

Abstract
Current options for influenza antiviral therapy are limited to the neuraminidase inhibitors, and knowledge that high levels of oseltamivir resistance have been seen amongst previously circulating H1N1 viruses increases the urgency to find new influenza therapeutics. To feed this pipeline, assays that are appropriate for use in high-throughput screens are being developed and are discussed in this review. Particular emphasis is placed on cell-based assays that capture both inhibitors of viral functions as well as the host functions that facilitate optimal influenza virus replication. Success in this area has been fueled by a greater understanding of the genome structure of influenza viruses and the ability to generate replication-competent recombinant viruses that carry a reporter gene, allowing for easy monitoring of viral infection in a high-throughput setting. This article forms part of a symposium in Antiviral Research on “Treatment of influenza: targeting the virus or the host.”
Keywords: Influenza virus, antiviral drug, high-throughput screen, cell-based assay, multi-cycle virus replication
1. Introduction
The use of high-throughput screening (HTS) technology for antiviral discovery is a fairly recent endeavor, first undertaken exclusively by the pharmaceutical industry and now also performed by academic scientists. The development of HTS has been driven by increasing advances in automation and the ability to handle large datasets. It has also expanded the types of target that can be explored and consequently assay development, particularly of cell-based assays, is a major part of all antiviral HTS campaigns.
As a small RNA virus, influenza virus encodes a limited number of proteins and thus there are only a few viral functions that are considered to be tractable drug targets by traditional standards. This essentially means that the target must have a function that is amenable to inhibition by a small molecule. The current two classes of approved antivirals for influenza target either the ion channel function of the M2 protein or the neuraminidase function of the NA protein. The neuraminidase inhibitors (NAI) were developed through the rational design of small molecules that mimic sialic acid and bind with high affinity to the active site of NA (Gubareva et al., 2000). The adamantanes are an interesting example of an antiviral whose approval preceded knowledge of the target (M2) or the function of the target as an ion channel. Moreover, the precise mechanism of action is still under debate following publication of structures showing different placement of the drug relative to M2 (Cady and Hong, 2008; Cady et al., 2010; Pielak and Chou, 2010; Stouffer et al., 2008). Other well-characterized viral functions that should be druggable are the RNA-dependent RNA polymerase activity of PB1 and the endonuclease function of PA. Apart from the fact that the description of PA endonuclease activity was only made in 2009 (Dias et al., 2009; Yuan et al., 2009), the major reason that these targets have not been explored fully is the inability to produce purified, full-length and active polymerase proteins, which severely limits the development of biochemical screening assays.
The examples above refer to viral functions that are considered to be validated targets, as it is known a priori that they are essential for influenza virus growth, and biochemical assays can (or could) be developed to screen for specific inhibitors of that function. Alternatively, one can cast a wider net by not requiring knowledge of the target or function upfront and instead using a phenotypic readout such as virus replication. This approach requires a cell-based assay and it is in this area that we have seen most development in the influenza virus HTS field. The advantages are: i) that it potentially allows one to capture all stages of the virus life-cycle in one assay, ii) it detects inhibitors of cellular functions that are required for virus replication, and iii) it may reveal unknown functions of viral proteins that are susceptible to small molecule inhibition. This review will focus on the new tools that have been developed for influenza antiviral drug discovery, with an emphasis on the use of fluorescent or luminescent reporters and the development of novel cell-based assays.
2. Suitable HTS assays for influenza antiviral discovery
The type of assay chosen for a screen depends on the question being asked and what tools are available. If the purpose is to identify inhibitors of as many different steps of the influenza virus life-cycle as possible, then an assay involving virus infection of cells must be used, preferably under conditions of multi-cycle replication (see 3.1). The readout for this type of assay can vary from antibody-based detection of viral proteins, to expression of reporter genes encoded by the virus (see 3.2), to indirect measurements such as cytopathic effect (see 3.4). Cell-based assays with reporter readouts can also be used to assess specific stages of the virus life-cycle (e.g. entry or replication phases, see 3.3), whereas if the purpose of the screen is to find inhibitors of a specific protein it is preferable to analyze this target in isolation using a biochemical assay that provides a readout of the protein function (see 4.1). In cases where a crystal structure of the protein target is available it may be possible to use an in silico approach where large libraries of small molecules are computationally docked onto the structure to identify those with potential binding properties (see 4.2). These predicted hits can then be validated in a functional assay, either biochemical or cell-based. The design of such assays obviously requires extensive prior knowledge of the functional properties of the protein target and of how this property affects virus replication, as well as the availability of appropriate tools e.g. purified protein. In many cases this information or the tools (or both) are lacking and increasingly antiviral screens are being conducted using cell-based assays without any knowledge of a specified target. Rather, the objective is to identify small molecules that have an overall phenotypic effect on virus replication and to then employ secondary assays to characterize the mechanism of action and identify the target protein. Increased accessibility to the required automation and to small molecule libraries for those outside the pharmaceutical industry has facilitated the design of new tools for use in cell-based virus assays for HTS.
A successful HTS assay must be robust, have an easy and quantifiable readout and be amenable to miniaturization and the use of robotic machinery. At a very minimum, the assay should function in 96-well format but in most cases further miniaturization to 384-well format is required for compatibility with library plates and pin tools. The smaller, 1536-well format is sometimes used, but it can be more challenging to maintain the assay quality in this format, especially with cell-based assays. The advantage of the smaller format is speed (more compounds screened per day) and reduced costs due to the lower volumes, which can be an important factor if an expensive reagent is required. The assay must be highly reproducible with a large window between the positive and negative controls. A statistical measurement of this is provided by the Z′-factor (Z′ = 1 − 3(STDpos + STDneg)/(MEANpos − MEANneg)) and a robust assay that is suitable for use in a screen should have a Z′-factor >0.5 (Zhang et al., 1999). To achieve this, the number of manipulations during the course of the assay should be minimized and it is a common rule that nothing is ever removed from the plate, only added, which helps to reduce variability. If available, reference compounds with known mechanisms of action should be examined in the assay to ascertain assay sensitivity and one should also be aware of possible false positives that may arise from the screen. Another factor to consider is DMSO compatibility as the library compounds will be delivered in 100% DMSO. In general the assay should be able to withstand a range of 0.1–1% DMSO. Finally, in an optimal assay the distribution of signal across the plate will be even with no evidence of edge effects (often due to evaporation from the outside wells) or drifting signal from left-to-right or top-to-bottom.
3. Cell-based assays for measuring influenza virus infection
3.1. Single versus multi-cycle viral replication assays
When designing an assay to monitor influenza virus replication it is important to understand the concept of single cycle vs. multi-cycle replication as this affects the stages of the virus life-cycle that can be captured by the assay. In a single cycle assay, 100% of cells are infected in the first round and thus this type of assay is performed with a high multiplicity of infection (MOI). If the assay readout is viral gene expression, this assay will capture all steps from virus attachment through to gene expression, but inhibitors that act at subsequent steps will not be detected because there are no more uninfected cells in the culture (Figure 1A, Table 1). Thus a neuramindase inhibitor such as oseltamivir, which targets virus release, will not have any effect in a single cycle assay. In contrast if the assay is performed with low MOI conditions this allows for multi-cycle virus growth. For example, an MOI of 0.05 will theoretically result in 5% of cells infected in the first round. In the presence of oseltamivir there will be no effect on viral gene expression in these cells but the drug will prevent the release of new virus particles and thus limit secondary rounds of infection which will be seen as an overall decrease in signal compared to the untreated control (Figure 1B, C). In this sense, oseltamivir is a particularly useful control as the ability of the assay to detect inhibition by oseltamivir implies that the assay is operating under multi-cycle conditions and that every step of the influenza virus life-cycle is being captured. Another tool that can be used in this regard is trypsin, which is usually included in post-infection media. Trypsin is required to cleave the hemagglutinin (HA) proteins of most influenza viruses (those that have a monobasic cleavage site) such that the newly-made virus particles are capable of entering cells in the second round of infection. In the absence of trypsin the infection will stall after the first round. An exception to this is the WSN influenza virus strain, which can replicate in tissue culture independently of trypsin (Appleyard and Maber, 1974). In fact, this can be an advantage as too much trypsin can obviously be detrimental to cell attachment and must be titrated carefully (Table 1). For assays that use imaging as a readout (i.e. high-content screens), conditions that maintain optimal cell morphology may be preferable.
Figure 1. Schematic overview of single- and multi-cycle replication assays.
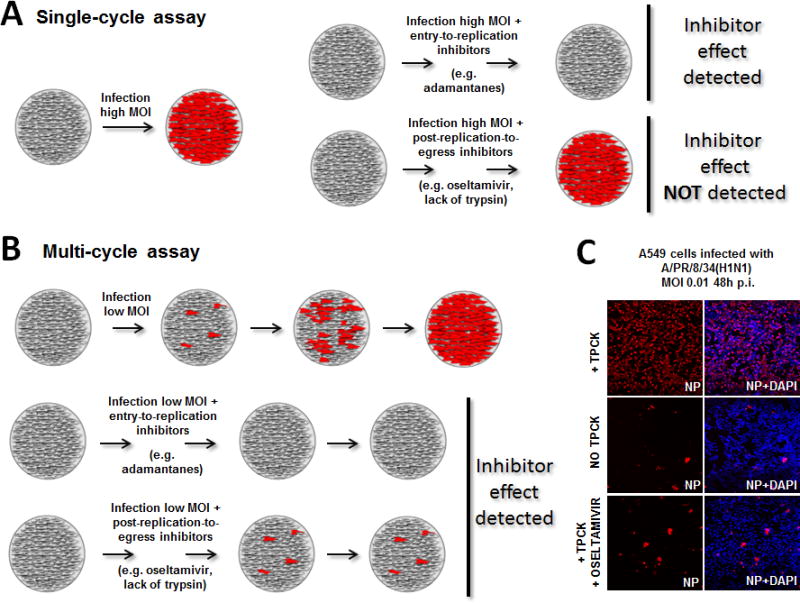
A) Single-cycle replication assay. Cell monolayers are infected at a high multiplicity of infection (MOI) and viral gene expression is detected in the majority of cells (left). Inhibitors acting on viral entry or replication stages are detected, while inhibitors affecting subsequent stages such as viral egress are not detected, as no uninfected cells remain in the culture. B) Multi-cycle replication assay. Cell monolayers are infected at a low MOI to allow newly generated virions to infect neighboring cells over several cycles, increasing the signal for viral gene expression over time. This type of assay allows detection of antiviral effects affecting any step of the virus life-cycle. C) A multi-cycle replication assay on A549 cell monolayers. Cells were infected with an influenza A/PR/8/34 reporter virus encoding a fusion NS1-mCherry protein (MOI of 0.01). In the presence of trypsin (TPCK) most of the culture has been infected by 48h post-infection. In the absence of exogenous trypsin newly generated viruses are fusion incompetent and the reporter signal is limited to the cells infected in the first round. A similar result can be observed when the cells are treated with the NA inhibitor oseltamivir carboxylate, which prevents the release of virions from infected cells.
