

SUMMARY
A more complete understanding of how fear extinction alters neuronal activity and connectivity within fear circuits may aid in the development of strategies to treat human fear disorders. Using a c-fos based transgenic mouse, we found that contextual fear extinction silenced basal amygdala (BA) excitatory neurons that had been previously activated during fear conditioning. We hypothesized that the silencing of BA fear neurons was caused by an action of extinction on BA inhibitory synapses. In support of this hypothesis, we found extinction-induced target-specific remodeling of BA perisomatic inhibitory synapses originating from parvalbumin and cholecystokinin-positive interneurons. Interestingly, the predicted changes in the balance of perisomatic inhibition matched the silent and active states of the target BA fear neurons. These observations suggest that target-specific changes in perisomatic inhibitory synapses represent a mechanism through which experience can sculpt the activation patterns within a neural circuit.
INTRODUCTION
Exposure therapy is widely used to treat fear disorders, but it rarely leads to a complete and permanent loss of maladaptive fear. A deeper understanding of the neurobiological mechanisms that underlie exposure therapy can be achieved by studying fear extinction in animal models (Graham et al., 2011) and may be useful for the development of more effective therapies. Over the past decades studies on the neurobiological basis of fear extinction have discovered that multiple brain regions are recruited by fear extinction (Corcoran and Maren, 2001; Falls et al., 1992; Morgan et al., 1993; Vianna et al., 2001). These brain regions include both cortical and subcortical areas that are reciprocally connected, thereby forming a distributed extinction circuit that can be recruited by behavioral extinction training and which, upon its recruitment, can lead to the loss or suppression of fear (Orsini and Maren, 2012). In addition to the extinction circuit, a fear circuit has been characterized that is responsible for the storage and expression of fear memories and that is also distributed over multiple brain regions (Orsini and Maren, 2012). Important for using rodents as model organisms, both the extinction and fear circuits are highly conserved between rodents and humans (Hartley and Phelps, 2010). In this study, we address the question of the precise anatomical and functional connection between the extinction circuit and the fear circuit towards the aim of gaining a greater understanding of how they interact during fear extinction.
One potential strategy for identifying the interface between the extinction circuit and the fear circuit is to identify neurons within the fear circuit that are silenced by extinction, and then use these neurons as a starting point for determining which upstream events within the extinction circuit cause their silencing. The first step towards applying this strategy was made using electrophysiological recordings of neurons in the amygdala, a brain region known as a central hub within the fear circuit (Orsini and Maren, 2012). Electrophysiological recordings revealed that neurons in the lateral amygdala and the basal amygdala can increase their firing in response to fear conditioning and, subsequently, can be silenced in response to fear extinction (Amano et al., 2011; Herry et al., 2008; Hobin et al., 2003; Livneh and Paz, 2012; Repa et al., 2001). However, the precise mechanisms through which the extinction circuit achieves the extinction-induced silencing of amygdala fear neurons are not fully understood. Modification of synaptic input, either by decreasing excitatory input or increasing inhibitory input, is a candidate mechanism. The importance of inhibitory synaptic plasticity is increasingly being appreciated (Kullmann et al., 2012), and inhibitory plasticity has been implicated in fear extinction (Ehrlich et al., 2009; Makkar et al., 2010).
In this study we employed an imaging approach to identify the precise location of basal amygdala (BA) fear neurons that are silenced by contextual fear extinction and determine how these fear neurons are silenced. We previously imaged BA fear neurons with a transgenic mouse that uses tetracycline-controlled tagging (TetTag) of neurons activated during fear conditioning (Tayler et al., 2013; Reijmers et al., 2007). Here, we utilize the TetTag mouse to image BA fear neurons that are silenced by extinction. We find evidence for structural plasticity of perisomatic inhibitory synapses originating from parvalbumin positive interneurons after silencing of BA fear neurons by fear extinction. Importantly, these parvalbumin positive synapses were located immediately around the soma of the silenced BA fear neurons, revealing an anatomical and functional connection between the extinction circuit and the fear circuit. In addition, fear extinction altered the presence of perisomatic endocannabinoid receptors around the soma of BA fear neurons that remained active after fear extinction. Our findings provide insight into the mechanisms underlying the silencing of fear circuits and, more generally, add to our knowledge of how behavior can sculpt activity and connectivity within a neural circuit.
RESULTS
Fear extinction silences the basal amygdala fear memory circuit
Prior electrophysiological studies have revealed that fear extinction can decrease the firing of basal amygdala (BA) fear neurons (Amano et al., 2011; Herry et al., 2008; Livneh and Paz, 2012), but the underlying mechanisms are not fully understood. We took advantage of a c-fos-based reporter mouse, the TetTag mouse (Reijmers et al., 2007), to image the effect of contextual fear extinction on BA fear neuron activation. The TetTag mouse expresses long-lasting nuclear green fluorescent protein (GFP) under control of the c-fos promoter (Figure 1A), which enabled us to tag excitatory neurons activated during fear conditioning (i.e. fear neurons, Figures 1B and S1A- S1B). The expression of the immediate-early gene zif268/egr1 (Zif) served as a marker for neurons activated during a later retrieval test (Okuno, 2011; Reijmers et al., 2007) (Figures 1C and S1C). Two groups of TetTag mice (FC, n=15; FC+EXT, n=17) were subjected to contextual fear conditioning (Figures 1C and 1D). As expected, similar numbers of BA fear neurons were tagged with GFP in both groups (Figure 1E). The next 2 days, only one group (FC+EXT) was subjected to extinction trials, while the other group (FC) remained in the home cage. Extinction caused the loss of fear expression as indicated by the total suppression of freezing at the end of the extinction procedure (Figure 1F), and during retrieval on day 4 (Figure 1G). Extinction had no significant effect on the activation of BA neurons that were not tagged during fear conditioning (GFP-Zif+, Figure 1H). We analyzed the BA fear memory circuit by defining 2 types of fear neurons (i.e. tagged during fear conditioning): silent fear neurons (GFP+Zif-, fear neurons not reactivated during retrieval) and active fear neurons (GFP+Zif+, fear neurons reactivated during retrieval) (Figure 1I). We found that fear extinction caused a 2.3-fold decrease in the number of active BA fear neurons, with a subgroup of fear neurons remaining active after extinction (GFP+Zif+; Figures 1J and S1D). This extinction-induced silencing of the BA fear memory circuit, caused by contextual fear extinction, is similar to that previously found in electrophysiological studies on tone fear extinction (Amano et al., 2011; Herry et al., 2008; Livneh and Paz, 2012). In addition, the observed concomitant reduction in active BA fear neurons and freezing replicates previous studies (Herry et al., 2008; Reijmers et al., 2007), and reflects the causative role of the basal amygdala in the behavioral expression of contextual fear (Maren, 1998). Therefore, our results provide further support for a model where extinction decreases fear by silencing the fear memory circuit within the BA.
Figure 1. Fear extinction silences the basal amygdala fear memory circuit.
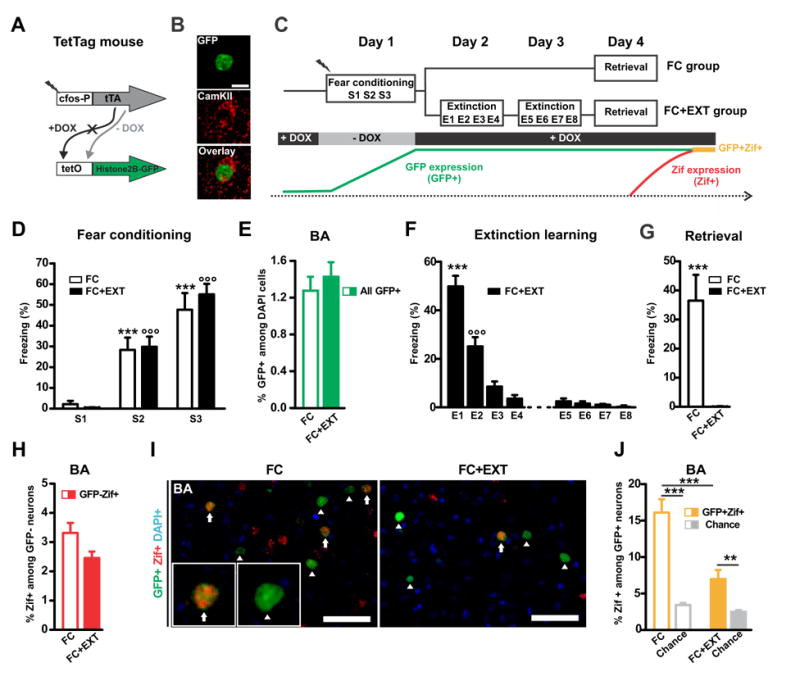
(A) A double transgenic TetTag mouse line was used that expresses the tetracycline transcriptional activator (tTA) under control of the activity-regulated c-fos promoter. In the absence of doxycycline (- DOX), tTA binds to the tet operator (tetO) in the second transgene and induces expression of a long-lasting histone2B-GFP fusion protein. (B) Image of an excitatory basal amygdala (BA) neuron tagged with long-lasting histone2B/GFP (green) and immunolabeled with a Calcium/calmodulin-dependent kinase II (CamKIIa) antibody (red). (C) Schema of the experimental procedure. “-DOX” opened a time-window for tagging fear conditioning activated neurons (GFP+; i.e. fear neurons) in two experimental groups (FC, n=15; FC+EXT, n=17). Zif expression during retrieval (Zif+) was used to detect the reactivation of tagged fear neurons (GFP+Zif+; i.e. active fear neurons). The absence of Zif was used to identify silent fear neurons (GFP+Zif-). (D) As training progressed, FC and FC+EXT mice showed an increase in their level of freezing (between sessions: p<0.001 *** for FC and °°° for FC+EXT). (E) FC and FC+EXT groups had similar percentages of GFP+ neurons (sum of GFP+Zif- and GFP+Zif+) in the BA (p=0.49). (F) The FC+EXT group showed a significant decrease in freezing during the extinction sessions on day 2 and 3 (p<0.001: *** E1 vs. others and °°° E2 vs. others). (G) Freezing during retrieval on day 4 in the FC+EXT group was absent and was significantly lower compared to the FC group (p=0.00014). (H) There was no significant difference in the percentage of Zif+ among GFP- neurons between the two groups (p=0.064). (I) Representative image of GFP-positive (GFP+, green) and Zif-positive (Zif+, red) nuclei in the basal amygdala (BA) of a FC (left) and FC+EXT mouse (right). Arrows indicate active fear neurons (GFP+Zif+) and arrowheads indicate silent fear neurons (GFP+Zif-). (J) Extinction decreased the number of active BA fear neurons, as indicated by FC+EXT mice having less GFP+Zif+ neurons than FC mice (p=0.00023). In both groups the percentage of GFP+ZIF+ neurons was higher than chance level (FC: p=0.0000098; FC+EXT p= 0.0014). Chance level was determined by using the % of Zif+ among GFP- neurons as shown in (H). Scale bar, 50 μm. Graphs show means ± SEM. **P < 0.01, ***P < 0.001.
