

Abstract
Carbonyl–carbonyl (C=O···C′=O′) interactions are ubiquitous in both small and large molecular systems. This interaction involves delocalization of a lone pair (n) of a donor oxygen into the antibonding orbital (π*) of an acceptor carbonyl group. Analyses of high-resolution protein structures suggest that these carbonyl–carbonyl interactions prefer to occur in pairs, that is, one donor per acceptor. Here, the reluctance of the acceptor carbonyl group (C′=O′) to engage in more than one n→π* electron delocalization is probed using imidazolidine-based model systems with one acceptor carbonyl group and two equivalent donor carbonyl groups. The data indicate that the electrophilicity of the acceptor carbonyl group is reduced when it engages in n→π* electron delocalization. This diminished electrophilicity discourages a second n→π* interaction with the acceptor carbonyl group.
Intimate carbonyl–carbonyl (C=O···C′=O′) interactions occur frequently in both small molecules and large biomolecular assemblies. In this “n→π* interaction”, a lone pair (n) of a donor oxygen (O) overlaps with the antibonding orbital (π*) of an acceptor carbonyl group (C′=O′).1–11 The ensuing van der Waals contact between the donor oxygen and the acceptor carbon is reminiscent of the Bürgi–Dunitz trajectory.12
In a recent statistical survey of high-resolution proteins in the Protein Data Bank (PDB), 57.8% of 1,210,099 residues had one donor oxygen engaged in an n→π* interaction with the backbone carbonyl carbon. Only 2.8% of these residues had two or more donor oxygens engaging in an n→π* interaction with the same backbone carbonyl carbon.13 The preference for one donor–one acceptor over multiple donors–one acceptor could arise due to protein topology. Alternatively, the intrinsic electrophilicity of the acceptor carbon could be reduced when it engages in an n→π* interaction, presumably due to an increase in the energy of the π* orbital.10,14 This reduction in electrophilicity would discourage multiple n→π*electronic delocalization to a single acceptor carbonyl carbon. We sought to determine if an n→π* interaction alters the electrophilicity of the acceptor carbonyl.
Previously, we and others have used an AcProOMe model system for the study of n→π* interactions (Fig. 1A).1,2,4,5,8,15–18 In this system, the trans conformation is stabilized differentially over the cis conformation by an n→π* interaction. The equilibrium constant K1 reports on the strength of the n→π* interaction and can be measured readily with NMR spectroscopy. We envisioned comparing the K1 of this system, which has one amide donor and one acceptor, with K2 of an imidazolidine-based system with two amide donors and one acceptor (Fig. 1B, X = O). If an n→π* interaction does indeed reduce the electrophilicity of the acceptor carbonyl, then K1 should be greater than K2.
Fig. 1.

Conformational equilibria of compounds (A) 1 (X = O), (B) 2 (X = O) and 4 (X = S), and (C) 3 (X = O) and 5 (X = S). Equilibrium constants K1, K2, and K3 refer to compounds 1, 2, and 3, respectively.
We synthesized compounds 1–5 by known routes19,5 and measured the equilibrium constants K1 and K2 in water using NMR spectroscopy. In accord with our hypothesis, we found that K1 = 5.1 is much greater than K2 = 1.7. This finding is made more striking by the electrophilicity of the acceptor carbonyl in 2 being enhanced by inductive electron-withdrawal from two (rather than one) amido groups.
The value of K2 could be diminished for a reason unrelated to the electrophilicity of the acceptor carbonyl group. In the trans,trans conformation of compound 2, the dipoles of the donor carbonyl groups have a syn alignment; whereas in the trans,cis conformation, they have more favorable anti alignment. To assess this contribution, we explored the conformational preferences of compound 3. As this compound lacks an acceptor carbonyl group, the value of K3 reports solely on the intrinsic orientational preferences of the donor carbonyl groups. In accord with our hypothesis, we found that trans,cis is indeed the most stable conformation in water, followed by trans,trans. The value of K3 = 0.6 is, however, close to unity. Thus, dipole–dipole interactions diminish the value of K1, but not dramatically.
We suspected that the observed inequivalency in the strength of the two carbonyl–carbonyl (C=O···C′=O′) interactions should be observable in the trans,trans conformation of 2 in the solid and gas phases. Our previous studies indicated that the strength of a carbonyl–carbonyl interaction correlates inversely with the distance (d; Fig. 3) between the donor oxygen (O) and the acceptor carbon (C′).10 Accordingly, we sought insight from X-ray diffraction analysis and gas-phase geometry optimization using hybrid density functional theory (DFT).
