photopolymerization of poly(ethylene glycol) diacrylate upon porcine islets. Biotechnol Bioeng. 1998;57:655–65. doi: 10.1002/(sici)1097-0290(19980320)57:6<655::aid-bit3>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 43.Turturro MV, Sokic S, Larson JC, Papavasiliou G. Effective tuning of ligand incorporation and mechanical properties in visible light photopolymerized poly(ethylene glycol) diacrylate hydrogels dictates cell adhesion and proliferation. Biomed Mater. 2013:8. doi: 10.1088/1748-6041/8/2/025001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin-Gibson S, Bencherif S, Cooper JA, Wetzel SJ, Antonucci JM, Vogel BM, et al. Synthesis and characterization of PEG dimethacrylates and their hydrogels. Biomacromolecules. 2004;5:1280–7. doi: 10.1021/bm0498777. [DOI] [PubMed] [Google Scholar]
- 45.Rydholm AE, Reddy SK, Anseth KS, Bowman CN. Development and characterization of degradable thiol-allyl ether photopolymers. Polymer. 2007;48:4589–600. doi: 10.1016/j.polymer.2007.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shih H, Lin CC. Cross-linking and degradation of step-growth hydrogels formed by thiol-ene photoclick chemistry. Biomacromolecules. 2012;13:2003–12. doi: 10.1021/bm300752j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anderson SB, Lin CC, Kuntzler DV, Anseth KS. The performance of human mesenchymal stem cells encapsulated in cell-degradable polymer-peptide hydrogels. Biomaterials. 2011;32:3564–74. doi: 10.1016/j.biomaterials.2011.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bahney CS, Lujan TJ, Hsu CW, Bottlang M, West JL, Johnstone B. Visible light photoinitiation of mesenchymal stem cell-laden bioresponsive hydrogels. Eu Cells Mater. 2011;22:43–55. doi: 10.22203/ecm.v022a04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kizilel S. Mathematical model for microencapsulation of pancreatic islets within a biofunctional PEG hydrogel. Macromol Theory Simulat. 2010;19:514–31. [Google Scholar]
- 50.Kizilel R, Kizilel S. Application of the numerical fractionation approach to the design of biofunctional PEG hydrogel membranes. Macromol React Eng. 2012;6:160–79. [Google Scholar]
- 51.Colak S, Tew GN. Synthesis and Solution Properties of Norbornene Based Polybetaines. Macromolecules. 2008;41:8436–40. [Google Scholar]
- 52.DeForest CA, Anseth KS. Cytocompatible click-based hydrogels with dynamically tunable properties through orthogonal photoconjugation and photocleavage reactions. Nat Chem. 2011;3:925–31. doi: 10.1038/nchem.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lutolf MP, Tirelli N, Cerritelli S, Cavalli L, Hubbell JA. Systematic modulation of Michael-type reactivity of thiols through the use of charged amino acids. Bioconjug Chem. 2001;12:1051–6. doi: 10.1021/bc015519e. [DOI] [PubMed] [Google Scholar]
- 54.Lutolf MP, Hubbell JA. Synthesis and physicochemical characterization of end-linked poly(ethylene glycol)-co-peptide hydrogels formed by Michael-type addition. Biomacromolecules. 2003;4:713–22. doi: 10.1021/bm025744e. [DOI] [PubMed] [Google Scholar]
- 55.Levesque SG, Shoichet MS. Synthesis of enzyme-degradable, peptide-cross-linked dextran hydrogels. Bioconjug Chem. 2007;18:874–85. doi: 10.1021/bc0602127. [DOI] [PubMed] [Google Scholar]
- 56.Risbud M, Hardikar A, Bhonde R. Growth modulation of fibroblasts by chitosan-polyvinyl pyrrolidone hydrogel: Implications for wound management? J Biosci. 2000;25:25–31. doi: 10.1007/BF02985178. [DOI] [PubMed] [Google Scholar]
- 57.Lopes CMA, Felisberti MI. Mechanical behaviour and biocompatibility of poly(1-vinyl-2-pyrrolidinone)-gelatin IPN hydrogels. Biomaterials. 2003;24:1279–84. doi: 10.1016/s0142-9612(02)00448-9. [DOI] [PubMed] [Google Scholar]
- 58.Smith LE, Rimmer S, MacNeil S. Examination of the effects of poly(N-vinylpyrrolidinone) hydrogels in direct and indirect contact with cells. Biomaterials. 2006;27:2806–12. doi: 10.1016/j.biomaterials.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 59.Raza A, Lin C. The Influence of Matrix Degradation and Functionality on Cell Survival and Morphogenesis in PEG-Based Hydrogels. Macromol Biosci. 2013 doi: 10.1002/mabi.201300044. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS, Burdick JA. Degradation-mediated cellular traction directs stem cell fate in covalently crosslinked three-dimensional hydrogels. Nat Mater. 2013;12:458–65. doi: 10.1038/nmat3586. [DOI] [PMC free article] [PubMed] [Google Scholar]