See also Figure S1.
Activation of brain regions upstream of the basal amygdala is not altered after fear extinction
We next explored where extinction acted to cause a silencing of the BA fear memory circuit. We first addressed the possibility that contextual fear extinction might act on brain regions upstream of the BA, and thereby indirectly silence fear neurons in the BA. The BA receives inputs from the CA1 area of the hippocampus and from the infralimbic prefrontal cortex, brain regions that have been implicated in fear extinction (Hartley and Phelps, 2010; Orsini and Maren, 2012). We therefore tested if fear extinction altered the activation of excitatory neurons in the CA1 region of the hippocampus (both dorsal and ventral: dCA1 and vCA1), and in the infralimbic prefrontal cortex (IL). We analyzed the same brains that were used for the BA analysis, since the TetTag mouse enables the tagging of neurons recruited by fear conditioning throughout the whole brain (Deng et al., 2013; Garner et al., 2012; Liu et al., 2012; Matsuo et al., 2008; Reijmers et al., 2007; Tayler et al., 2011; Tayler et al., 2013). As expected, the FC and FC+EXT groups had similar percentages of neurons tagged in these brain regions during contextual fear conditioning (Figures 2A and 2B). Contextual fear extinction did not alter the activation of non-tagged dCA1 and vCA1 neurons during retrieval (Figure 2C), nor the reactivation of tagged dCA1 and vCA1 neurons during retrieval (Figures 2B and 2D). These results are consistent with a previous study reporting that contextual fear extinction acts on a population of CA1 neurons that is segregated from the CA1 neurons recruited during fear conditioning (Tronson et al., 2009), and indicate that the memory of the context is retained after contextual fear extinction (Rudy et al., 2004). Similar to the hippocampal CA1 neurons, the activation of non-tagged IL neurons (Figure 2C) and the reactivation of tagged IL neurons (Figures 2B and 2D) were not affected by contextual fear extinction. Overall, we did not detect extinction-induced functional changes in two important brain structures upstream of the BA. We therefore shifted our focus to potential local changes within the BA that might have caused the silencing of the BA fear memory circuit.
Figure 2. Activation of brain regions upstream of the basal amygdala is not altered after fear extinction.
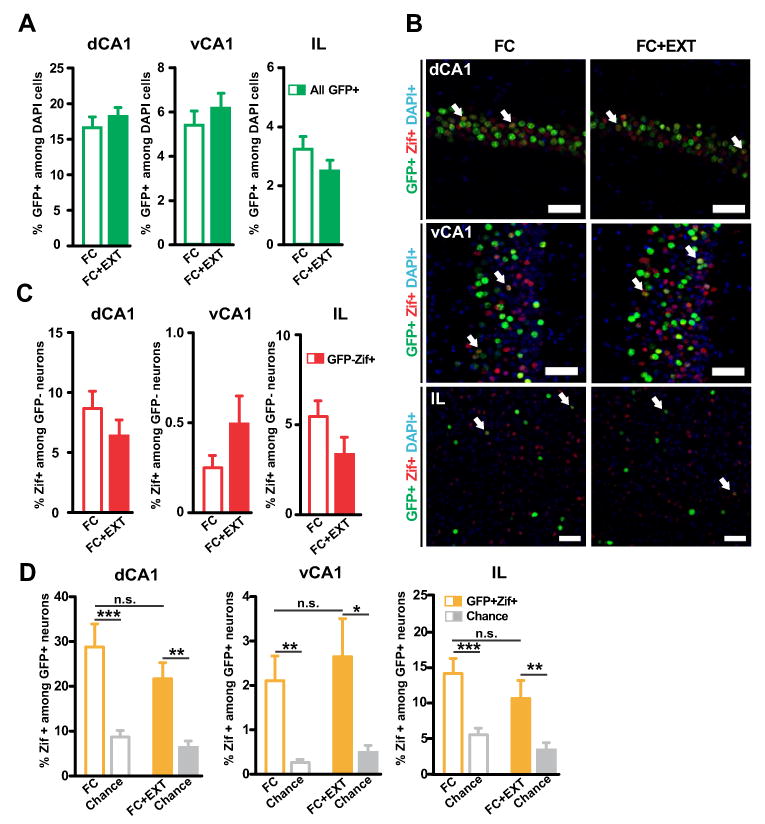
(A) The FC and FC+EXT groups had similar percentages of GFP+ neurons in the dCA1, vCA1 and IL (dCA1: p=0.44; vCA1: p=0.50; IL: p=0.17). (B) Representative image of GFP+ neurons (green) and Zif+ neurons (red) in the dorsal CA1 (dCA1, top), ventral CA1 (vCA1, middle) and infralimbic cortex (IL, bottom) of a FC mouse (left) and a FC+EXT mouse (right). Blue: DAPI. Arrows indicate active fear neurons (GFP+Zif+). Scale bar, 50 um. (C) No significant differences in the percentage of Zif+ among GFP- neurons were found (dCA1: p=0.25; vCA1: p=0.21; IL: p=0.056). (D) Extinction had no effect on reactivation of dCA1, vCA1 and IL. The number of GFP+Zif+ neurons was similar between the two groups (dCA1: p=0.26; vCA1: p=0.61; IL: p=0.27). The number of GFP+Zif+ neurons was above chance level in the dCA1, vCA1 and IL of both groups (dCA1: FC vs. chance, p=0.00017; FC-EXT vs. chance, p=0.000017; vCA1: FC vs. chance, p=0.0027; FC-EXT vs. chance, p=0.0072; IL: FC vs. chance, p=0.000062; FC-EXT vs. chance, p=0.0026). Chance level was determined by using the % of Zif+ among GFP- neurons as shown in (C). Graphs show means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Fear extinction causes target-specific remodeling of perisomatic inhibitory synapses in the basal amygdala
Around 85% of the neuronal cell population within the BA consists of excitatory projection neurons, whereas the remaining 15% are local interneurons that make inhibitory synapses onto the projection neurons (McDonald, 1992). Because BA inhibitory interneurons have been implicated in fear extinction (Ehrlich et al., 2009; Heldt and Ressler, 2007), we addressed the possibility that structural changes involving inhibitory circuits in the BA might have caused the extinction-induced silencing of BA fear neurons by increasing local inhibition. We first examined the expression of 67-kDa glutamic acid decarboxylase (GAD67), a key enzyme in GABA synthesis. Both GAD67 and the smaller isoform GAD65 have been implicated in fear extinction, but a specific role within the amygdala has so far only been established for GAD67 (Heldt et al., 2012; Sangha et al., 2009). We did not find evidence for increased GAD67 expression in either the complete BA or in the soma of BA interneurons (Figures 3A and 3B and 3C), consistent with a recent study (Sangha et al., 2012). We hypothesized that fear extinction might act on a synaptic site where local interneurons interface with the BA fear neurons. We tested this hypothesis by imaging a special type of inhibitory synapses called perisomatic synapses. Perisomatic inhibitory synapses are a plausible candidate for silencing BA fear neurons, since they are well-positioned to modulate the functional activation of excitatory neurons (Miles et al., 1996). Consistent with our hypothesis, we found that silent fear neurons had increased GAD67 around their soma after extinction (Figure 3D). Interestingly, this increase in perisomatic GAD67 was not observed around active fear neurons (Figure 3E). The selective increase in perisomatic GAD67 around silent fear neurons seemed to be caused by a selective increase in the number of inhibitory synapses (Figures S2A and S2B). Thus, our data reveal that extinction can cause the target-specific remodeling of perisomatic inhibitory synapses in the BA, with extinction-induced changes in perisomatic GAD67 matching the activation states of the postsynaptic fear neurons.