Fig. 3.
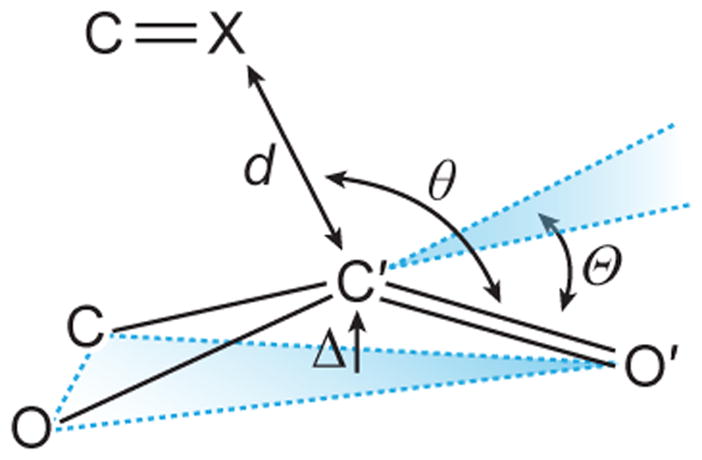
Parameters used to characterize an n→π*interaction.
Structural analyses indicated that the strength of the two carbonyl–carbonyl interactions in the trans,trans conformation of 2 are inequivalent. In the crystal structure (Fig. 2A; Table 1), one donor oxygen is in van der Waals contact with the acceptor carbon (d = 2.84 Å), whereas the other is not (d′ = 3.26 Å). The strength of carbonyl–carbonyl interactions also depends on the angle (θ) made by the donor oxygen (O) and the acceptor carbonyl (C′=O′). The donor oxygen (θ = 109.9°) that is within van der Waals contact is closer to the Bürgi–Dunitz trajectory than is the other oxygen (θ′ = 126.5°). Natural Bond Orbital (NBO) analysis20–22 of the optimized trans,trans conformation of 2 gave the two n→π* interaction energies (En→π*) as 0.75 and 0.01 kcal/mol (Fig. 2C and 2D), again inequivalent.
Fig. 2.
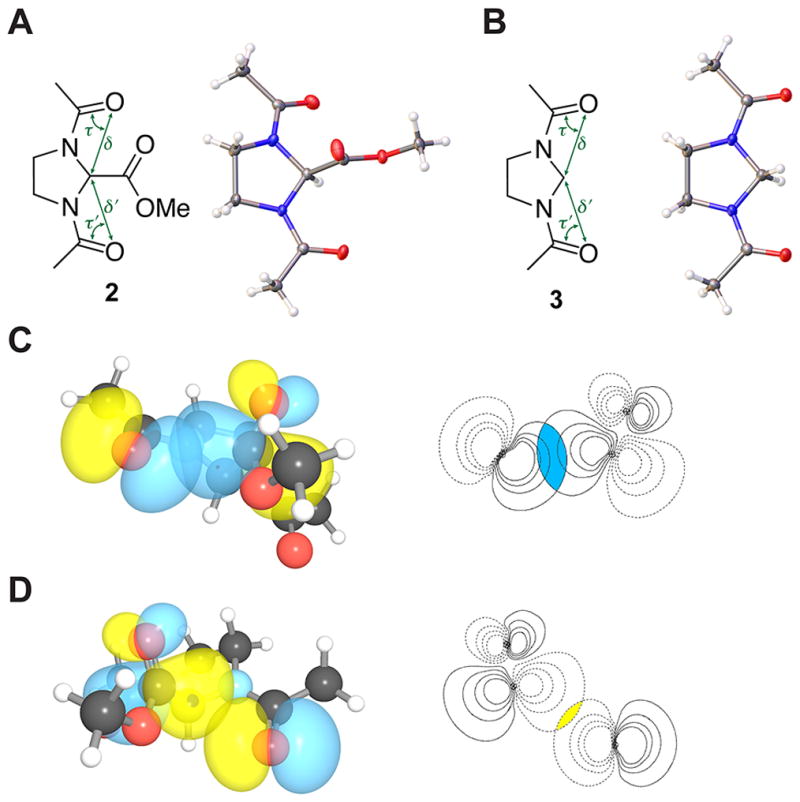
Structures of N,N′-bisacylimidazolidine compounds 2 and 3. (A) Crystal structure of 2. (B) Crystal structure of 3. (C) Strong n→π* interaction in 2; magnitude of the overlap integral: 0.0873. (D) Weak n→π* interaction in 2; magnitude of the overlap integral: 0.0370.