Table 1.
Challenges encountered with influenza virus HTS assays
| HTS Assay | Challenges | |
|---|---|---|
| Cell-based assay (section 3) |
|
|
|
| ||
| Single vs. multi-cycle assays (3.1) |
|
|
|
| ||
| Virus reporter assays (3.2) | ||
| First generation (3.2.1) |
|
|
| Second generation (3.2.2) |
|
|
| Third generation (3.2.3) |
|
|
|
| ||
| Assays for specific viral stages (3.3) |
|
|
|
| ||
| Indirect readouts (3.4) |
|
|
|
| ||
| Cell-free assay (section 4) | ||
|
| ||
| Biochemical assays (4.1) |
|
|
|
| ||
| In silico assays (4.2) |
|
|
It is estimated that influenza virus has an eight-hour life-cycle, so three cycles of infection are possible within 24 hours. Therefore the MOI and the time of the assay readout are two important parameters that need to be optimized during assay development. The drawback with low MOI infections and longer times for assay readout is that they are often associated with increased error and therefore there can be a trade-off between the degree of multi-cycle replication and assay quality (Table 1). For the same reason there is a limit to how far a multi-cycle replication assay can be miniaturized. In 96-well format an MOI of 0.01 would require approximately 320 virus particles per well, whereas in 384-well or 1536-well this would require 80 or 20 virus particles, respectively to achieve the same MOI. The ability to transfer 20 virus particles into each well is prone to error, so one would either use a higher MOI in the 1536 well format or switch to a larger plate format to maintain a robust assay.
3.2. Development of reporter assays for direct measurements of influenza virus infection
The majority of cell-based high-throughput screens that assess influenza virus replication have used cytopathic effect (CPE) as the readout, which is essentially a measure of virus-induced cell death. This indirect measurement of virus growth will be discussed in more detail in section 3.4, but assays that provide a direct measurement of a viral product are easier to interpret and less likely to yield false positives or false negatives. The measurement of neuraminidase activity in the supernatant of infected cells has been used successfully as a screen assay and is amenable to multiple subtypes of influenza A virus as well as influenza B virus (Eichelberger et al., 2008; Gerritz et al., 2011). Immunostaining for viral antigens followed by high-content imaging is another direct approach that has been employed in RNAi screens for influenza, using antibodies either for HA or NP (Brass et al., 2009; Chin and Brass, 2013; Karlas et al., 2010; Prusty et al., 2011). The downside to this method is the expense of reagents and the extra time needed for the staining procedure. Where possible, the latest screening assays are trying to take advantage of the ease and speed of luminescent and fluorescent reporters and the next sections will focus on these developments in the influenza field.
3.2.1. First generation: influenza virus-activated reporter
This reporter assay is based on the established mini-genome system for influenza virus, which is used to monitor the transcription and replication steps of the viral life-cycle (Luytjes et al., 1989; Seong and Brownlee, 1992). A reporter gene such as firefly luciferase is cloned in the reverse orientation and complementary sense between the 5′ and 3′ non-coding regions of one of the viral RNA segments. This cassette is inserted into a plasmid between a human RNA pol I promoter and either a pol I terminator sequence or a hepatitis delta ribozyme sequence (Figure 2A). When transfected into human cells the cellular RNA pol I produces a transcript off the plasmid that is unmodified on both 5′ and 3′ ends, thus faithfully mimicking a viral RNA segment. The incoming viral polymerase machinery (PB1, PB2, PA and NP) delivered by influenza virus infection recognizes the non-coding regions on the reporter transcript and initiates both replication and mRNA transcription, resulting in expression of the reporter protein. Luciferase activity can be measured in cell lysates using commercial substrates and the resulting signal is detected and quantified on a luminometer. Fluorescent reporters, such as green fluorescent protein (GFP), are also amenable to easy detection by fluorescence microscopy or FACS analysis. Stable cell lines that carry the influenza reporter have been generated successfully and the assay has been shown to provide a rapid and sensitive measurement of virus replication that is capable of detecting the inhibitory effects of antiviral compounds and antibodies (Lutz et al., 2005). As such, the assay has been used in high-throughput screens for the purposes of identifying small molecules with antiviral activity (Hoffmann et al., 2011; Hoffmann et al., 2008; Zhang et al., 2012) and also for identifying critical host genes in RNAi screens (Karlas et al., 2010; Prusty et al., 2011; Shapira et al., 2009).
Figure 2. Three generations of HTS reporter assays for influenza virus infections.

A) Influenza virus-activated reporter. Cells are transfected with a plasmid that will generate a virus-like RNA, which encodes a reporter gene under control of the viral polymerase regulatory sequences. Infection with a helper, wild-type influenza virus delivers the viral polymerase, which drives expression of the reporter. As the reporter plasmid will not spread together with the helper virus, this system can only be used for single-cycle assays (unless using modified procedures as discussed in 3.2.1). B) Reporter-encoding influenza virus. A recombinant virus in which the HA coding region has been substituted with a reporter gene while maintaining the required packaging signals for the HA segment. This virus cannot undergo multi-cycle replication unless HA is supplied in trans (e.g. using an HA-expressing cell line that pseudotypes the viral particle, as depicted in red on the virus schematic). Therefore wild-type cells can support single-cycle replication of this virus, but a HA-complementing cell line is required for multi-cycle assays. C) Replication-competent reporter-encoding influenza virus. This virus contains an NS segment that has been engineered to express a reporter while maintaining expression of the viral products, NS1 and NEP. The wild-type splicing mechanism has been disrupted to allow for bicistronic expression of a chimeric, functional NS1-reporter fusion protein and a full length, wild-type NEP protein. This third generation of reporter influenza virus, with their full set of genes, is replication competent and can be used for multi-cycle assays without the need of for specialized cells.
When the reporter is activated by the incoming polymerase following virus infection it allows for the capture of all steps from the point of virus attachment through to viral gene (or reporter) expression. Due to the fact that a high MOI is usually necessary to activate the reporter in a reproducible manner, the HTS assay is run under single-cycle conditions and will therefore be biased for inhibitors of viral entry and replication steps (Table 1). However, in the case of the RNAi screens the assay has been used to quantify the production of virus from cells impacted by the RNAi, i.e. in lieu of an infectivity assay (Karlas et al., 2010; Prusty et al., 2011; Shapira et al., 2009). Briefly, target cells are treated with siRNAs and infected with influenza virus. The supernatants from these cells, containing progeny virus, are transferred to cells containing the influenza reporter gene and activation of the reporter is reflective of the amount (or infectivity) of the progeny virus produced in the presence of each siRNA. When used in this context, all steps of the influenza life-cycle are captured but the two-step process is obviously more cumbersome (Table 1).
3.2.2. Second generation: reporter-encoding influenza virus
The generation of an influenza virus that encodes a reporter gene is preferable to virus activation of an exogenous reporter as the assay is less sensitive to non-specific signals. For a long time this development was hindered by the segmented nature of the genome and the inability to stably incorporate additional open reading frames (ORFs) into the influenza virus genome. The realization that each viral segment possesses unique signals on its 5′ and 3′ ends that ensure specific packaging of that segment into progeny virions opened the door to the generation of reporter viruses (Marsh et al., 2007; Watanabe et al., 2003). By incorporating these packaging signals onto the ends of the reporter construct it is possible to retain the recombinant segment over multiple virus passages. However, one problem remains in that packaging of the reporter segment will compete with packaging of the viral segment that contains the same packaging sequences. And as all eight viral segments are necessary to make an infectious influenza virus, a strategy must be used that allows for complementation of the missing viral function. Such an approach has been used for two influenza RNAi screens. In Hao et al. (Hao et al., 2008) the authors replaced the NA coding region with that of Renilla luciferase and retained the NA packaging signals. They also replaced the HA ORF with that of the G-protein of vesicular stomatitis virus (VSV), while retaining the HA packaging signals. The presence of VSV-G allows the recombinant virus to infect Drosophila cells, which was their model system for the RNAi screen, and because this virus no longer relies on the HA-sialic acid interaction for attachment, it does not require the neuraminidase activity of NA. Clearly there are disadvantages to this system as the recombinant influenza virus now lacks both major glycoproteins, yet it is the first report of a luciferase-expressing virus that is suitable for use in HTS assays. König et al. used another variation of this approach for their RNAi screen assay (Konig et al., 2010). They generated a recombinant influenza virus where the HA ORF was replaced with Renilla luciferase but retained the HA packaging signals on the ends (Figure 2B). The virus must be grown in a cell-line that stably expresses HA to complement the loss of the viral segment encoding HA. In these cells, the virus can undergo multi-cycle replication and this assay has been used to screen small-molecule libraries for identification of influenza antiviral compounds (Bottini et al., 2012). If the screen assay involves infection of non-HA expressing cells, as was done for the RNAi screen (Konig et al., 2010), only single-cycle replication is possible (Figure 2B). Thus the need to engineer specialized cells in order to recapitulate the full influenza virus replication cycle is a restriction when using this recombinant virus (Table 1).
A related approach that requires complementation of two portions of GFP to reconstitute fluorescence has also been described and may be adaptable to a high-throughput screen assay. In this split GFP system, the PB2 ORF is fused to residues 215–230 of GFP, followed by the 5′ PB2 packaging signal (Avilov et al., 2012). This recombinant PB2-GFP11 virus grows to similar titers as wild-type virus. When it is used to infect cells expressing the remaining portion of GFP (residues 1–214), GFP fluorescence can be visualized and corresponds with the expression and localization of the PB2 protein, thereby providing a means to easily monitor viral gene expression.