Figure 3. Fear extinction causes target-specific remodeling of perisomatic inhibitory synapses in the basal amygdala.
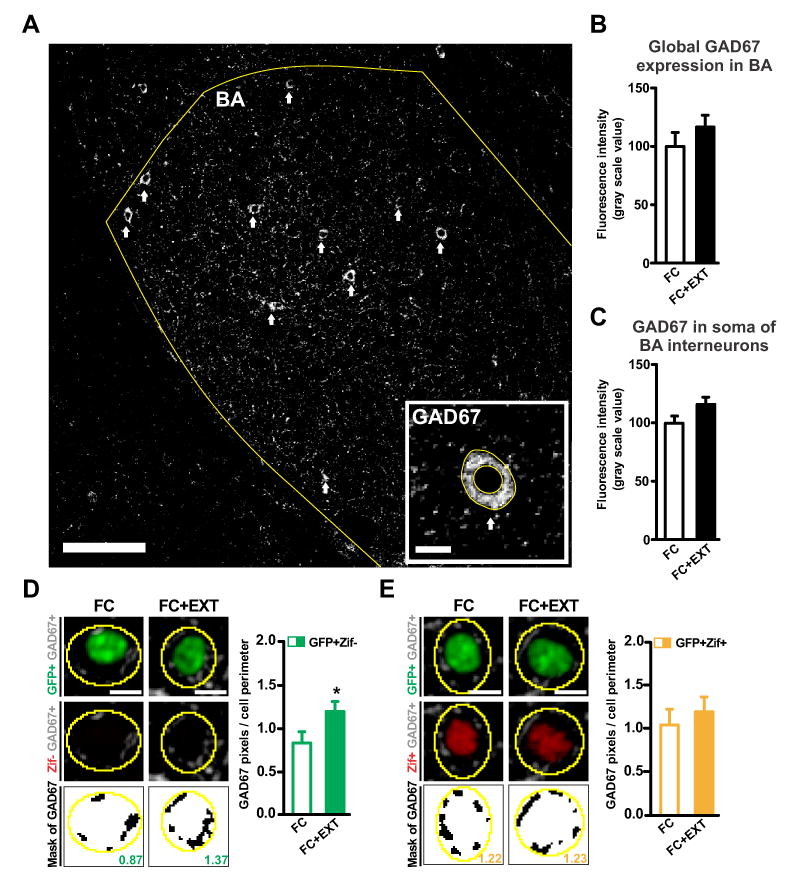
(A) Image of glutamic acid decarboxylase-67 (GAD67) immunolabeling of the basal amygdala (BA; scale bar: 100μm). Arrows indicate GAD67+ soma in the BA. The inset shows a representative image of a GAD67-positive (GAD67+) interneuron soma (scale bar: 10μm). (B) Extinction had no effect on GAD67 expression in the total basal amygdala (p=0.30). (C) Extinction had no effect on somatic expression of GAD67 (p=0.16). (D-E left) Representative images of perisomatic GAD67 immunolabeling around silent BA fear neurons (D, GFP+Zif-) and active BA fear neurons (E, GFP+Zif+). Three images are shown for each fear neuron (top: GFP in green, GAD67 in grey; middle: Zif in red, GAD67 in grey; bottom: GAD67 mask in black, value for that neuron obtained by dividing black pixels by perimeter of yellow outline). Scale bars, 10 μm. (D-E right) Extinction increased perisomatic GAD67 around silent fear neurons (D, p=0.047) but not around active fear neurons (E, p=0.46). Graphs show means ± s.e.m. *P < 0.05.
See also Figure S2.
Fear extinction increases perisomatic parvalbumin around silent BA fear neurons
We decided to further investigate the nature of the extinction-induced remodeling of perisomatic inhibitory synapses in the BA. Parvalbumin-positive interneurons (PV+) are the predominant interneuron type in the BA, and form perisomatic synapses around BA excitatory projection neurons (McDonald and Betette, 2001). Extinction did not change the expression of PV in the soma of BA interneurons (Figures 4A and 4B). Next, we analyzed the presence of PV around the soma of BA fear neurons. We verified that our perisomatic PV immunolabeling represented perisomatic synapses (Figure S3A). Consistent with the extinction-induced increase in perisomatic GAD67, extinction also increased perisomatic PV around the silent fear neurons (Figure 4C). Again, there was no significant increase around the active fear neurons (Figure 4D). The effects of extinction on perisomatic PV seemed to reflect changes in synapse numbers (Figures S3B and S3C). Importantly, the increase in perisomatic PV that we detected with image analysis is similar to that reported to increase perisomatic inhibition using electrophysiological analysis (Gittis et al., 2011; Kohara et al., 2007). Thus, our data suggest an extinction-induced increase in perisomatic inhibition underlies the decreased number of active BA fear neurons and the resulting silencing of the fear memory circuit. This reveals a direct connection between extinction-induced structural and functional changes in the BA.
Figure 4. Fear extinction increases perisomatic parvalbumin around silent BA fear neurons.
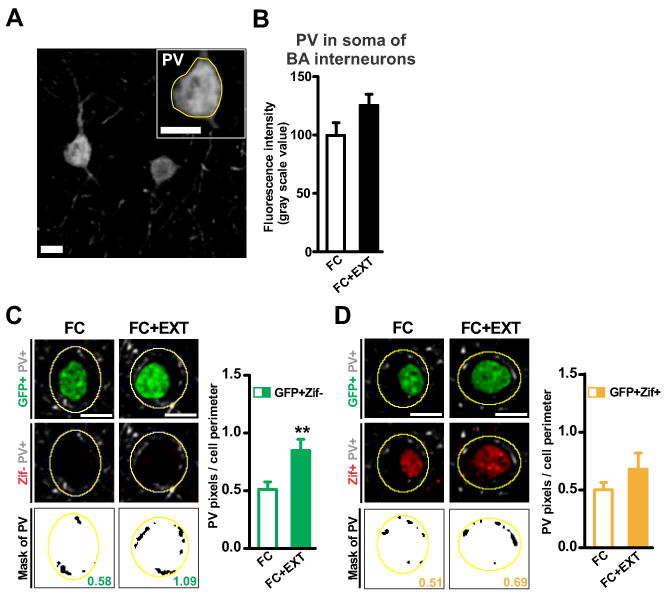
(A) Representative image of a parvalbumin-positive (PV+) interneuron soma in the basal amygdala (BA; scale bar: 10μm). (B) Extinction had no effect on somatic expression of PV (p=0.064). (C-D left) Representative images of perisomatic PV immunolabeling around silent BA fear neurons (C, GFP+Zif-) and active BA fear neurons (D, GFP+Zif+). Three images are shown for each fear neuron (top: GFP in green, PV in grey; middle: Zif in red, PV in grey; bottom: PV mask in black, value for that neuron obtained by dividing black pixels by perimeter of yellow outline). Scale bars, 10 μm. (C-D right) Extinction increased perisomatic PV around silent fear neurons (C, p=0.0061) but not around active fear neurons (D, p=0.18). Graphs show means ± s.e.m. **P < 0.01.
See also Figure S3.
The extinction-induced increase in perisomatic parvalbumin reflects new learning
We asked whether the extinction-induced increase in perisomatic PV might have reversed any fear conditioning-induced changes in those synapses, which would indicate that BA perisomatic inhibitory synapses were part of the original fear circuit. To address this question, we performed a separate experiment where we compared a fear conditioned group (FC) with a home cage group (HC) (Figures 5A and 5B). Consistent with our previous study (Reijmers et al., 2007), BA neurons activated during fear conditioning were tagged with long-lasting expression of GFP (Figure 5C). During retrieval on day 4, the FC group showed significant freezing (Figure 5D). The retrieval of contextual fear caused activation of both untagged (GFP-Zif+; Figure 5E) and tagged (GFP+Zif+; Figure 5F) neurons in the BA, with a preferential reactivation of the tagged BA fear neurons (Figures 5E and 5F). Importantly, we did not find fear conditioning-induced changes in perisomatic PV around silent or active fear neurons (Figures 5G and 5H). These data strongly suggest that the extinction-induced changes in PV+ perisomatic synapses constituted a new form of learning that occurred within the extinction circuit.
Figure 5. The extinction-induced increase in perisomatic parvalbumin reflects new learning.