Table 1.
Structural and energetic parameters of compounds 2–5
| Crystallographic Data | Computational Data | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||||
| d (Å) | θ (°) | d′ (Å) | θ ′ (°) | δ (Å) | τ (°) | δ ′ (Å) | τ ′ (°) | d (Å) | θ (°) | d′ (Å) | θ ′ (°) | δ (Å) | τ (°) | δ ′ (Å) | τ ′ (°) | En→π* (kcal/mol) | E′n→π* (kcal/mol) | |
| 2 | 2.84 | 109.9 | 3.26 | 126.5 | 2.69 | 64.3 | 2.73 | 63.4 | 2.94 | 108.8 | 3.34 | 129.3 | 2.72 | 63.9 | 2.77 | 62.9 | 0.75 | 0.01 |
| 3 | – | – | – | – | 2.74 | 63.1 | 2.74 | 63.1 | – | – | – | – | 2.74 | 63.6 | 2.74 | 63.6 | – | – |
| 4 | 3.27 | 113.4 | 3.69 | 135.0 | 3.00 | 54.6 | 3.08 | 53.5 | 3.30 | 108.4 | 3.78 | 141.4 | 3.03 | 54.3 | 3.10 | 53.2 | 1.14 | 0.00 |
| 5 | – | – | – | – | 2.95 | 54.5 | 2.95 | 54.5 | – | – | – | – | 3.02 | 54.5 | 3.02 | 54.5 | – | – |
The inequivalency in the disposition of the two donor carbonyls with respect to the acceptor carbonyl in the solid and gas phases could arise from the inherent asymmetry of the imidazolidine ring. To clarify the origin of this inequivalency, we examined the trans,trans conformation of compound 3. If the asymmetry in the carbonyl disposition in 2 arose from an n→π* interaction, then it should be lost in 3. If, however, the inequivalent disposition of the donor carbonyl oxygens arose due to asymmetry in the imidazolidine ring, then it should be preserved in 3. We found that the oxygen donors are disposed symmetrically in 3 (δ = δ′, τ = τ′) but asymmetrically in 2 (δ ≠ δ′, τ ≠ τ′) in both the solid and gas phases (Table 1).
We have reported that n→π* electron delocalization induces pyramidalization of the acceptor carbonyl carbon in a manner reminiscent of nucleophilic addition to carbonyl groups.5,9,14 In 2, the acceptor carbonyl is positioned to interact with two donor oxygens. We reasoned that the direction of any acceptor carbonyl pyramidalization would designate the stronger n→π* interaction. As X-ray crystallography and hybrid density functional theory revealed that the van der Waals surface of only one of the donor oxygens overlaps to a significant extent with that of the acceptor carbonyl carbon (Fig. 2), we expected the acceptor carbonyl carbon to be displaced towards this donor. That was indeed observed in both the crystal structure of 2 (Δ = 0.01 Å, Θ = 1.36°) as well as its computationally optimized geometry. This finding affirms the inequivalency of the two n→π* interactions.
The sulphur of a thioamide is a stronger donor for an n→π* interaction than is the oxygen of an amide.5,11 The poor solubility of thioamide 4 in water prevented examination of its conformational preferences in solution. The inequivalency of its thiocarbonyl groups was, however, observed in its trans,trans conformation in the solid and gas phases (Fig. 3). The sulphur donors are disposed asymmetrically (δ ≠ δ′, τ ≠ τ′; Table 1), and the acceptor carbonyl carbon is displaced from its plane towards the stronger donor (Δ = 0.01 Å, Θ = 1.18°). NBO analyses indicated the two n→π* interactions to be worth 1.14 and 0.00 kcal/mol. Thiocarbonyl equivalency was restored in the trans,trans conformation of compound 5 (δ = δ′, τ = τ′). We note that the diminished symmetry in compounds 2 and 4 is reminiscent of Jahn–Teller distortion (Fig. 2 and 4).10,23
Fig. 4.