3.2.3. Third generation: replication-competent reporter-encoding influenza virus
The hurdle in developing influenza reporter viruses has been the generation of a virus that does not require engineered cells to achieve multi-cycle growth and therefore more accurately mimics a wild-type influenza virus. Two recent approaches have succeeded in this regard. The first strategy involves the re-engineering of segment 8 of influenza A virus which normally expresses the NS1 protein from an unspliced transcript and the NEP protein from an overlapping spliced transcript. The GFP ORF was fused to the C-terminus of NS1 and, to maintain expression of NEP, the splice acceptor site was removed and the NEP ORF was duplicated after GFP (Manicassamy et al., 2010; Perez et al., 2013) (Figure 2C). The sequence encoding the 19-residue self-cleaving 2A peptide from porcine teschovirus-1 was inserted between the GFP and NEP ORFs in order to separate NEP from NS1-GFP. The resultant PR8/NS1-GFP virus shows slightly attenuated growth properties compared to wild-type PR8 virus under multi-cycle conditions (possibly due to the longer segment 8) but it stably incorporates the recombinant segment and the NS1-GFP fusion protein is fully functional (Manicassamy et al., 2010). Most importantly, it does not require any specialized cells for growth. Furthermore, the PR8/NS1-GFP virus retains the ability to infect mice and the GFP signal allows for easy visualization of virus infection in vivo (Manicassamy et al., 2010).
In the second strategy an influenza virus encoding Gaussia luciferase (GLuc) on its PB2 segment was generated (Heaton et al., 2013). The GLuc ORF was inserted after the PB2 ORF, which contained a mutated packaging signal on its 3′ end. A functional PB2 packaging signal was then repeated on the 3′ end of the GLuc ORF. The self-cleaving 2A peptide from foot and mouth disease virus was inserted between PB2 and GLuc so that they would be co-translationally cleaved into separate proteins. Normally GLuc is a secreted protein, which may not be ideal for certain applications, so the authors also added an ER retention signal (KDEL) at the C-terminus of GLuc. Cells infected with the PR8-GLuc virus express GLuc intracellularly and the signal is shown to increase over time with a low multiplicity infection, indicative of multi-cycle replication (Heaton et al., 2013). Thus, GLuc activity in cell lysates can be used as an easy readout of viral gene expression in any cell susceptible to influenza virus infection. As with the PR8/NS1-GFP virus, the PR8-GLuc virus can also be used to monitor influenza virus infection in an animal model (Heaton et al., 2013), with the advantage that whole animal bioluminescent imaging is more sensitive than fluorescent imaging and allows for non-invasive, real-time visualization of the course of virus infection in individual animals.
Through better knowledge and understanding of the constraints of the influenza virus genome, most importantly the identification of packaging signals, it has become possible to engineer replication-competent influenza viruses that stably incorporate a reporter gene. While there are still limitations to the size of reporter that can be accommodated (Table 1), the two examples above demonstrate feasibility and the numerous potential applications, including their use in cell-based HTS assays for antiviral discovery.
3.3. Cell-based HTS assays for specific stages of the influenza virus life-cycle
The influenza virus life-cycle can be divided into three distinct stages, each of which can be targeted by small molecule inhibitors: entry (attachment and fusion), replication (transcription, translation, and replication), and egress (virion formation, budding and release) (Figure 3). In addition to assays that cover either the first half of the life-cycle or the entire life-cycle (as described in 3.2) (Figure 3), there are several cell-based assays that focus on the specific stages of entry, replication and egress. Cell-based assays, while relatively time consuming and constrained by cytotoxicity (Table 1), do offer advantages in selecting for small molecules that are membrane permeable, stable, and functional in a cellular environment. Assays specific to certain stages have value as secondary assays for characterization of hits from primary screens (which target viral replication more broadly), however they may also be used as primary screen assays if inhibitors of a specific step are being sought (Table 1). For the latter, the advantage of a more focused screen is that downstream efforts to determine mechanism of action are more efficient and this ultimately decreases the time from hit identification to drug development phase.
Figure 3. Three stages of the influenza virus life-cycle and their detection by different reporter assays.

First-, second- and third-generation reporters can all detect viral entry and replication stages within the infected cell. The final stage, viral egress, can only be detected in the context of multi-cycle assays by using replication competent viruses; either second-generation reporter viruses used together with complementing cell lines or the third-generation of replication competent reporter viruses.
3.3.1. Viral Entry
Two excellent cell-based assays have been established for the study of influenza entry and are adaptable for high-throughput screening. The first is a replication deficient HIV-based pseudo-typed particle assay, which mimics influenza virus entry mediated by the HA glycoprotein. These particles are generated by transfection of producer cells with plasmids encoding: i) a replication deficient HIV provirus expressing a reporter gene, ii) HIV Gag-Pol, iii) influenza virus HA, and iv) influenza virus NA. The M2 protein can be included for maximal downstream transduction of target cells. Infection of target cells with these pseudo-typed particles occurs through HA-mediated attachment and fusion and results in the delivery of the provirus and replication machinery, which drives expression of the reporter gene. Various reporters such as GFP, beta-Gal (Ao et al., 2008), or luciferase (Wang et al., 2009) can be used and all are amenable to high-throughput screening. A second option available for screening entry inhibitors is an influenza virus-like particle (VLP) assay (Tscherne and Garcia-Sastre, 2011). Typically, expression of the HA, NA, and M1 matrix protein alone are sufficient for the budding of VLPs from producer cells. Because these VLPs do not contain any genetic material, the matrix protein was fused to a beta-lactamase (Bla) reporter protein as a readout with sufficient sensitivity for screening. Upon HA-mediated entry into the cytoplasm of a target cell, this M1-Bla reporter is released from the VLP into the cytoplasm. The beta-lactamase activity of the released M1-Bla can then be detected by the commercially available FRET substrate CCF2. Cleavage of the substrate by M1-Bla leads to a shift in emission fluorescence from 520nM to 447nM, easily detectable on a fluorometric plate reader. These two systems offer simple, robust assays for detecting inhibitors of the membrane binding and fusion steps of influenza virus entry in a high-throughput manner, with each offering distinct advantages. The HIV pseudo-typed system uses a less expensive and more sensitive readout (e.g. luciferase), while the M1-Bla VLPs do not rely on gene expression and so do not suffer from potential false positives arising from inhibition of the replication machinery.
3.3.2. Viral Replication
Viral replication is a critically important stage of the influenza life-cycle for which there are no currently approved antiviral drugs. The influenza mini-genome assay has been the gold standard for the study of viral replication for many years and has recently been employed in secondary screening (Su et al., 2010). The mini-genome assay functions through a luciferase reporter gene as described in section 3.2.1, but rather than having the polymerase machinery provided by viral infection, PB1, PB2, PA and NP expression plasmids are co-transfected to drive the reporter. Therefore, only the transcription/replication process is reconstituted in this mini-genome assay and an inhibitor of entry will not be detected, as is the case when viral infection drives the system. Additionally, a new cell line has been generated which stably expresses vRNA-GFP, PB1, PB2, PA, and NP to constitutively express a reporter vRNP complex, which removes the need for transfection (Ozawa et al., 2013). This should improve the efficiency and reproducibility of future screens using the influenza mini-genome system.
A more targeted approach for discovery of replication inhibitors would be to screen for small molecules that interrupt protein-protein interactions which are critical to viral transcription/replication (Ghanem et al., 2007). Two assays have been developed, based on similar split-reporter systems, for detection of inhibitors of specific viral protein-protein interactions. Firstly, Bimolecular Fluorescence Complementation (BiFC) has been established as a technique for detection of protein interactions within cells and has recently been used to characterize the PA-PB2 interaction in influenza virus infection (Hemerka et al., 2009). BiFC is based on the principle that, when split into two halves, fluorescent proteins (GFP or one of its derivatives) will lose their fluorescent properties. If these two halves are brought within close proximity via a set of conjugated interacting proteins, fluorescence is restored. A screen for inhibitors would then detect the disruption of this interaction and subsequent decrease in fluorescence. BiFC has the added benefit of being able to track the localization of an interaction within live cells, which can be visualized in a high-throughput manner using a high-content imager. Using similar principles, a new split-Renilla luciferase reporter system has been developed in the study of the PA-PB1 interaction (Deng et al., 2011). In this case, the paired interacting proteins reinstate luciferase activity and any inhibitors of the PA-PB1 interaction would be detected as a reduction in luciferase signal. Although the Renilla luciferase system cannot be used for localization studies as described for BiFC, luciferase does generally offer a more sensitive readout and therefore should produce a more robust screen in a high-throughput environment. It should also be noted that these split-reporter systems are not limited to the two viral protein interactions discussed here, and relevant viral-cellular or cellular-cellular protein interactions should be considered as viable screening targets.
3.3.3. Viral Egress
Although influenza virus egress is the primary target of the approved neuraminidase inhibitors, there are no current cell-based assays specifically designed to detect inhibitors of this process in a HTS format. Despite this, there are several assays available as candidates to be repurposed for HTS viral egress assays. A well-known requirement for virion formation is RNP export from the nucleus (where replication occurs) to the cytoplasm (reviewed in (Palese and Shaw, 2007)). Therefore a screen could be developed that would track the translocation of NP by visualizing a NP-GFP fusion construct (Ketha and Atreya, 2008) transfected in the presence or absence of viral infection. An inhibitor that causes aberrant localization could then be detected using a high-content imager. An alternative approach would be to track endogenous NP during infection by immunostaining with a fluorescent-labeled antibody at time points corresponding to the nuclear and cytoplasmic phases of NP localization and then quantifying the ratio of nuclear:cytoplasmic NP (Alamares-Sapuay et al., 2013).
Similarly, a less influenza specific assay is available utilizing a construct containing GFP fused to a generic Nuclear Localization Signal (NLS) and Nuclear Export Signal (NES) (Abkallo et al., 2011). Although this could be employed as described, one could envision altering this construct to contain the NLS of influenza virus NP and the NES of the Nuclear Export Protein (NEP) for greater specificity. Another candidate for a HTS to study late-stage viral egress would be a VLP budding assay. In this case the HA, NA, and M1 proteins can be expressed in human cells for efficient budding of VLPs containing the viral glycoproteins. Inhibitors such as oseltamivir have already been proven to block the release of a similar pseudo-typed particle system expressing HA and NA (Ao et al., 2008). HTS-appropriate readouts for such an assay include testing the supernatant with a commercial neuraminidase activity kit (NA-Star) (Eichelberger et al., 2008) or, if using Bla-M1 VLPs as discussed in the viral entry section, quantifying beta-lactamase activity.