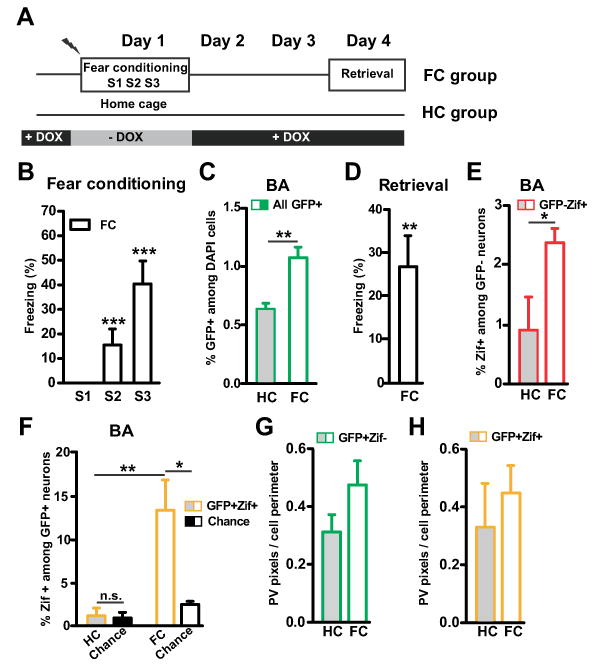
(A) Design of an experiment with a home cage (HC, n=8) and a fear conditioned group (FC, n=8). (B) Freezing during contextual fear conditioning on day 1. As training progressed, FC mice showed an increase in their level of freezing (p=0.0006). (C) The FC group had a larger number of GFP+ neurons than the HC group in the basal amygdala (BA, p=0.0049). (D) FC mice showed significant freezing during retrieval on day 4 (p=0.0077). (E) The FC group had a larger number of GFP-Zif+ neurons than the HC group (p=0.036). (F) The FC group had a larger number of GFP+Zif+ neurons than the HC group (p=0.0043). Only in the FC group was the percentage of GFP+ZIF+ neurons higher than chance level, confirming that a subset of BA fear neurons was reactivated during retrieval (HC: p=0.23; FC: p=0.016). Chance level was determined by using the % of Zif+ among GFP- neurons as shown in (E). (G-H) Fear conditioning had no effect on perisomatic PV around either type of tagged BA neuron (G: GFP+Zif-, p=0.10; H: GFP+Zif+, p=0.49). Graphs show means ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001.
Fear extinction increases perisomatic CB1R around active BA fear neurons
In addition to PV+ perisomatic synapses, the BA also contains perisomatic inhibitory synapses that originate from cholecystokinin (CCK) interneurons (Yoshida et al., 2011). We therefore examined if fear extinction also affected perisomatic CCK+ synapses. Extinction did not change the expression of CCK in the soma of BA interneurons (Figures 6A and 6B). In addition, perisomatic CCK around fear neurons, either silent or active, was not altered by fear extinction (Figures 6C, 6D, S4A and S4B). Fear conditioning itself did also not change perisomatic CCK in the BA (Figures S4C and S4D). We next examined if extinction might have affected CCK+ perisomatic synapses without changing CCK levels. Recently, it was discovered that BA perisomatic synapses positive for CCK, but not PV, contain a unique enrichment of proteins involved in endocannabinoid signaling, including cannabinoid receptor type 1 (CB1R) (Yoshida et al., 2011). Since CB1R in the BA have been implicated in fear extinction (Marsicano et al., 2002), we examined whether perisomatic CB1R presence was modulated by extinction and whether this modulation was target-specific. We first confirmed that in the BA our CB1R immunolabeling colocalized with our CCK, but not PV, immunolabeling (Figure 6E), and that CB1R and CCK colocalized directly adjacent to the soma (Figure S4E). We did not observe labeling of CB1R in the soma of BA interneurons, so somatic expression of CB1R was not quantified. Perisomatic CB1R presence around the silent fear neurons was similar in the FC and FC+EXT groups (Figures 6F and S4F). Intriguingly, extinction increased perisomatic CB1R around the active fear neurons (Figures 6G and S4G). This effect of extinction appeared to constitute a new form of learning, as fear conditioning itself did not change perisomatic CB1R in the BA (Figures S4H and S4I). Since CB1R inhibit the release of γ-aminobutyric acid (GABA) (Katona et al., 2001), these results suggest that the extinction-induced upregulation of perisomatic CB1R facilitated the persistence of a small subset of active BA fear neurons in the extinction group (Figure 1J).
Figure 6. Fear extinction increases perisomatic CB1R around active BA fear neurons.
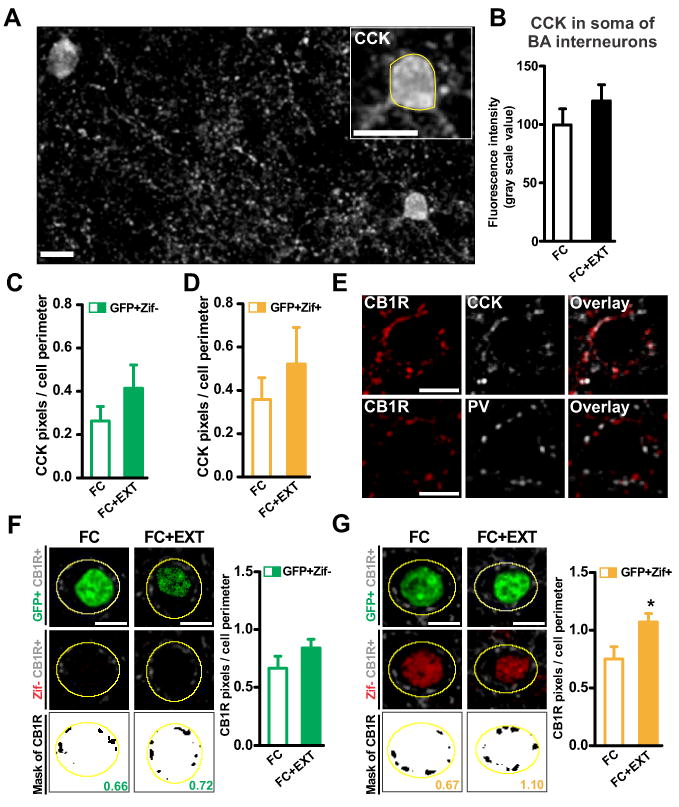
(A) Representative image of cholecystokinin-positive (CCK+) interneuron soma in the basal amygdala (BA; scale bar: 10μm). (B) Extinction had no effect on somatic expression of CCK (p=0.25). (C-D) Extinction had no effect on perisomatic CCK around silent (C: GFP+Zif-, p=0.25) and active BA fear neurons (D: GFP+Zif+, p=0.42). (E) Top: colocalization of perisomatic CB1R (red) and perisomatic CCK (grey) in the BA. Bottom: no colocalization of perisomatic CB1R (red) and perisomatic PV (grey) was detected in the BA. Scale bars: 10μm. (F-G left) Representative images of perisomatic CB1R immunolabeling around silent BA fear neurons (F, GFP+Zif-) and active BA fear neurons (G, GFP+Zif+). Three images are shown for each fear neuron (top: GFP in green, CB1R in grey; middle: Zif in red, CB1R in grey; bottom: CB1R mask in black, value for that neuron obtained by dividing black pixels by perimeter of yellow outline). Scale bars, 10 μm. (F-G right) Extinction increased perisomatic CB1R expressed in CCK terminals around active fear neurons (G: p=0.036), but not around silent fear neurons (F: p=0.19). Graphs show means ± s.e.m. *P < 0.05.
See also Figure S4.
Fear extinction might alter perisomatic inhibition outside of the fear circuit
The results of our analysis of the effects of extinction on silent (GFP+Zif-) versus active (GFP+Zif+) fear neurons are summarized in Figure 7. To explore if fear extinction might also alter perisomatic inhibition outside of the fear circuit, we quantified perisomatic markers around GFP-Zif+ neurons in the BA. We found that extinction increased PV and CB1R around GFPZif+ cells, but had no significant effect on perisomatic GAD67 and CCK (Figure S5A). This result has two possible explanations that are not mutually exclusive. First, it suggests that fear extinction might alter perisomatic inhibition around BA neurons that are not part of the fear circuit. One possibility is that fear extinction also changes perisomatic inhibition of BA neurons that are part of the extinction circuit (Herry et al., 2008). These extinction neurons were reported to be silent during fear conditioning and subsequently activated during extinction, so they would not be tagged with GFP in our experimental design. Second, some GFP-Zif+ neurons might actually be fear neurons that were not tagged with GFP. For example, neurons with a relatively low level of c-fos promoter activation during fear conditioning might express c-fos protein and tTA protein, but the tTA protein level might be too low to activate the tetO promoter and trigger GFP expression. We tried to find support in favor of one of these two explanations by correlating the number of GFP-Zif+ neurons with freezing levels during the extinction trials. If a significant portion of the GFP-Zif+ neurons were extinction neurons, then a negative correlation with freezing during extinction might be observed. On the other hand, if a significant portion of the GFP-Zif+ neurons were non-tagged active fear neurons, then a positive correlation with freezing during extinction might be observed similar to the positive correlation found for GFP+Zif+ neurons (Figure S1D). We did not find a significant correlation, either positive or negative (Figure S5B). This suggests that GFP-Zif+ neurons might consist of a mix of neurons with varying functions. Table S1 summarizes the extinction-induced perisomatic changes observed around the three types of BA neurons, showing that the changes around GFP-Zif+ neurons differ from the changes around fear neurons, either silent (GFP+Zif-) or active (GFP+Zif+). The different perisomatic profiles around the three BA cell types illustrate the target-specific nature of fear-extinction induced perisomatic synapse remodeling.