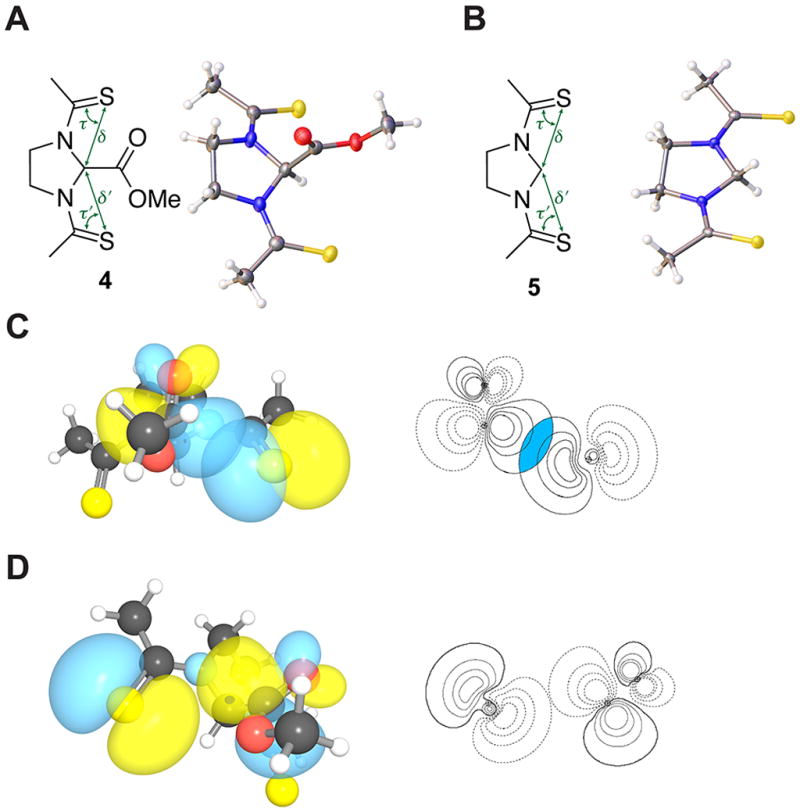
Structures of N,N′-bisthioacylimidazolidine compounds 4 and 5. (A) Crystal structure of 4. (B) Crystal structure of 5. (C) Strong n→π* interaction in 4; magnitude of the overlap integral: 0.1088. (D) Weak n→π* interaction in 4; magnitude of the overlap integral: 0.0305.
The reduction in electrophilicity of an acceptor carbonyl due to an n→π* interaction had been suggested previously. For example, an n→π* interaction has been invoked to explain the slow rate of phosphate-mediated hydrolysis of a nucleoside in a putative prebiotic synthetic route.7 More recently, Kent and coworkers attributed the reduced rate of native chemical ligation at a proline thioester to an n→π* interaction.24 This reduction in electrophilicity could have ramifications for protein folding. The development of protein secondary structures can occur concomitantly with the development of n→π* interactions.6,25 Byweakening interactions with the other face of a carbonyl, an n→π* interaction could deter a folding protein from veering towards off-pathway intermediates and thus contribute to the steepness of the walls of the folding funnel. Finally, our results suggest that n→π* interactions, in addition to providing thermodynamic stability, endow chemical stability upon protein secondary structures by deterring attack by water and other nucleophiles on backbone carbonyl groups. This deterrence could contribute to the remarkable longevity of the strands in a collagen triple helix. These strands have resisted chemical degradation for 80 million years,26 and each peptide bond (other than that of the N-terminal residue) accepts an n→π* interaction.27
Supplementary Material
Acknowledgments
We are grateful to Dr. I. A. Guzei for assistance with X-ray crystallography. K.J.K. was supported by a Barry M. Goldwater Scholarship and a Hilldale Undergraduate/Faculty Research Fellowship. This work was supported by grant R01 AR044276 (NIH).