3.4. Indirect measurements of influenza virus infection
Some cell-based assays rely on measuring the effects of virus replication on the cell, rather than direct detection of virus. The advantage to such assays is that wild-type virus can be used, so there is more flexibility in the choice of virus strain, however these readouts are more likely to yield false positives (hits unrelated to virus growth) and there may be restrictions on the cell types that are amenable to the readout (Table 1).
3.4.1 Cytopathic Effect
Cytopathic effect (CPE) can be used as a measure of influenza virus infection and was in fact considered the gold standard in influenza virus antiviral screens before the development of the direct methods described above. In its simplest form the cell monolayer is stained with crystal violet or neutral red (Sidwell and Smee, 2000) and lack of staining is indicative of virus-induced CPE or cell death. Some stains can more selectively differentiate between viable and non-viable cells through dye exclusion at the cell membrane (Propidium iodide, Evans blue, trypan blue or Ethidium homodimers) while others rely on enzymatic functions of viable cells (fluorescein diacetate, resazurin or MTT and XTT) (Kepp et al., 2011). Of these, MTT-based assays in particular have been used to screen for influenza virus inhibitors (Hsu et al., 2012; Kao et al., 2010). Many of these stains are particularly well suited to flow cytometry which can be scaled for high-throughput applications, making screening of beyond 100 000 samples per day possible (Kuckuck et al., 2001). Advances in computational image processing make it possible to track the fate of individual cells using fluorescence microscopy and common stains such as Hoechst 33342 and DAPI. In these systems, nuclear counts are used as a readout for cell viability and the method is easily amenable to high-throughput purposes (Kepp et al., 2011).
Cellular metabolism is also used as a measure of viability, with the most obvious example being the detection of cellular ATP using a luminescence-based assay. CellTiter-Glo® from Promega is a well-established commercial solution that has been widely used for influenza HTS assays (Maddry et al., 2011; Noah et al., 2007; Severson et al., 2008) and other cell biology applications. A visible measure of CPE is the rounding up and detachment of cells in culture and this too can be used to measure cell death. Microelectrodes that measure surface impedance across the well surface are used to track cell viability in real time. This method is simpler as it does not employ stains or dyes, however it is sensitive to changes in cell morphology and loses sensitivity when the cells become confluent. The technique allows real-time measurements to be taken, facilitating temporal tracking of cell death (Solly et al., 2004).
3.4.2. Interferon induction
A classic indirect readout for viral infection of a host cell is type-I(α/β) interferon (IFN) production. IFN-α/β is induced by all viruses and has broad and potent antiviral activity through induction of the PKR and RNase L pathways, as well as many other Interferon Stimulated Genes (ISGs) (reviewed in (Takaoka and Yanai, 2006)). These properties have led to recombinant IFN being used as an antiviral treatment for decades (reviewed in (George et al., 2012)). There have also been attempts to use synthetic IFN-inducers, such as poly I:C, as antiviral therapy (Field et al., 1971). More recently, small-molecule screens have been performed to identify compounds that stimulate either IFN production or ISG expression (Bedard et al., 2012; Martinez-Gil et al., 2012; Patel et al., 2012). For monitoring IFN production, the assay used a cell line stably transfected with a firefly luciferase reporter driven by the IFN-beta promoter, and hits were selected based on an increase in luciferase signal (Martinez-Gil et al., 2012). A general induction of IFN in this manner may lead to undesirable side effects similar to those experienced with exogenous IFN treatments. One strategy for avoiding this issue is to screen for compounds that specifically stimulate IFN only in infected cells to limit widespread IFN toxicity. Influenza virus infection typically does not induce a strong IFN-α/β response due to the actions of its NS1 protein (reviewed in (Ehrhardt et al., 2010)), therefore if an inhibitor increases IFN production in the presence of influenza virus it can be indicative of dysregulation of viral anti-IFN functions. Successful screens and subsequent secondary characterization from this laboratory have shown that induction of IFN by a small molecule in a virus dependent manner is possible and that NS1 does not need to be the direct target of the inhibitor to show this effect ((Ortigoza et al., 2012) and K. White, unpublished). It should also be noted that the IFN induction from these compounds may not be the primary cause of viral inhibition in vitro, but may augment the antiviral effect in an in vivo context.
4. Cell-free assays for viral proteins
4.1 Biochemical assays
Cell-free biochemical assays for drug discovery come with the benefits of shorter duration, the absence of toxicity issues, simpler experimental conditions (there is no need for sterile technique, for example) and amenability to HTS and automation. For these reasons cell-free systems have often been the starting point in drug-screening projects, however not all biological activities can be studied in this way.
4.1.1 Assays for enzymatic viral functions
Studying enzymatic function in a cell-free biochemical format is generally the simplest approach. The biochemical measurement of any particular viral enzymatic function often correlates with viral fitness. Influenza virus has eight single-stranded, negative sense RNA segments, which each encode one, two or three viral proteins. Of these viral proteins, only PB1, PA, NA and M2 have a confirmed enzymatic function. However, other influenza virus proteins, for example HA, PB2 and NS1, have specific binding activities that are also subject to inhibition.
When the current M2 inhibitors (amantadine and rimantadine) were discovered, their mode of action was unknown. Due to the emergence of resistance, the CDC no longer recommends these drugs for clinical use and thus the need for novel drugs in this class has grown and new M2 inhibitors are being sought. The patch-clamp technique is the gold standard for assessing ion channel activity, however the technique is labor intensive, slow and expensive and has thus had limited use in screening assays. Novel solid-supported membrane (SSM) technologies have simplified the technique, and have subsequently allowed for suitable scaling to a HTS platform for influenza A virus M2 inhibitors (Balannik et al., 2010); SSM was shown to be efficient and reliable when compared to conventional methods. It has been observed that expression of M2 in yeast is toxic, resulting in decreased growth kinetics. Furthermore, this effect is rescued by the addition of amantadine. These findings were exploited in an assay for novel M2 inhibitors, which uses the rescue of yeast growth by an M2 inhibitor as a readout for the efficacy of that inhibitor (Kurtz et al., 1995; Stevens et al., 2006b).
The existing neuraminidase inhibitors (NAIs) were developed using a combination of rational drug design and in silico optimization. Numerous assays for measuring the activity of NAIs exist and are amenable to HTS. There are chemiluminescent and fluorescence based assays available, which use either a 1,2-dioxetane derivative or methyl umbelliferone N-acetyl neuraminic acid (MUNANA) as substrates (Buxton et al., 2000; Potier et al., 1979), and several commercial systems are available (QFlu™ from Cellex and the NA-Fluor Influenza NA Assay and NA-Star® Influenza NA Inhibitor Resistance Detection Kits from Applied Biosystems). While these have typically been used to screen clinical samples for NAI resistance, a 96-well format has been developed for drug screening, which uses recombinant baculovirus-expressed NA (Kongkamnerd et al., 2012). Lawn based methods have potential for massive scale-up, as the platform makes use of continuous biological test matrices (such as agarose gel) onto which the enzyme under study (NA) is attached. The compound array is imposed through the action of compound delivery (for example, on coated beads), then a substrate gel (using the chemiluminescent or fluorescent technologies described) is overlaid and the resulting reaction is recorded using an imager (Marron and Jayawickreme, 2003).
The polymerase complex, consisting of PB1, PB2 and PA, has been studied extensively in order to elucidate the functions of the various subunits. The PB1 subunit is responsible for the RNA replicase functionality, PB2 has cap-binding activity and so functions to recruit the cellular mRNA primer used in viral transcription, and the endonuclease activity of PA serves to cleave the RNA primer from host mRNA. Furthermore, non-covalent interactions between these subunits are crucial for polymerase activity. Lastly, the binding of the polymerase complex with NP and the nascent vRNA segments plays a role in nuclear export, cellular trafficking and viral packaging of the segments. These many and varied functions present a suite of potential targets for drug development, however a lack of reagents (specifically the lack of full length recombinant proteins) and methods has been a barrier to progress.
An assay to measure the enzymatic activity of influenza polymerase using purified RNP complexes has been developed (Hooker et al., 2001), although examples of its use in drug discovery could not be found. Purified polymerase complexes expressed in insect cells using baculovirus expression vectors have been shown to synthesize short stretches of vRNA and cRNA in vitro (Newcomb et al., 2009) and this could be extended to a scintillation proximity assay (SPA) (Glickman et al., 2008; Sidwell and Smee, 2000) to examine replication initiation, which is suitable for high-throughput screening. An HTS-ready fluorescence polarization assay was developed using the purified recombinant N-terminal of PA to screen for endonuclease inhibitors, although this assay only detects inhibitors that prevent pre-mRNA from binding in the active site of PA, not cleavage of the pre-mRNA (Baughman et al., 2012; Kepp et al., 2011). Noble et al. (Noble et al., 2012) developed a FRET assay using full-length PA purified from insect cells, which they suggest will be more biologically relevant for the endonuclease function of PA. This system has been used to characterize the substrate requirements of PA endonuclease and has been proposed as a potential drug-screening tool. A radiometric cap-dependent endonuclease activity assay (Dias et al., 2009; Kuckuck et al., 2001) and an electrophoretic endonuclease assay (Iwai et al., 2010; Solly et al., 2004) have also been described, however these are more suitable for characterization rather than screening. At this point, there are no systems in place to probe cap recognition and binding or RNA extension.
There are crystal structures for fragments of the polymerase complex (reviewed in (Kepp et al., 2011; Resa-Infante et al., 2011; Ruigrok et al., 2010)), however it remains unclear how these subunits form the functional polymerase and interact with host proteins. Recent findings have elucidated more on the macro structure of the RNP complex involving the NP protein (Arranz et al., 2012; Balannik et al., 2010; Moeller et al., 2012). However more detailed structure and function information on the polymerase complex itself would significantly advance the search for polymerase inhibitors.
4.1.2 Assays for non-enzymatic viral functions
Several influenza virus proteins bear no enzymatic function but are still vital components of the viral life-cycle. Many of these act through protein-protein interactions, either with components of the virus or host machinery. HA, NS1 and NP are such proteins.