Figure 7. Model connecting extinction-induced functional and structural changes in the basal amygdala.
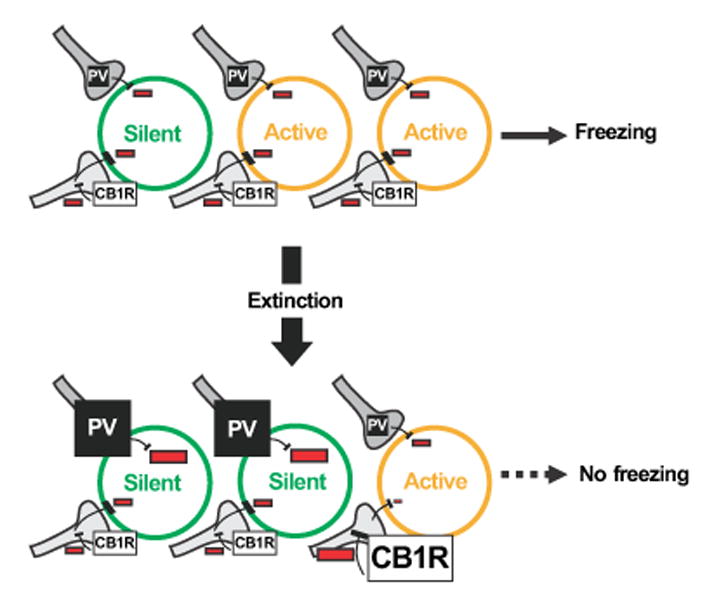
Extinction increases the ratio of silent (GFP+Zif-) versus active (GFP+Zif+) fear neurons in the basal amygdala (BA), thereby causing the elimination of behavioral expression of fear as indicated by the absence of freezing during retrieval. In this model the increased number of silent fear neurons is caused by increased perisomatic inhibition from PV neurons. The increase in CB1R occurs around a selective subset of BA fear neurons that as a result remain active because of increased CB1R mediated disinhibition. Inhibition is represented by the “-“ sign.
See also Figure S5 and Table S1
DISCUSSION
Our findings reveal that remodeling of perisomatic inhibitory synapses located immediately around fear neurons in the basal amygdala occurs during fear extinction. These perisomatic synapses reveal a site where the circuits for fear extinction and fear storage connect. The direct anatomical and functional relationship between the perisomatic synapses and the fear neurons suggests a straightforward mechanism for the silencing of fear circuits. Perisomatic inhibitory synapses therefore provide an attractive therapeutic target for improving the efficacy of fear extinction in humans treated with exposure therapy. In addition, we found that extinction might alter perisomatic inhibition outside of the fear circuit, possibly contributing to the behavioral effects of extinction by altering perisomatic inhibition of extinction neurons (Herry et al., 2008). The fine-tuned nature of the observed perisomatic synapse remodeling provides an important insight into how behavior can sculpt the flow of information in the brain.
Notably, the extinction-induced remodeling of perisomatic synapses was interneuron and target-neuron specific, and the predicted changes in the balance of perisomatic inhibition matched the state of the target fear neurons in two ways (Figure 7). First, the silent state of fear neurons (GFP+Zif-) corresponded to an extinction-induced increase in perisomatic PV, which is predicted to increase perisomatic inhibition (Gittis et al., 2011; Kohara et al., 2007). Second, the active state of fear neurons (GFP+Zif+) corresponded to an extinction-induced increase in perisomatic CB1R. We propose that the CB1R increase prevented a subset of active fear neurons from switching into silent fear neurons by decreasing GABA release from CCK terminals (Katona et al., 2001). The CB1R-mediated inhibition of GABA release around active fear neurons could strengthen their future activation by facilitating long-term potentiation of excitatory synapses (Carlson et al., 2002). Therefore, an intriguing possibility is that BA fear neurons that remain active after contextual fear extinction might, over time, reawaken the fear circuit and limit the effectiveness of exposure therapy by triggering spontaneous recovery (Myers and Davis, 2007).
The subset of fear neurons that remained active after extinction did not trigger freezing during the retrieval test (Figures 1G and 1J). This suggests that, in addition to BA perisomatic synapses, an additional downstream site exists where the extinction circuit inhibits the fear circuit. This downstream site might be located in the central amygdala, which contains neurons that mediate the effects of BA fear neurons on freezing. Previous studies support a model where central amygdala neurons are inhibited by intercalated interneurons, which become more active as a result of infralimbic prefrontal cortex activation during extinction (Amano et al., 2010; Likhtik et al., 2008; Milad and Quirk, 2002). Though we did not observe extinction-induced changes in the activation of brain regions upstream of the basal amygdala (Figure 2), a role for these upstream brain regions in fear extinction remains probable. For example, recent studies have found that projections from the prefrontal cortex and the hippocampus to the basal and lateral amygdala can regulate to what extent an extinguished fear memory is retrieved (Knapska et al, 2012, Orsini et al., 2011). It needs to be determined how the numerous neural circuits involved in fear extinction, located in various brain regions such as the prefrontal cortex, hippocampus, and amygdala, work together to silence the fear circuit. We propose that the approach used in this study can be more widely applied for this purpose. Identifying additional elements of the fear circuit that are silenced by extinction might enable the reconstruction of multiple functional extinction circuits, each responsible for silencing a specific element of the fear circuit. The discovery of structural changes in BA perisomatic synapses lays the foundation for reconstructing at least one coherent functional extinction circuit, with future studies determining which neural circuits need to be recruited during extinction to enable the target-specific remodeling of perisomatic synapses around BA fear neurons.
Does fear extinction decrease fear by suppressing or erasing the fear memory circuit? If extinction-induced changes in perisomatic inhibitory synapses constitute a form of erasure, then they should reverse changes induced by fear conditioning at these synapses. We did not find evidence for this, as fear conditioning itself did not change the perisomatic presence of PV, CCK, and CB1R. This strongly suggests that the observed changes in perisomatic synapses constituted a new form of learning that led to suppression of the fear memory circuit. This leaves open the possibility that in our study the fear memory circuit remained completely intact, for example by retaining a pattern of strengthened and weakened excitatory synapses induced by fear conditioning. In contrast with our findings, two recent papers reported examples of possible erasure of components of the fear memory circuit. One study using mice found that extinction reversed changes in dendritic spines that were induced by fear conditioning (Lai et al., 2012). It should be noted that the reported spine dynamics occurred in the frontal association cortex, a brain region that has not been firmly established yet as an essential component of the fear memory circuit. Nevertheless, this study provides an important first step towards identifying a mechanism by which fear memory circuits can be erased. Another recent study using human subjects reported that a certain behavioral extinction protocol, where extinction follows a retrieval trial, can erase a memory trace in the amygdala (Agren et al., 2012). However, in this study the erasure of the memory trace was inferred from changes in the activation state of the complete basolateral amygdala. Our data illustrate how extinction-induced changes in local inhibition within the basal amygdala might alter the activation state of the complete brain region without erasing the fear memory circuit, in which case it should be considered suppression. The question of suppression versus erasure has important implications for the treatment of fear disorders, as a treatment based on a form of erasure might make the return of debilitating fear less likely. Future studies using animal models will be invaluable to address the suppression versus erasure distinction, because validating a true mechanism for fear memory erasure will require more data collected at the cellular, subcellular, and ultimately the molecular level.
Our findings shed light on two proposed molecular mechanisms of extinction. Studies in humans and rodents have found that both CB1R (Gunduz-Cinar et al., 2012; Heitland et al., 2012; Marsicano et al., 2002; Rabinak et al., 2012), and brain-derived neurotrophic factor (BDNF; Andero et al., 2011; Chhatwal et al., 2006; Soliman et al., 2010) signaling in the BA support fear extinction. CB1R and BDNF signaling can both occur at inhibitory and excitatory synapses, and it is unclear which synapse type mediates their effects on fear extinction. In the case of CB1R signaling, the perisomatic CCK+ inhibitory synapses provide a plausible site of action, since the major components of CB1R signaling in the BA are highly enriched and colocalized in these synapses (Yoshida et al., 2011). However, the increase in perisomatic CB1R around the remaining active fear neurons seems in contradiction with a potential role for perisomatic CB1R signaling in the reduction of fear. We found that extinction might also increase CB1R outside of the fear circuit. If this increase occurred around extinction neurons (Herry et al., 2008), then it might have contributed to the increased activation of extinction neurons. A fear extinction role for CB1R at perisomatic inhibitory synapses would be in agreement with a recent finding that expression of CB1R at excitatory synapses is not sufficient to support fear extinction (Ruehle et al., 2013). In the case of BDNF, it is interesting to note that postsynaptic release of BDNF promotes the formation of perisomatic PV+ synapses in the cortex (Hong et al., 2008; Huang et al., 1999; Jiao et al., 2011; Kohara et al., 2007). We therefore propose that BDNF signaling in the BA supports fear extinction by increasing the number of perisomatic PV+ synapses around BA fear neurons, which is predicted to increase perisomatic inhibition (Gittis et al., 2011; Kohara et al., 2007). A better understanding of the molecular mechanisms used by BDNF to increase PV+ perisomatic synapse numbers could lead to new therapeutic targets for the treatment of fear disorders. Though BDNF acts on many types of synapses, both inhibitory and excitatory, it seems to use different signaling pathways within each type of synapse (Gottmann et al., 2009; Matsumoto et al., 2006). It is therefore feasible that targets will be identified that specifically modulate the effect of BDNF on perisomatic inhibitory synapses.