Notes and references
- 1.Bretscher LE, Jenkins CL, Taylor KM, DeRider ML, Raines RT. J Am Chem Soc. 2001;123:777–778. doi: 10.1021/ja005542v. [DOI] [PubMed] [Google Scholar]
- 2.DeRider ML, Wilkens SJ, Waddell MJ, Bretscher LE, Weinhold F, Raines RT, Markley JL. J Am Chem Soc. 2002;124:2497–2505. doi: 10.1021/ja0166904. [DOI] [PubMed] [Google Scholar]
- 3.Hinderaker MP, Raines RT. Protein Sci. 2003;12:1188–1194. doi: 10.1110/ps.0241903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hodges JA, Raines RT. Org Lett. 2006;8:4695–4697. doi: 10.1021/ol061569t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choudhary A, Gandla D, Krow GR, Raines RT. J Am Chem Soc. 2009;131:7244–7246. doi: 10.1021/ja901188y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartlett GJ, Choudhary A, Raines RT, Woolfson DN. Nat Chem Biol. 2010;6:615–620. doi: 10.1038/nchembio.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choudhary A, Kamer KJ, Powner MW, Sutherland JD, Raines RT. ACS Chem Biol. 2010;5:655–657. doi: 10.1021/cb100093g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jakobsche CE, Choudhary A, Raines RT, Miller SJ. J Am Chem Soc. 2010;132:6651–6653. doi: 10.1021/ja100931y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choudhary A, Raines RT. Protein Sci. 2011;20:1077–1087. doi: 10.1002/pro.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choudhary A, Kamer KJ, Raines RT. J Org Chem. 2011;76:7933–7937. doi: 10.1021/jo201389d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newberry RW, VanVeller B, Guzei IA, Raines RT. J Am Chem Soc. 2013;135:7843–7846. doi: 10.1021/ja4033583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bürgi HD, Dunitz JD, Shefter E. Acta Crystallogr. 1974;B30:1517–1527. [Google Scholar]
- 13.Fufezan C. Proteins. 2010;78:2831–2838. doi: 10.1002/prot.22800. [DOI] [PubMed] [Google Scholar]
- 14.Kamer KJ, Choudhary A, Raines RT. J Org Chem. 2013;78:2099–2103. doi: 10.1021/jo302265k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sonntag LS, Schweizer S, Ochsenfeld C, Wennemers H. J Am Chem Soc. 2006;128:14697–14703. doi: 10.1021/ja0654938. [DOI] [PubMed] [Google Scholar]
- 16.Kuemin M, Nagel YA, Schweizer S, Monnard FW, Ochsenfeld C, Wennemers H. Angew Chem Int Ed. 2010;49:6324–6327. doi: 10.1002/anie.201001851. [DOI] [PubMed] [Google Scholar]
- 17.Erdmann RS, Wennemers H. J Am Chem Soc. 2012;134:17117–17124. doi: 10.1021/ja3066418. [DOI] [PubMed] [Google Scholar]
- 18.Pandey AK, Naduthambi D, Thomas KM, Zondlo NJ. J Am Chem Soc. 2013;135:4333–4363. doi: 10.1021/ja3109664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coldham I, Houdayer PMA, Judkins RA, Witty DR. Synlett. 1996;1996:1109–1111. [Google Scholar]
- 20.Weinhold F. In: Encyclopedia of Computational Chemistry. Schleyer PvR, Allinger NL, Clark T, Gasteiger J, Kollman PA, Shaefer HF, III, Schreiner PR., editors. Vol. 3. John Wiley & Sons; Chichester, UK: 1998. pp. 1792–1811. [Google Scholar]
- 21.Glendening ED, et al. NBO 5.0. Theoretical Chemistry Institute, University of Wisconsin–Madison; Madison, Wisconsin, USA: 2001. [Google Scholar]
- 22.Weinhold F, Landis CR. Valency and Bonding: A Natural Bond Orbital Donor–Acceptor Perspective. Cambridge University Press; Cambridge, UK: 2005. [Google Scholar]
- 23.Jahn H, Teller E. Proc R Soc London, Ser A. 1937;161:220–235. [Google Scholar]
- 24.Pollock SB, Kent SBH. Chem Commun. 2011;47:2342–2344. doi: 10.1039/c0cc04120c. [DOI] [PubMed] [Google Scholar]
- 25.Shi Z, Kallenbach NR. Proc Natl Acad Sci USA. 2011;108:3–4. doi: 10.1073/pnas.1017021108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schweitzer MH, Zheng W, Organ CL, Avci R, Suo Z, Freimark LM, Lebleu VS, Duncan MB, Vander Heiden MG, Neveu JM, Lane WS, Cottrell JS, Horner JR, Cantley LC, Kalluri R, Asara JM. Science. 2009;324:626–631. doi: 10.1126/science.1165069. [DOI] [PubMed] [Google Scholar]
- 27.Shoulders MD, Raines RT. Annu Rev Biochem. 2009;78:929–958. doi: 10.1146/annurev.biochem.77.032207.120833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.