The HA protein on the viral surface is responsible for cell-surface receptor binding and (in a distinct step) viral fusion. Glycan microarrays have been used to define receptor specificity and may be employed to screen for binding inhibitors (Kurtz et al., 1995; Stevens et al., 2006a; Stevens et al., 2006b). The first use of fluorescence polarization to study HA came in 1992 and used a fluorescent α-sialoside that could bind to HA. Molecules that could out-compete α-sialoside for binding to HA at the glycan-binding site would change the fluorescence intensity or polarization of emitted light (Kongkamnerd et al., 2012; Weinhold and Knowles, 1992). However the affinity of HA for these sialosides is low and presented a hurdle in the application of these assays. In a recent advancement, novel fluorescent nanoparticles consisting of quantum dots with sialylated N-glycan chains present a feasible option for the screening of influenza attachment inhibitors in FP-type high-throughput assays (Marron and Jayawickreme, 2003; Okamatsu et al., 2013). Surface plasmon resonance (SPR) has been shown to be amenable to HTS (Piliarik et al., 2005) and in 1996, SPR was used to probe the interaction between HA and its receptor (Palese and Shaw, 2007; Takemoto et al., 1996), while more recently it has been used to screen for RNA aptamers that bind HA (Hooker et al., 2001; Misono and Kumar, 2005). The fusion process is mediated by a pH dependent conformational change in HA, and this process also serves as a potential drug target in the viral life-cycle. Calorimetry has been employed to monitor specific events of viral fusion (Nebel et al., 1995) however this assay does not seem amenable to HTS.
The NS1 protein has several functions, most crucial of which is modulation of the host innate immune response in which double stranded RNA (dsRNA) binding by NS1 plays a major role (Hale et al., 2008). The key residues involved in RNA binding (Donelan et al., 2003; Pan et al., 2011) have been identified, and several conserved sites that could serve as drug targets to block this effect have been identified (Darapaneni et al., 2009). The RNA-binding domain has been affinity-purified from bacteria for use in HTS. A radioisotope FlashPlate® assay has been developed to measure NS1-RNA binding, wherein the amount of radiolabelled RNA bound to recombinant NS1 (which is fixed to the plate) is measured (Maroto et al., 2008). A FP assay for NS1-RNA binding has also been used (You et al., 2011). Based on the finding that NS1 expression in yeast is toxic and that this effect can be mapped to the C-terminal NLS and the N-terminal RNA binding domain (Ward et al., 1994), the Engel group devised an assay to screen for NS1 inhibitors wherein yeast cells would recover their normal growth characteristic if NS1 activity was inhibited (Basu et al., 2009).
The NP protein is an important viral structural protein that binds non-specifically to viral RNA forming the viral ribonucleoprotein (RNP) complex (Portela and Digard, 2002). Photo-cross-linked chemical arrays have been used with some success to detect NP inhibitors. A library of small molecules is cross-linked to a glass slide and a solution of red fluorescent protein (RFP)-fused NP is used to probe for binding ability. Following washing, prospective hits are detected by fluorescent imaging and software analysis. One of the reports describing the antiviral activity of nucleozin used this assay (Hagiwara et al., 2010). This approach is not specific for NP and can be adapted to screen for inhibitors against various proteins so long as a readout is possible (i.e. a fluorescent fusion protein or perhaps immunofluorescence based detection) (Kanoh et al., 2006). In another assay to screen for NP inhibitors, the inherent fluorescence of the tryptophan residues in NP is quenched upon drug binding (Hung et al., 2012), which holds potential as a high-throughput screen for novel NP inhibitors.
4.1.3 Assays for host proteins involved in the influenza life-cycle
While there are no drugs currently targeting influenza virus host factors, one can presume that as these factors are elucidated and validated the need for assays to screen for small molecules to inhibit these interactions will emerge. TMPRSS, a serine protease, was found to be crucial for influenza replication (Bottcher et al., 2006) and Meyer et al. (Meyer et al., 2013) developed an in vitro assay to screen for inhibitors of recombinant TMPRSS using fluorogenic and chromogenic substrates. Several putative inhibitors were found, and some of these were confirmed in viral replication assays, so this study provides proof of concept that targeting host factors may lead to the discovery of novel small molecules that inhibit influenza virus.
4.2 In silico studies
In silico refers to scientific discoveries that are made using computer simulation instead of biological studies. As computer technology and processing power have increased in recent years, the scale at which these types of projects can be applied has also increased massively. A distinct advantage of in silico study is the lower cost, however hits still require biological validation before they are fully accepted. In silico experiments generally go hand-in-hand with advances in structural biology because without accurate structural information for the influenza virus proteins there could be no such study (Table 1).
In a drug development context, in silico studies can be used to i) screen a library for compounds using molecular docking techniques (virtual screening), ii) rationally design novel drugs and analogues of existing drugs based on knowledge of protein interactions or their active sites, and iii) investigate the mode of action of a drug or study drug-resistant mutants.
There is extensive structural knowledge for full-length HA, NA (reviewed in (Gamblin and Skehel, 2010)) and NP (Ng et al., 2008; Ye et al., 2006), while structural information for the remaining influenza proteins is more limited. The structure of the transmembrane region of the M2 protein is well understood, however the external region has not been resolved (Pielak and Chou, 2011). The drug-binding sites of M2 are in this transmembrane region, and provide models for the mechanisms of drug activity and resistance (Cross et al., 2012). Similarly, only structures of select domains of other influenza proteins are know: the N- and C-termini of PB1, the cap-binding site of PB2, the N- and C-termini of PA (reviewed in (Resa-Infante et al., 2011; Ruigrok et al., 2010)) and the N-terminal region of M1 (Arzt et al., 2001; Harris et al., 2001; Sha and Luo, 1997). There is a single full structure of NS1 (Bornholdt and Prasad, 2008), with many complementary structures of the N-terminal RNA-binding domain and the effector domain (reviewed by (Hale et al., 2008)). There are no structures available for PB1-F2 at present. Complementing some of these partial structures, 3D structures of the polymerase holoenzyme (Coloma et al., 2009; Martin-Benito et al., 2001) and the viral RNP have been generated by electron microscopy (Area et al., 2004; Moeller et al., 2012; Torreira et al., 2007). Virtual screening of a library of compounds has been carried out for HA (Li et al., 2011; Tang et al., 2010), NA (Durrant and McCammon, 2010), M2 (Li et al., 2011; Lin et al., 2011), NP (Fedichev et al., 2011; Shen et al., 2011) and the PB1-PA interaction (Muratore et al., 2012).
Rational drug design was perhaps most successfully employed in the case of NA inhibitors zanamivir and oseltamivir. It was known that sialic acid derivatives could bind and inhibit NA, although their efficacy was too low. When the structure of the NA protein became available, a rational approach was used to generate sialic acid derivatives, which resulted in the eventual development of these two drugs (reviewed in (von Itzstein, 2007)). This has been tried for the other influenza proteins too, such as disruption of the NP-NP interaction by a rationally designed peptide that binds at the protein-protein interface (Shen et al., 2011), or the rational design of small molecules against the RNA-binding groove of NP (Fedichev et al., 2011). Examples exist for other influenza proteins as well (reviewed in (Du et al., 2012)). The residues lining the pore of the M2 protein have been shown to be critical for pH dependent proton shuttling (Sharma et al., 2010), and thus these residues present an attractive target for rational drug design that has not yet been explored.
Structural information about protein-protein or protein-small molecule interactions may also lead to rational design strategies for inhibitors. A co-crystal structure of the inhibitor TBHQ and the HA protein found the binding site to be in a pre-fusion trimeric interface of HA, which stabilizes the pre-fusion conformation and thus inhibits viral fusion (Russell et al., 2008). This opens the door to rational design of other fusion inhibitors that may target this region. Another example is that of the PB1-PA interaction, in which partial structures of PA reveal a “dragon-like head” with a mouth into which the N-terminal of PB1 inserts (He et al., 2008; Obayashi et al., 2008). This structure was used as the basis for an in silico screen to identify small molecules that potentially disrupt the PB1-PA interaction (Muratore et al., 2012).
5. Summary
As described above, there have been significant advances in the development of tools for influenza virus HTS assays in recent years. In particular, the generation of recombinant, reporter-expressing viruses that are replication competent allows for the design of cell-based assays that capture all stages of the virus life-cycle. With these viruses there is greater flexibility in the choice of cells for the assay, so together this provides increased potential for identifying inhibitors of both viral and cellular functions that are critical for optimal virus replication. A screen performed with this type of assay must be supported by secondary assays (some of which may also be HTS-compatible) that assist in categorizing the primary screen hits. This may involve cell-based assays such as those described in section 3.3, which can be used to identify the stage of the virus life-cycle that is being affected by the inhibitor. Once the target is known, more specific assays such as those described in section 4 can be employed to investigate the precise mechanism of action and to explore options for optimizing the compound-target interaction.
It is also advisable to determine the breadth of antiviral activity across multiple influenza viruses (or even non-influenza viruses) at an early stage. It is not uncommon for small molecules to show specificity for the virus strain used in the primary screen, which is perhaps less interesting from a drug-development perspective, and can be due to a single amino acid difference. Currently, the majority of influenza screens tend to use common laboratory strains due to the availability of reagents and established reverse genetics systems for these viruses. The generation of reporter viruses in backgrounds covering different influenza virus subtypes or viruses more representative of recent human strains, such that broader spectrum inhibitors can be quickly identified, is definitely an area to explore in the future. Also, we may begin to perform assays that have more than one readout. For example, an assay with a direct readout of viral gene expression could be combined with an assay using an indirect readout (CPE or interferon induction). In this way we may be able to capture small molecules whose actions probe the relationship between virus replication and the cellular response. As more tools are developed, the options for exploring the influenza virus-host relationship under HTS settings will likely expand and we can expect this to support new discoveries in the basic research arena as well as drug discovery efforts.
SHAW Highlights.
There is a need for new influenza drugs.
New assays are being developed to support drug discovery efforts.
Recombinant influenza viruses that express reporters are one of the new tools available.