A potential role for inhibitory synapse plasticity in shaping patterns of neural circuit activation has recently become more appreciated (Kullmann et al., 2012). Inhibitory interneurons can be highly interconnected, resulting in synchronized firing (Bartos et al., 2007), and are in many brain regions outnumbered by excitatory neurons, with a single interneuron contacting as many as a thousand excitatory neurons (Miles et al., 1996). These traits make inhibitory interneurons seem ill-suited to exert finely targeted effects on individual excitatory neurons. The discovery of various forms of inhibitory synapse plasticity has made clear how inhibitory interneurons can specifically modulate the activation of individual target neurons (Kullmann et al., 2012). Perisomatic inhibitory synapses are especially well-positioned to enable this “personalized inhibition” by using their ability to suppress action potentials in the target neuron (Miles et al., 1996), thereby functioning as a brake that keeps the excitatory “gas pedal” in check. If perisomatic synapses indeed participate in the fine-tuned sculpting of patterns of neural circuit activation, then they should be subjected to forms of target-specific plasticity so that two excitatory neurons receiving perisomatic synapses from the same cluster of interneurons can be differentially inhibited. Recently, target-specific properties have been reported for perisomatic PV+ synapses in the striatum (Gittis et al., 2011), and for perisomatic CCK+ synapses in the entorhinal cortex (Varga et al., 2010). Our study adds to the understanding of perisomatic synapse dynamics in three ways. First, our study shows that target-specific changes can differentially affect PV and CCK perisomatic synapses around the same type of neuron. Second, we demonstrate target-specific modulation of perisomatic CB1R. Last and most important, our study reveals that behavior can trigger target-specific changes in perisomatic synapses. Behavior-induced target-specific plasticity of perisomatic synapses may be a central feature of neural circuits across the brain.
In summary, we discovered that contextual fear extinction causes the remodeling of perisomatic inhibitory synapses located directly around fear neurons in the basal amygdala. This discovery provides an anatomical and functional connection between the extinction circuit and the fear circuit. Since perisomatic synapses directly impinge on the fear circuit, they provide an attractive target for modulating maladaptive fear. In addition, our study reveals a mechanism by which behavior can use inhibitory synapse plasticity to alter the flow of information through the neural circuits. An important goal for future studies will be to determine the extent to which silencing of BA fear neurons is achieved by changes in perisomatic inhibitory synapses versus changes in other inhibitory and excitatory synapses and changes in neuronal excitability.
EXPERIMENTAL PROCEDURES
Animals
All animal procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Tufts University Institutional Animal Care and Use Committee. The TetTag mouse line used in this study was heterozygous for 2 transgenes: c-fos promoter driven tetracycline transactivator (cfosP-tTA; Jackson laboratory stock number 008344) and a tet operator driven fusion of histone2B and GFP (tetO-His2BGFP; Jackson laboratory stock number 005104). TetTag mice were backcrossed to a C57Bl6/J background. Thy1-YFP mice were obtained from Jackson laboratory (line H; stock number 003782). Mice had food and water ad libitum and were socially housed (3-5 animals per cage) until the start of the experiment, which was at an age of at least 12 weeks. Mice were kept on a regular light-dark cycle, and all experimental manipulations were done during the light phase. Mice were raised on food with doxycycline (40 mg doxycycline / 1 kg chow). One week before fear conditioning all mice were individually housed, and four days before fear conditioning doxycycline was removed from the food. After the last fear conditioning trial on day 1, mice were put on food with a high dose of doxycycline (1 g/kg) to rapidly block the tagging of neurons activated after fear conditioning. On day 2 mice were put back on the regular dose of doxycycline (40 mg/kg).
Behavior
A total of 48 TetTag mice were used for the study. Experiment 1 consisted of a fear conditioning group (FC, n=15), and a fear conditioning followed by extinction group (FC+EXT, n=17). Experiment 2 consisted of a home cage group (HC, n=8), and a fear conditioning group (FC, n=8).
Experiment 1
On day 1, mice were subjected to contextual fear conditioning consisting of 3 training trials (S1, S2 and S3) with 3 hours between each trial. The total duration of each training trial was 500 sec. A training trial started with placing the mouse in a square chamber with grid floor (Coulbourn Instruments; H10-11RTC, 120W x 100D x 120H). At 198sec., 278 sec., 358 sec., and 438 sec. a foot shock was delivered (2 sec., 0.70 mA). On day 2 and 3, FC+EXT mice were subjected to 4 extinction trials per day (day 2: E1 to E4; day 3: E5 to E8). Each extinction trial lasted 1800 sec with a trial interval of 2 hours. For each extinction trial, mice were placed in the same box used for fear conditioning without receiving foots shocks. On day 4, FC and FC+EXT mice were tested over 500 sec during a single retrieval test. For the retrieval test, mice were placed in the same box used for fear conditioning without receiving foots shocks.
Experiment 2
The FC group in experiment 2 was subjected to the same protocol as the FC group in experiment 1. The HC group consisted of mice that stayed in their home cage during the entire experiment. HC mice were perfused at the same time as the FC mice.
Quantification of freezing
Freezing behavior was measured using a digital camera connected to a computer with Actimetrics FreezeFrame software. The bout length was 1 second and the threshold for freezing behavior was determined by an experimenter blind to experimental conditions and animal group. Freezing scores were obtained by averaging freezing during minute 2 and 3 of each trial.
Tissue preparation and immunohistochemistry
Ninety minutes after retrieval testing, mice were deeply anesthetized with ketamine/xylazine and transcardially perfused with Phosphate Buffer (PB) 0.1M followed by 4% paraformaldehyde (PFA 4%) dissolved in PB 0.1M. Brains were extracted and post-fixed in PFA 4% for 24 h. Brains were transferred to 30% sucrose for 48–72 h before slicing 20 μm coronal sections of the entire brain using a cryostat. Sections were stored in cryoprotectant at -20°C until use. Free-floating sections were rinsed extensively in Phosphate Buffer Saline with 0.25% Triton X-100 (PBS-T). Sections were blocked for 1h at room temperature in PBS-T with 10% normal goat serum (or 3% donkey serum for CB1R). Sections were incubated in rabbit anti-Zif268 (Santa-Cruz; polyclonal; 1:3000) with either mouse anti-GAD67 (Millipore; monoclonal; 1:10000), mouse anti-PV (Millipore; monoclonal; 1:2000), mouse anti-CCK/Gastrin (Center for Ulcer Research and Education UCLA; monoclonal; 1:1000) or goat anti-CB1 (kind gift of Dr. K. Mackie; polyclonal; 1:2000) (Harkany et al, 2005). Additional primary antibodies used were rabbit anti-CamKII (kind gift of Dr. M. Jacob; polyclonal; 1:2000), and rabbit anti-Rab3b (kind gift of Dr. T. Südhof; polyclonal; 1:4000). Primary antibodies were diluted in the blocking solution, incubated at 4°C for 72 hours, and rinsed 3 times for 15 minutes in PBS-T. Secondary antibodies (Jackson Immuno Research; goat anti-rabbit 549 1/1500, goat anti-mouse 647 1/500, Donkey anti-rabbit 649 1/500, Donkey anti-goat 549 1/500) were diluted in the blocking solution and were then applied to the sections for 2 hours at room temperature followed by 3 rinses for 15 minutes in PBS-T. Sections were mounted on slides and coverslipped using DAPI mounting media to label cell nuclei and stored at 4°C.
Confocal Microscopy
A confocal laser-scanning microscope was used for all image acquisition (Nikon A1R). The settings for PMT, laser power, gain and offset were identical between experimental groups. Detection of Zif in GFP neurons was assessed for the basal amygdala (BA) (minimum of 7 sections per mouse), hippocampal CA1 (dCA1 and vCA1, 4 sections per mouse) and the infralimbic prefrontal cortex (IL, 4 sections per mouse). For the detection of perisomatic GAD67 a 20x objective was used and image stacks were collected with a 2μm step. For the detection of the other perisomatic markers (PV, CCK and CB1) a 40x objective was used and image stacks were collected with a 1 μm step. For the detection of PV, Rab3b, CCK and CB1 around neurons from Thy1-YFP mice a 60x objective was used and image stacks were collected with a 1 μm step.