Cell-based assays for influenza virus can identify inhibitors of viral and host functions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abkallo HM, Kawano H, Watanabe K, Kobayashi N. A new cell-based reporter system for sensitive screening of nuclear export inhibitors. Drug Discov Ther. 2011;5:286–292. doi: 10.5582/ddt.2011.v5.6.286. [DOI] [PubMed] [Google Scholar]
- Alamares-Sapuay JG, Martinez-Gil L, Stertz S, Miller MS, Shaw ML, Palese P. Serum- and glucocorticoid-regulated kinase 1 is required for nuclear export of the ribonucleoprotein of influenza A virus. J Virol. 2013;87:6020–6026. doi: 10.1128/JVI.01258-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ao Z, Patel A, Tran K, He X, Fowke K, Coombs K, Kobasa D, Kobinger G, Yao X. Characterization of a trypsin-dependent avian influenza H5N1-pseudotyped HIV vector system for high throughput screening of inhibitory molecules. Antiviral Res. 2008;79:12–18. doi: 10.1016/j.antiviral.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleyard G, Maber HB. Plaque formation by influenza viruses in the presence of trypsin. The Journal of general virology. 1974;25:351–357. doi: 10.1099/0022-1317-25-3-351. [DOI] [PubMed] [Google Scholar]
- Area E, Martin-Benito J, Gastaminza P, Torreira E, Valpuesta JM, Carrascosa JL, Ortin J. 3D structure of the influenza virus polymerase complex: localization of subunit domains. Proc Natl Acad Sci U S A. 2004;101:308–313. doi: 10.1073/pnas.0307127101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arranz R, Coloma R, Chichon FJ, Conesa JJ, Carrascosa JL, Valpuesta JM, Ortin J, Martin-Benito J. The structure of native influenza virion ribonucleoproteins. Science. 2012;338:1634–1637. doi: 10.1126/science.1228172. [DOI] [PubMed] [Google Scholar]
- Arzt S, Baudin F, Barge A, Timmins P, Burmeister WP, Ruigrok RW. Combined results from solution studies on intact influenza virus M1 protein and from a new crystal form of its N-terminal domain show that M1 is an elongated monomer. Virology. 2001;279:439–446. doi: 10.1006/viro.2000.0727. [DOI] [PubMed] [Google Scholar]
- Avilov SV, Moisy D, Munier S, Schraidt O, Naffakh N, Cusack S. Replication-competent influenza A virus that encodes a split-green fluorescent protein-tagged PB2 polymerase subunit allows live-cell imaging of the virus life cycle. J Virol. 2012;86:1433–1448. doi: 10.1128/JVI.05820-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balannik V, Obrdlik P, Inayat S, Steensen C, Wang J, Rausch JM, DeGrado WF, Kelety B, Pinto LH. Solid-supported membrane technology for the investigation of the influenza A virus M2 channel activity. Pflugers Archiv: European journal of physiology. 2010;459:593–605. doi: 10.1007/s00424-009-0760-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu D, Walkiewicz MP, Frieman M, Baric RS, Auble DT, Engel DA. Novel influenza virus NS1 antagonists block replication and restore innate immune function. J Virol. 2009;83:1881–1891. doi: 10.1128/JVI.01805-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman BM, Jake Slavish P, DuBois RM, Boyd VA, White SW, Webb TR. Identification of influenza endonuclease inhibitors using a novel fluorescence polarization assay. ACS chemical biology. 2012;7:526–534. doi: 10.1021/cb200439z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard KM, Wang ML, Proll SC, Loo YM, Katze MG, Gale M, Jr, Iadonato SP. Isoflavone agonists of IRF-3 dependent signaling have antiviral activity against RNA viruses. J Virol. 2012;86:7334–7344. doi: 10.1128/JVI.06867-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornholdt ZA, Prasad BV. X-ray structure of NS1 from a highly pathogenic H5N1 influenza virus. Nature. 2008;456:985–988. doi: 10.1038/nature07444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottcher E, Matrosovich T, Beyerle M, Klenk HD, Garten W, Matrosovich M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol. 2006;80:9896–9898. doi: 10.1128/JVI.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottini A, De SK, Baaten BJ, Wu B, Barile E, Soonthornvacharin S, Stebbins JL, Bradley LM, Chanda SK, Pellecchia M. Identification of small molecules that interfere with H1N1 influenza A viral replication. ChemMedChem. 2012;7:2227–2235. doi: 10.1002/cmdc.201200453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxton RC, Edwards B, Juo RR, Voyta JC, Tisdale M, Bethell RC. Development of a sensitive chemiluminescent neuraminidase assay for the determination of influenza virus susceptibility to zanamivir. Analytical biochemistry. 2000;280:291–300. doi: 10.1006/abio.2000.4517. [DOI] [PubMed] [Google Scholar]
- Cady SD, Hong M. Amantadine-induced conformational and dynamical changes of the influenza M2 transmembrane proton channel. Proc Natl Acad Sci U S A. 2008;105:1483–1488. doi: 10.1073/pnas.0711500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady SD, Schmidt-Rohr K, Wang J, Soto CS, Degrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CR, Brass AL. A genome wide RNA interference screening method to identify host factors that modulate influenza A virus replication. Methods. 2013;59:217–224. doi: 10.1016/j.ymeth.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coloma R, Valpuesta JM, Arranz R, Carrascosa JL, Ortin J, Martin-Benito J. The structure of a biologically active influenza virus ribonucleoprotein complex. PLoS Pathog. 2009;5:e1000491. doi: 10.1371/journal.ppat.1000491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross TA, Dong H, Sharma M, Busath DD, Zhou HX. M2 protein from influenza A: from multiple structures to biophysical and functional insights. Current opinion in virology. 2012;2:128–133. doi: 10.1016/j.coviro.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darapaneni V, Prabhaker VK, Kukol A. Large-scale analysis of influenza A virus sequences reveals potential drug target sites of non-structural proteins. The Journal of general virology. 2009;90:2124–2133. doi: 10.1099/vir.0.011270-0. [DOI] [PubMed] [Google Scholar]
- Deng Q, Wang D, Xiang X, Gao X, Hardwidge PR, Kaushik RS, Wolff T, Chakravarty S, Li F. Application of a split luciferase complementation assay for the detection of viral protein-protein interactions. J Virol Methods. 2011;176:108–111. doi: 10.1016/j.jviromet.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias A, Bouvier D, Crepin T, McCarthy AA, Hart DJ, Baudin F, Cusack S, Ruigrok RW. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature. 2009;458:914–918. doi: 10.1038/nature07745. [DOI] [PubMed] [Google Scholar]
- Donelan NR, Basler CF, Garcia-Sastre A. A recombinant influenza A virus expressing an RNA-binding-defective NS1 protein induces high levels of beta interferon and is attenuated in mice. J Virol. 2003;77:13257–13266. doi: 10.1128/JVI.77.24.13257-13266.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Cross TA, Zhou HX. Recent progress in structure-based anti-influenza drug design. Drug discovery today. 2012;17:1111–1120. doi: 10.1016/j.drudis.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant JD, McCammon JA. Potential drug-like inhibitors of Group 1 influenza neuraminidase identified through computer-aided drug design. Computational biology and chemistry. 2010;34:97–105. doi: 10.1016/j.compbiolchem.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt C, Seyer R, Hrincius ER, Eierhoff T, Wolff T, Ludwig S. Interplay between influenza A virus and the innate immune signaling. Microbes Infect. 2010;12:81–87. doi: 10.1016/j.micinf.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Eichelberger MC, Hassantoufighi A, Wu M, Li M. Neuraminidase activity provides a practical read-out for a high throughput influenza antiviral screening assay. Virol J. 2008;5:109. doi: 10.1186/1743-422X-5-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedichev P, Timakhov R, Pyrkov T, Getmantsev E, Vinnik A. Structure-based drug design of a new chemical class of small molecules active against influenza A nucleoprotein in vitro and in vivo. PLoS currents. 2011;3:RRN1253. doi: 10.1371/currents.RRN1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field AK, Young CW, Krakoff IH, Tytell AA, Lampson GP, Nemes MM, Hilleman MR. Induction of interferon in human subjects by poly I:C. Proc Soc Exp Biol Med. 1971;136:1180–1186. doi: 10.3181/00379727-136-35454. [DOI] [PubMed] [Google Scholar]
- Gamblin SJ, Skehel JJ. Influenza hemagglutinin and neuraminidase membrane glycoproteins. The Journal of biological chemistry. 2010;285:28403–28409. doi: 10.1074/jbc.R110.129809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George PM, Badiger R, Alazawi W, Foster GR, Mitchell JA. Pharmacology and therapeutic potential of interferons. Pharmacol Ther. 2012;135:44–53. doi: 10.1016/j.pharmthera.2012.03.006. [DOI] [PubMed] [Google Scholar]
- Gerritz SW, Cianci C, Kim S, Pearce BC, Deminie C, Discotto L, McAuliffe B, Minassian BF, Shi S, Zhu S, Zhai W, Pendri A, Li G, Poss MA, Edavettal S, McDonnell PA, Lewis HA, Maskos K, Mortl M, Kiefersauer R, Steinbacher S, Baldwin ET, Metzler W, Bryson J, Healy MD, Philip T, Zoeckler M, Schartman R, Sinz M, Leyva-Grado VH, Hoffmann HH, Langley DR, Meanwell NA, Krystal M. Inhibition of influenza virus replication via small molecules that induce the formation of higher-order nucleoprotein oligomers. Proc Natl Acad Sci U S A. 2011;108:15366–15371. doi: 10.1073/pnas.1107906108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem A, Mayer D, Chase G, Tegge W, Frank R, Kochs G, Garcia-Sastre A, Schwemmle M. Peptide-mediated interference with influenza A virus polymerase. J Virol. 2007;81:7801–7804. doi: 10.1128/JVI.00724-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman JF, Schmid A, Ferrand S. Scintillation proximity assays in high-throughput screening. Assay and drug development technologies. 2008;6:433–455. doi: 10.1089/adt.2008.135. [DOI] [PubMed] [Google Scholar]
- Gubareva LV, Kaiser L, Hayden FG. Influenza virus neuraminidase inhibitors. Lancet. 2000;355:827–835. doi: 10.1016/S0140-6736(99)11433-8. [DOI] [PubMed] [Google Scholar]
- Hagiwara K, Kondoh Y, Ueda A, Yamada K, Goto H, Watanabe T, Nakata T, Osada H, Aida Y. Discovery of novel antiviral agents directed against the influenza A virus nucleoprotein using photo-cross-linked chemical arrays. Biochemical and biophysical research communications. 2010;394:721–727. doi: 10.1016/j.bbrc.2010.03.058. [DOI] [PubMed] [Google Scholar]
- Hale BG, Randall RE, Ortin J, Jackson D. The multifunctional NS1 protein of influenza A viruses. The Journal of general virology. 2008;89:2359–2376. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- Hao L, Sakurai A, Watanabe T, Sorensen E, Nidom CA, Newton MA, Ahlquist P, Kawaoka Y. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature. 2008;454:890–893. doi: 10.1038/nature07151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A, Forouhar F, Qiu S, Sha B, Luo M. The crystal structure of the influenza matrix protein M1 at neutral pH: M1-M1 protein interfaces can rotate in the oligomeric structures of M1. Virology. 2001;289:34–44. doi: 10.1006/viro.2001.1119. [DOI] [PubMed] [Google Scholar]
- He X, Zhou J, Bartlam M, Zhang R, Ma J, Lou Z, Li X, Li J, Joachimiak A, Zeng Z, Ge R, Rao Z, Liu Y. Crystal structure of the polymerase PA(C)-PB1(N) complex from an avian influenza H5N1 virus. Nature. 2008;454:1123–1126. doi: 10.1038/nature07120. [DOI] [PubMed] [Google Scholar]
- Heaton NS, Leyva-Grado VH, Tan GS, Eggink D, Hai R, Palese P. In vivo bioluminescent imaging of influenza A virus infection and characterization of novel cross-protective monoclonal antibodies. J Virol. 2013 doi: 10.1128/JVI.00969-13. Ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemerka JN, Wang D, Weng Y, Lu W, Kaushik RS, Jin J, Harmon AF, Li F. Detection and characterization of influenza A virus PA-PB2 interaction through a bimolecular fluorescence complementation assay. J Virol. 2009;83:3944–3955. doi: 10.1128/JVI.02300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann HH, Kunz A, Simon VA, Palese P, Shaw ML. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc Natl Acad Sci U S A. 2011;108:5777–5782. doi: 10.1073/pnas.1101143108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann HH, Palese P, Shaw ML. Modulation of influenza virus replication by alteration of sodium ion transport and protein kinase C activity. Antiviral Res. 2008;80:124–134. doi: 10.1016/j.antiviral.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooker L, Strong R, Adams R, Handa B, Merrett JH, Martin JA, Klumpp K. A sensitive, single-tube assay to measure the enzymatic activities of influenza RNA polymerase and other poly(A) polymerases: application to kinetic and inhibitor analysis. Nucleic acids research. 2001;29:2691–2698. doi: 10.1093/nar/29.13.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JT, Yeh JY, Lin TJ, Li ML, Wu MS, Hsieh CF, Chou YC, Tang WF, Lau KS, Hung HC, Fang MY, Ko S, Hsieh HP, Horng JT. Identification of BPR3P0128 as an inhibitor of cap-snatching activities of influenza virus. Antimicrob Agents Chemother. 2012;56:647–657. doi: 10.1128/AAC.00125-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung HC, Liu CL, Hsu JT, Horng JT, Fang MY, Wu SY, Ueng SH, Wang MY, Yaw CW, Hou MH. Development of an anti-influenza drug screening assay targeting nucleoproteins with tryptophan fluorescence quenching. Analytical chemistry. 2012;84:6391–6399. doi: 10.1021/ac2022426. [DOI] [PubMed] [Google Scholar]
- Iwai Y, Takahashi H, Hatakeyama D, Motoshima K, Ishikawa M, Sugita K, Hashimoto Y, Harada Y, Itamura S, Odagiri T, Tashiro M, Sei Y, Yamaguchi K, Kuzuhara T. Anti-influenza activity of phenethylphenylphthalimide analogs derived from thalidomide. Bioorganic & medicinal chemistry. 2010;18:5379–5390. doi: 10.1016/j.bmc.2010.05.035. [DOI] [PubMed] [Google Scholar]
- Kanoh N, Asami A, Kawatani M, Honda K, Kumashiro S, Takayama H, Simizu S, Amemiya T, Kondoh Y, Hatakeyama S, Tsuganezawa K, Utata R, Tanaka A, Yokoyama S, Tashiro H, Osada H. Photo-cross-linked small-molecule microarrays as chemical genomic tools for dissecting protein-ligand interactions. Chemistry, an Asian journal. 2006;1:789–797. doi: 10.1002/asia.200600208. [DOI] [PubMed] [Google Scholar]
- Kao RY, Yang D, Lau LS, Tsui WH, Hu L, Dai J, Chan MP, Chan CM, Wang P, Zheng BJ, Sun J, Huang JD, Madar J, Chen G, Chen H, Guan Y, Yuen KY. Identification of influenza A nucleoprotein as an antiviral target. Nat Biotechnol. 2010;28:600–605. doi: 10.1038/nbt.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlas A, Machuy N, Shin Y, Pleissner KP, Artarini A, Heuer D, Becker D, Khalil H, Ogilvie LA, Hess S, Maurer AP, Muller E, Wolff T, Rudel T, Meyer TF. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010;463:818–822. doi: 10.1038/nature08760. [DOI] [PubMed] [Google Scholar]
- Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G. Cell death assays for drug discovery. Nature reviews. Drug discovery. 2011;10:221–237. doi: 10.1038/nrd3373. [DOI] [PubMed] [Google Scholar]
- Ketha KM, Atreya CD. Application of bioinformatics-coupled experimental analysis reveals a new transport-competent nuclear localization signal in the nucleoprotein of influenza A virus strain. BMC Cell Biol. 2008;9:22. doi: 10.1186/1471-2121-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kongkamnerd J, Milani A, Cattoli G, Terregino C, Capua I, Beneduce L, Gallotta A, Pengo P, Fassina G, Miertus S, De-Eknamkul W. A screening assay for neuraminidase inhibitors using neuraminidases N1 and N3 from a baculovirus expression system. Journal of enzyme inhibition and medicinal chemistry. 2012;27:5–11. doi: 10.3109/14756366.2011.568415. [DOI] [PubMed] [Google Scholar]
- Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Alamares JG, Tscherne DM, Ortigoza MB, Liang Y, Gao Q, Andrews SE, Bandyopadhyay S, De Jesus P, Tu BP, Pache L, Shih C, Orth A, Bonamy G, Miraglia L, Ideker T, Garcia-Sastre A, Young JA, Palese P, Shaw ML, Chanda SK. Human host factors required for influenza virus replication. Nature. 2010;463:813–817. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuckuck FW, Edwards BS, Sklar LA. High throughput flow cytometry. Cytometry. 2001;44:83–90. [PubMed] [Google Scholar]
- Kurtz S, Luo G, Hahnenberger KM, Brooks C, Gecha O, Ingalls K, Numata K, Krystal M. Growth impairment resulting from expression of influenza virus M2 protein in Saccharomyces cerevisiae: identification of a novel inhibitor of influenza virus. Antimicrob Agents Chemother. 1995;39:2204–2209. doi: 10.1128/aac.39.10.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XB, Wang SQ, Xu WR, Wang RL, Chou KC. Novel inhibitor design for hemagglutinin against H1N1 influenza virus by core hopping method. PLoS One. 2011;6:e28111. doi: 10.1371/journal.pone.0028111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Chang TT, Sun MF, Chen HY, Tsai FJ, Chang KL, Fisher M, Chen CY. Potent inhibitor design against H1N1 swine influenza: structure-based and molecular dynamics analysis for M2 inhibitors from traditional Chinese medicine database. Journal of biomolecular structure & dynamics. 2011;28:471–482. doi: 10.1080/07391102.2011.10508589. [DOI] [PubMed] [Google Scholar]
- Lutz A, Dyall J, Olivo PD, Pekosz A. Virus-inducible reporter genes as a tool for detecting and quantifying influenza A virus replication. J Virol Methods. 2005;126:13–20. doi: 10.1016/j.jviromet.2005.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luytjes W, Krystal M, Enami M, Parvin JD, Palese P. Amplification, expression, and packaging of foreign gene by influenza virus. Cell. 1989;59:1107–1113. doi: 10.1016/0092-8674(89)90766-6. [DOI] [PubMed] [Google Scholar]
- Maddry JA, Chen X, Jonsson CB, Ananthan S, Hobrath J, Smee DF, Noah JW, Noah D, Xu X, Jia F, Maddox C, Sosa MI, White EL, Severson WE. Discovery of novel benzoquinazolinones and thiazoloimidazoles, inhibitors of influenza H5N1 and H1N1 viruses, from a cell-based high-throughput screen. J Biomol Screen. 2011;16:73–81. doi: 10.1177/1087057110384613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, Garcia-Sastre A. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Natl Acad Sci U S A. 2010;107:11531–11536. doi: 10.1073/pnas.0914994107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroto M, Fernandez Y, Ortin J, Pelaez F, Cabello MA. Development of an HTS assay for the search of anti-influenza agents targeting the interaction of viral RNA with the NS1 protein. J Biomol Screen. 2008;13:581–590. doi: 10.1177/1087057108318754. [DOI] [PubMed] [Google Scholar]
- Marron BE, Jayawickreme CK. Going to the well no more: lawn format assays for ultra-high-throughput screening. Current opinion in chemical biology. 2003;7:395–401. doi: 10.1016/s1367-5931(03)00064-4. [DOI] [PubMed] [Google Scholar]
- Marsh GA, Hatami R, Palese P. Specific residues of the influenza A virus hemagglutinin viral RNA are important for efficient packaging into budding virions. J Virol. 2007;81:9727–9736. doi: 10.1128/JVI.01144-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Benito J, Area E, Ortega J, Llorca O, Valpuesta JM, Carrascosa JL, Ortin J. Three-dimensional reconstruction of a recombinant influenza virus ribonucleoprotein particle. EMBO reports. 2001;2:313–317. doi: 10.1093/embo-reports/kve063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Gil L, Ayllon J, Ortigoza MB, Garcia-Sastre A, Shaw ML, Palese P. Identification of small molecules with type I interferon inducing properties by high-throughput screening. PLoS One. 2012;7:e49049. doi: 10.1371/journal.pone.0049049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D, Sielaff F, Hammami M, Bottcher-Friebertshauser E, Garten W, Steinmetzer T. Identification of the first synthetic inhibitors of the type II transmembrane serine protease TMPRSS2 suitable for inhibition of influenza virus activation. The Biochemical journal. 2013;452:331–343. doi: 10.1042/BJ20130101. [DOI] [PubMed] [Google Scholar]