Image analysis
All quantification was performed blind to experimental groups.
Quantification of activated cells
Selection of GFP labeled cells was designed to only include excitatory neurons (Figures S1A and S1B). Image J software was used to select and count the total number of DAPI, GFP, and Zif positive nuclei and nuclei double positive for GFP and Zif (Figure S1C). In order to avoid bias, all three cell types (GFP+Zif-, GFP+Zif+, GFP-Zif+) were selected from the same pictures, and the threshold settings for GFP and Zif were identical across all mice.
Quantification of soma expression
To quantify expression of GAD67, PV, and CCK within the soma of basal amygdala interneurons (Figures 3C and 4B and 6B), approximately 20 soma were outlined for each mouse and average pixel intensity was calculated using Image J. Somatic expression of CB1R was not quantified since no labeling of CB1R was observed in the soma of basal amygdala interneurons.
Quantification of pixels and clusters positive for perisomatic markers
One mouse from the FC+EXT group was excluded from the perisomatic marker analysis, since no active fear neurons (GFP+Zif+) were found in the basal amygdala of this mouse. For each marker (GAD67, PV, CCK, and CB1R), tagged fear neurons (GFP+Zif- and GFP+Zif+) and non-tagged neurons (GFP-Zif+) were randomly selected in the basal amygdala, and confocal images were analyzed at the z-plane where the diameter of the nucleus was largest. The average number of analyzed cells per mouse for each perisomatic marker is summarized in Table S2. A mask for each perisomatic marker was generated by thresholding the image of the perisomatic marker. For each perisomatic marker we used the same threshold settings for all the counted cells in order to avoid any bias. This threshold was the same for both the pixel and cluster countings. Threshold settings only differed between the different perisomatic markers, since the signal intensity varied across different antibodies. The mask of the perisomatic marker was used to draw an oval-shaped outline that included all the pixels positive for the perisomatic marker around a single neuron. The number of positive pixels and positive clusters (groups of adjacent positive pixels) within the outline was counted using Image J. To normalize for variation in size of neurons, the numbers of pixels and clusters were divided by the outline perimeter.
Statistics
Data are presented as means +/- SEM and were analyzed using ANOVAs repeated measures and two-tailed t-test (unpaired or paired) for normally distributed variables to evaluate statistical significance with p<0.05 as level of statistical significance. See Table S2 for the average number of analyzed cells per mouse for each perisomatic marker, and Table S3 for detailed statistical results.
Supplementary Material
Acknowledgments
We thank K. Kan and M. Parakala for technical assistance, and M. Mayford for providing TetTag mice. We thank J. Aggleton, J. Ainsley, L. Drane, L. Feig, M. Jacob, K. Mackie, E. Perisse, and S. Waddell for critical reading of the manuscript. This work was supported by a NIH Director's New Innovator Award (L.G.R.; DP2 OD006446), a Fyssen Foundation Postdoctoral Fellowship, a Bettencourt-Schueller award, and a Philippe Foundation Award (S.T.), a Sackler Dean’s Graduate Fellowship (J.S.), the Synapse Neurobiology Training Program (J.S.; T32 NS061764; PI: K. Dunlap and M. Jacob), the Tufts Center for Neuroscience Research (P30 NS047243; PI: R. Jackson), and DA011322 (PI: K. Mackie).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agren T, Engman J, Frick A, Björkstrand J, Larsson EM, Furmark T, Fredrikson M. Disruption of Reconsolidation Erases a Fear Memory Trace in the Human Amygdala. Science. 2012;337:1550–1552. doi: 10.1126/science.1223006. [DOI] [PubMed] [Google Scholar]
- Amano T, Duvarci S, Popa D, Paré D. The Fear Circuit Revisited: Contributions of the Basal Amygdala Nuclei to Conditioned Fear. The Journal of Neuroscience. 2011;31:15481–15489. doi: 10.1523/JNEUROSCI.3410-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano T, Unal CT, Pare D. Synaptic correlates of fear extinction in the amygdala. Nat Neurosci. 2010;13:489–494. doi: 10.1038/nn.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andero R, Heldt SA, Ye K, Liu X, Armario A, Ressler KJ. Effect of 7,8-dihydroxyflavone, a small-molecule TrkB agonist, on emotional learning. Am J Psychiatry. 2011;168:163–172. doi: 10.1176/appi.ajp.2010.10030326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci. 2007;8:45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- Carlson G, Wang Y, Alger BE. Endocannabinoids facilitate the induction of LTP in the hippocampus. Nat Neurosci. 2002;5:723–724. doi: 10.1038/nn879. [DOI] [PubMed] [Google Scholar]
- Chhatwal JP, Stanek-Rattiner L, Davis M, Ressler KJ. Amygdala BDNF signaling is required for consolidation but not encoding of extinction. Nat Neurosci. 2006;9:870–872. doi: 10.1038/nn1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran KA, Maren S. Hippocampal inactivation disrupts contextual retrieval of fear memory after extinction. J Neurosci. 2001;21:1720–1726. doi: 10.1523/JNEUROSCI.21-05-01720.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Mayford M, Gage FH. Selection of distinct populations of dentate granule cells in response to inputs as a mechanism for pattern separation in mice. eLife Sciences. 2013 doi: 10.7554/eLife.00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Luthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Falls WA, Miserendino MJ, Davis M. Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci. 1992;12:854–863. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner AR, Rowland DC, Hwang SY, Baumgaertel K, Roth BL, Kentros C, Mayford M. Generation of a Synthetic Memory Trace. Science. 2012;335:1513–1516. doi: 10.1126/science.1214985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis Aryn H, Hang Giao B, LaDow Eva S, Shoenfeld Liza R, Atallah Bassam V, Finkbeiner S, Kreitzer Anatol C. Rapid Target-Specific Remodeling of Fast-Spiking Inhibitory Circuits after Loss of Dopamine. Neuron. 2011;71:858–868. doi: 10.1016/j.neuron.2011.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottmann K, Mittmann T, Lessmann V. BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Exp Brain Res. 2009;199:203–234. doi: 10.1007/s00221-009-1994-z. [DOI] [PubMed] [Google Scholar]
- Graham BM, Langton JM, Richardson R. Pharmacological enhancement of fear reduction: preclinical models. British Journal of Pharmacology. 2011;164:1230–1247. doi: 10.1111/j.1476-5381.2010.01175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunduz-Cinar O, Macpherson KP, Cinar R, Gamble-George J, Sugden K, Williams B, Godlewski G, Ramikie TS, Gorka AX, Alapafuja SO, et al. Convergent translational evidence of a role for anandamide in amygdala-mediated fear extinction, threat processing and stress-reactivity. Mol Psychiatry. 2012 doi: 10.1038/mp.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkany T, Dobszay MB, Cayetanot F, Hartig W, Siegemund T, Aujard F, Mackie K. Redistribution of CB1 cannabinoid receptors during evolution of cholinergic basal forebrain territories and their cortical projection areas: a comparison between the gray mouse lemur (Microcebus murinus, primates) and rat. Neuroscience. 2005;135:595–609. doi: 10.1016/j.neuroscience.2005.06.043. [DOI] [PubMed] [Google Scholar]
- Hartley CA, Phelps EA. Changing fear: the neurocircuitry of emotion regulation. Neuropsychopharmacology. 2010;35:136–146. doi: 10.1038/npp.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitland I, Klumpers F, Oosting R, Evers D, Kenemans JL, Baas J. Failure to extinguish fear and genetic variability in the human cannabinoid receptor 1. Translational Psychiatry. 2012 doi: 10.1038/tp.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldt SA, Ressler KJ. Training-induced changes in the expression of GABAA-associated genes in the amygdala after the acquisition and extinction of Pavlovian fear. Eur J Neurosci. 2007;26:3631–3644. doi: 10.1111/j.1460-9568.2007.05970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldt SA, Mou L, Ressler KJ. In vivo knockdown of GAD67 in the amygdala disrupts fear extinction and the anxiolytic-like effect of diazepam in mice. Transl Psychiatry 2. 2012:e181. doi: 10.1038/tp.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Muller C, Luthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- Hobin JA, Goosens KA, Maren S. Context-dependent neuronal activity in the lateral amygdala represents fear memories after extinction. J Neurosci. 2003;23:8410–8416. doi: 10.1523/JNEUROSCI.23-23-08410.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EJ, McCord AE, Greenberg ME. A Biological Function for the Neuronal Activity-Dependent Component of Bdnf Transcription in the Development of Cortical Inhibition. Neuron. 2008;60:610–624. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Zhang Z, Zhang C, Wang X, Sakata K, Lu B, Sun QQ. A key mechanism underlying sensory experience-dependent maturation of neocortical GABAergic circuits in vivo. Proc Natl Acad Sci U S A. 2011;108:12131–12136. doi: 10.1073/pnas.1105296108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Rancz EA, Acsády L, Ledent C, Mackie K, Hájos N, Freund TF. Distribution of CB1 Cannabinoid Receptors in the Amygdala and their Role in the Control of GABAergic Transmission. The Journal of Neuroscience. 2001;21:9506–9518. doi: 10.1523/JNEUROSCI.21-23-09506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohara K, Yasuda H, Huang Y, Adachi N, Sohya K, Tsumoto T. A local reduction in cortical GABAergic synapses after a loss of endogenous brain-derived neurotrophic factor, as revealed by single-cell gene knock-out method. J Neurosci. 2007;27:7234–7244. doi: 10.1523/JNEUROSCI.1943-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapska E, Macias M, Mikosz M, Nowak A, Owczarek D, Wawrzyniak M, Pieprzyk M, Cymerman IA, Werka T, Sheng M, et al. Functional anatomy of neural circuits regulating fear and extinction. Proceedings of the National Academy of Sciences. 2012;109:17093–17098. doi: 10.1073/pnas.1202087109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann Dimitri M, Moreau Alexandre W, Bakiri Y, Nicholson E. Plasticity of Inhibition. Neuron. 2012;75:951–962. doi: 10.1016/j.neuron.2012.07.030. [DOI] [PubMed] [Google Scholar]
- Lai CS, Franke TF, Gan WB. Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature. 2012;483:87–91. doi: 10.1038/nature10792. [DOI] [PubMed] [Google Scholar]
- Likhtik E, Popa D, Apergis-Schoute J, Fidacaro GA, Pare D. Amygdala intercalated neurons are required for expression of fear extinction. Nature. 2008;454:642–645. doi: 10.1038/nature07167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ramirez S, Pang PT, Puryear CB, Govindarajan A, Deisseroth K, Tonegawa S. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nature. 2012;484:381–385. doi: 10.1038/nature11028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livneh U, Paz R. Aversive-Bias and Stage-Selectivity in Neurons of the Primate Amygdala during Acquisition, Extinction, and Overnight Retention. The Journal of Neuroscience. 2012;32:8598–8610. doi: 10.1523/JNEUROSCI.0323-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makkar SR, Zhang SQ, Cranney J. Behavioral and Neural Analysis of GABA in the Acquisition, Consolidation, Reconsolidation, and Extinction of Fear Memory. Neuropsychopharmacology. 2010;35:1625–1652. doi: 10.1038/npp.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S. Overtraining does not mitigate contextual fear conditioning deficits produced by neurotoxic lesions of the basolateral amygdala. J Neurosci. 1998;18:3088–3097. doi: 10.1523/JNEUROSCI.18-08-03088.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Numakawa T, Yokomaku D, Adachi N, Yamagishi S, Numakawa Y, Kunugi H, Taguchi T. Brain-derived neurotrophic factor-induced potentiation of glutamate and GABA release: Different dependency on signaling pathways and neuronal activity. Molecular and Cellular Neuroscience. 2006;31:70–84. doi: 10.1016/j.mcn.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Matsuo N, Reijmers L, Mayford M. Spine-type-specific recruitment of newly synthesized AMPA receptors with learning. Science. 2008;319:1104–1107. doi: 10.1126/science.1149967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald AJ. Projection neurons of the basolateral amygdala: a correlative Golgi and retrograde tract tracing study. Brain Res Bull. 1992;28:179–185. doi: 10.1016/0361-9230(92)90177-y. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Betette RL. Parvalbumin-containing neurons in the rat basolateral amygdala: morphology and co-localization of Calbindin-D(28k) Neuroscience. 2001;102:413–425. doi: 10.1016/s0306-4522(00)00481-4. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Miles R, Toth K, Gulyas AI, Hajos N, Freund TF. Differences between somatic and dendritic inhibition in the hippocampus. Neuron. 1996;16:815–823. doi: 10.1016/s0896-6273(00)80101-4. [DOI] [PubMed] [Google Scholar]
- Morgan MA, Romanski LM, LeDoux JE. Extinction of emotional learning: contribution of medial prefrontal cortex. Neurosci Lett. 1993;163:109–113. doi: 10.1016/0304-3940(93)90241-c. [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M. Mechanisms of fear extinction. Mol Psychiatry. 2007;12:120–150. doi: 10.1038/sj.mp.4001939. [DOI] [PubMed] [Google Scholar]
- Okuno H. Regulation and function of immediate-early genes in the brain: beyond neuronal activity markers. Neurosci Res. 2011;69:175–186. doi: 10.1016/j.neures.2010.12.007. [DOI] [PubMed] [Google Scholar]
- Orsini CA, Kim JH, Knapska E, Maren S. Hippocampal and prefrontal projections to the basal amygdala mediate contextual regulation of fear after extinction. J Neurosci. 2011;31:17269–17277. doi: 10.1523/JNEUROSCI.4095-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini CA, Maren S. Neural and cellular mechanisms of fear and extinction memory formation. Neurosci Biobehav Rev. 2012;36:1773–1802. doi: 10.1016/j.neubiorev.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinak CA, Angstadt M, Sripada CS, Abelson JL, Liberzon I, Milad MR, Phan KL. Cannabinoid facilitation of fear extinction memory recall in humans. Neuropharmacology. 2012;64:396–402. doi: 10.1016/j.neuropharm.2012.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijmers LG, Perkins BL, Matsuo N, Mayford M. Localization of a stable neural correlate of associative memory. Science. 2007;317:1230–1233. doi: 10.1126/science.1143839. [DOI] [PubMed] [Google Scholar]
- Repa JC, Muller J, Apergis J, Desrochers TM, Zhou Y, LeDoux JE. Two different lateral amygdala cell populations contribute to the initiation and storage of memory. Nat Neurosci. 2001;4:724–731. doi: 10.1038/89512. [DOI] [PubMed] [Google Scholar]
- Rudy JW, Huff NC, Matus-Amat P. Understanding contextual fear conditioning: insights from a two-process model. Neuroscience and biobehavioral reviews. 2004;28:675–685. doi: 10.1016/j.neubiorev.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Ruehle S, Remmers F, Romo-Parra H, Massa F, Wickert M, Wörtge S, Häring M, Kaiser N, Marsicano G, Pape HC, et al. Cannabinoid CB1 receptor in dorsal telencephalic glutamatergic neurons: distinctive sufficiency for hippocampus-dependent and amygdala-dependent synaptic and behavioral functions. J Neurosci. 2013;33:10264–10277. doi: 10.1523/JNEUROSCI.4171-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangha S, Narayanan RT, Bergado-Acosta JR, Stork O, Seidenbecher T, Pape HC. Deficiency of the 65 kDa isoform of glutamic acid decarboxylase impairs extinction of cued but not contextual fear memory. J Neurosci. 2009;29:15713–15720. doi: 10.1523/JNEUROSCI.2620-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangha S, Ilenseer J, Sosulina L, Lesting J, Pape HC. Differential regulation of glutamic acid decarboxylase gene expression after extinction of a recent memory vs. intermediate memory. Learn Mem. 2012;19:194–200. doi: 10.1101/lm.025874.112. [DOI] [PubMed] [Google Scholar]
- Soliman F, Glatt CE, Bath KG, Levita L, Jones RM, Pattwell SS, Jing D, Tottenham N, Amso D, Somerville L, et al. A Genetic Variant BDNF Polymorphism Alters Extinction Learning in Both Mouse and Human. Science. 2010;327:863–866. doi: 10.1126/science.1181886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayler KK, Lowry E, Tanaka K, Levy B, Reijmers L, Mayford M, Wiltgen BJ. Characterization of NMDAR-Independent Learning in the Hippocampus. Front Behav Neurosci. 2011;5:28. doi: 10.3389/fnbeh.2011.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayler KK, Tanaka KZ, Reijmers LG, Wiltgen BJ. Reactivation of Neural Ensembles during the Retrieval of Recent and Remote Memory. Curr Biol. 2013;23:99–106. doi: 10.1016/j.cub.2012.11.019. [DOI] [PubMed] [Google Scholar]
- Tronson NC, Schrick C, Guzman YF, Huh KH, Srivastava DP, Penzes P, Guedea AL, Gao C, Radulovic J. Segregated populations of hippocampal principal CA1 neurons mediating conditioning and extinction of contextual fear. The Journal of neuroscience. 2009;29:3387–3394. doi: 10.1523/JNEUROSCI.5619-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga C, Lee SY, Soltesz I. Target-selective GABAergic control of entorhinal cortex output. Nat Neurosci. 2010;13:822–824. doi: 10.1038/nn.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vianna MR, Szapiro G, McGaugh JL, Medina JH, Izquierdo I. Retrieval of memory for fear-motivated training initiates extinction requiring protein synthesis in the rat hippocampus. Proc Natl Acad Sci U S A. 2001;98:12251–12254. doi: 10.1073/pnas.211433298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Uchigashima M, Yamasaki M, Katona I, Yamazaki M, Sakimura K, Kano M, Yoshioka M, Watanabe M. Unique inhibitory synapse with particularly rich endocannabinoid signaling machinery on pyramidal neurons in basal amygdaloid nucleus. Proceedings of the National Academy of Sciences. 2011;108:3059–3064. doi: 10.1073/pnas.1012875108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.