- Misono TS, Kumar PK. Selection of RNA aptamers against human influenza virus hemagglutinin using surface plasmon resonance. Analytical biochemistry. 2005;342:312–317. doi: 10.1016/j.ab.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Moeller A, Kirchdoerfer RN, Potter CS, Carragher B, Wilson IA. Organization of the influenza virus replication machinery. Science. 2012;338:1631–1634. doi: 10.1126/science.1227270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratore G, Goracci L, Mercorelli B, Foeglein A, Digard P, Cruciani G, Palu G, Loregian A. Small molecule inhibitors of influenza A and B viruses that act by disrupting subunit interactions of the viral polymerase. Proc Natl Acad Sci U S A. 2012;109:6247–6252. doi: 10.1073/pnas.1119817109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebel S, Bartoldus I, Stegmann T. Calorimetric detection of influenza virus induced membrane fusion. Biochemistry. 1995;34:5705–5711. doi: 10.1021/bi00017a001. [DOI] [PubMed] [Google Scholar]
- Newcomb LL, Kuo RL, Ye Q, Jiang Y, Tao YJ, Krug RM. Interaction of the influenza a virus nucleocapsid protein with the viral RNA polymerase potentiates unprimed viral RNA replication. J Virol. 2009;83:29–36. doi: 10.1128/JVI.02293-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng AK, Zhang H, Tan K, Li Z, Liu JH, Chan PK, Li SM, Chan WY, Au SW, Joachimiak A, Walz T, Wang JH, Shaw PC. Structure of the influenza virus A H5N1 nucleoprotein: implications for RNA binding, oligomerization, and vaccine design. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2008;22:3638–3647. doi: 10.1096/fj.08-112110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noah JW, Severson W, Noah DL, Rasmussen L, White EL, Jonsson CB. A cell-based luminescence assay is effective for high-throughput screening of potential influenza antivirals. Antiviral Res. 2007;73:50–59. doi: 10.1016/j.antiviral.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Noble E, Cox A, Deval J, Kim B. Endonuclease substrate selectivity characterized with full-length PA of influenza A virus polymerase. Virology. 2012;433:27–34. doi: 10.1016/j.virol.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi E, Yoshida H, Kawai F, Shibayama N, Kawaguchi A, Nagata K, Tame JR, Park SY. The structural basis for an essential subunit interaction in influenza virus RNA polymerase. Nature. 2008;454:1127–1131. doi: 10.1038/nature07225. [DOI] [PubMed] [Google Scholar]
- Okamatsu M, Feng F, Ohyanagi T, Nagahori N, Someya K, Sakoda Y, Miura N, Nishimura S, Kida H. Fluorescence polarization-based assay using N-glycan-conjugated quantum dots for screening in hemagglutinin blockers for influenza A viruses. J Virol Methods. 2013;187:390–394. doi: 10.1016/j.jviromet.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Ortigoza MB, Dibben O, Maamary J, Martinez-Gil L, Leyva-Grado VH, Abreu P, Jr, Ayllon J, Palese P, Shaw ML. A novel small molecule inhibitor of influenza A viruses that targets polymerase function and indirectly induces interferon. PLoS Pathog. 2012;8:e1002668. doi: 10.1371/journal.ppat.1002668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa M, Shimojima M, Goto H, Watanabe S, Hatta Y, Kiso M, Furuta Y, Horimoto T, Peters NR, Hoffmann FM, Kawaoka Y. A cell-based screening system for influenza A viral RNA transcription/replication inhibitors. Sci Rep. 2013;3:1106. doi: 10.1038/srep01106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palese P, Shaw ML. Orthomyxoviridae: The viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 1647–1689. [Google Scholar]
- Pan D, Sun H, Shen Y, Liu H, Yao X. Exploring the molecular basis of dsRNA recognition by NS1 protein of influenza A virus using molecular dynamics simulation and free energy calculation. Antiviral Res. 2011;92:424–433. doi: 10.1016/j.antiviral.2011.09.009. [DOI] [PubMed] [Google Scholar]
- Patel DA, Patel AC, Nolan WC, Zhang Y, Holtzman MJ. High throughput screening for small molecule enhancers of the interferon signaling pathway to drive next-generation antiviral drug discovery. PLoS One. 2012;7:e36594. doi: 10.1371/journal.pone.0036594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez JT, Garcia-Sastre A, Manicassamy B. Insertion of a GFP Reporter Gene in Influenza Virus. Curr Protoc Microbiol. 2013;Chapter 15(Unit15G):14. doi: 10.1002/9780471729259.mc15g04s29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielak RM, Chou JJ. Flu channel drug resistance: a tale of two sites. Protein Cell. 2010;1:246–258. doi: 10.1007/s13238-010-0025-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielak RM, Chou JJ. Influenza M2 proton channels. Biochimica et biophysica acta. 2011;1808:522–529. doi: 10.1016/j.bbamem.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piliarik M, Vaisocherova H, Homola J. A new surface plasmon resonance sensor for high-throughput screening applications. Biosensors & bioelectronics. 2005;20:2104–2110. doi: 10.1016/j.bios.2004.09.025. [DOI] [PubMed] [Google Scholar]
- Portela A, Digard P. The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. The Journal of general virology. 2002;83:723–734. doi: 10.1099/0022-1317-83-4-723. [DOI] [PubMed] [Google Scholar]
- Potier M, Mameli L, Belisle M, Dallaire L, Melancon SB. Fluorometric assay of neuraminidase with a sodium (4-methylumbelliferyl-alpha-D-N-acetylneuraminate) substrate. Analytical biochemistry. 1979;94:287–296. doi: 10.1016/0003-2697(79)90362-2. [DOI] [PubMed] [Google Scholar]
- Prusty BK, Karlas A, Meyer TF, Rudel T. Genome-wide RNAi screen for viral replication in mammalian cell culture. Methods Mol Biol. 2011;721:383–395. doi: 10.1007/978-1-61779-037-9_24. [DOI] [PubMed] [Google Scholar]
- Resa-Infante P, Jorba N, Coloma R, Ortin J. The influenza virus RNA synthesis machine: advances in its structure and function. RNA biology. 2011;8:207–215. doi: 10.4161/rna.8.2.14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruigrok RW, Crepin T, Hart DJ, Cusack S. Towards an atomic resolution understanding of the influenza virus replication machinery. Current opinion in structural biology. 2010;20:104–113. doi: 10.1016/j.sbi.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Russell RJ, Kerry PS, Stevens DJ, Steinhauer DA, Martin SR, Gamblin SJ, Skehel JJ. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc Natl Acad Sci U S A. 2008;105:17736–17741. doi: 10.1073/pnas.0807142105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong BL, Brownlee GG. A new method for reconstituting influenza polymerase and RNA in vitro: a study of the promoter elements for cRNA and vRNA synthesis in vitro and viral rescue in vivo. Virology. 1992;186:247–260. doi: 10.1016/0042-6822(92)90079-5. [DOI] [PubMed] [Google Scholar]
- Severson WE, McDowell M, Ananthan S, Chung DH, Rasmussen L, Sosa MI, White EL, Noah J, Jonsson CB. High-throughput screening of a 100,000-compound library for inhibitors of influenza A virus (H3N2) J Biomol Screen. 2008;13:879–887. doi: 10.1177/1087057108323123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha B, Luo M. Structure of a bifunctional membrane-RNA binding protein, influenza virus matrix protein M1. Nature structural biology. 1997;4:239–244. doi: 10.1038/nsb0397-239. [DOI] [PubMed] [Google Scholar]
- Shapira SD, Gat-Viks I, Shum BO, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE, Hill DE, Regev A, Hacohen N. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139:1255–1267. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath DD, Zhou HX, Cross TA. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science. 2010;330:509–512. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen YF, Chen YH, Chu SY, Lin MI, Hsu HT, Wu PY, Wu CJ, Liu HW, Lin FY, Lin G, Hsu PH, Yang AS, Cheng YS, Wu YT, Wong CH, Tsai MD. E339...R416 salt bridge of nucleoprotein as a feasible target for influenza virus inhibitors. Proc Natl Acad Sci U S A. 2011;108:16515–16520. doi: 10.1073/pnas.1113107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidwell RW, Smee DF. In vitro and in vivo assay systems for study of influenza virus inhibitors. Antiviral Res. 2000;48:1–16. doi: 10.1016/s0166-3542(00)00125-x. [DOI] [PubMed] [Google Scholar]
- Solly K, Wang X, Xu X, Strulovici B, Zheng W. Application of real-time cell electronic sensing (RT-CES) technology to cell-based assays. Assay and drug development technologies. 2004;2:363–372. doi: 10.1089/adt.2004.2.363. [DOI] [PubMed] [Google Scholar]
- Stevens J, Blixt O, Glaser L, Taubenberger JK, Palese P, Paulson JC, Wilson IA. Glycan microarray analysis of the hemagglutinins from modern and pandemic influenza viruses reveals different receptor specificities. Journal of molecular biology. 2006a;355:1143–1155. doi: 10.1016/j.jmb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Stevens J, Blixt O, Paulson JC, Wilson IA. Glycan microarray technologies: tools to survey host specificity of influenza viruses. Nature reviews. Microbiology. 2006b;4:857–864. doi: 10.1038/nrmicro1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CY, Cheng TJ, Lin MI, Wang SY, Huang WI, Lin-Chu SY, Chen YH, Wu CY, Lai MM, Cheng WC, Wu YT, Tsai MD, Cheng YS, Wong CH. High-throughput identification of compounds targeting influenza RNA-dependent RNA polymerase activity. Proc Natl Acad Sci U S A. 2010;107:19151–19156. doi: 10.1073/pnas.1013592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Yanai H. Interferon signalling network in innate defence. Cell Microbiol. 2006;8:907–922. doi: 10.1111/j.1462-5822.2006.00716.x. [DOI] [PubMed] [Google Scholar]
- Takemoto DK, Skehel JJ, Wiley DC. A surface plasmon resonance assay for the binding of influenza virus hemagglutinin to its sialic acid receptor. Virology. 1996;217:452–458. doi: 10.1006/viro.1996.0139. [DOI] [PubMed] [Google Scholar]
- Tang G, Qiu Z, Lin X, Li W, Zhu L, Li S, Li H, Wang L, Chen L, Wu JZ, Yang W. Discovery of novel 1-phenyl-cycloalkane carbamides as potent and selective influenza fusion inhibitors. Bioorganic & medicinal chemistry letters. 2010;20:3507–3510. doi: 10.1016/j.bmcl.2010.04.141. [DOI] [PubMed] [Google Scholar]
- Torreira E, Schoehn G, Fernandez Y, Jorba N, Ruigrok RW, Cusack S, Ortin J, Llorca O. Three-dimensional model for the isolated recombinant influenza virus polymerase heterotrimer. Nucleic acids research. 2007;35:3774–3783. doi: 10.1093/nar/gkm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tscherne DM, Garcia-Sastre A. An enzymatic assay for detection of viral entry. Curr Protoc Cell Biol. 2011;Chapter 26(Unit 26):12. doi: 10.1002/0471143030.cb2612s51. [DOI] [PubMed] [Google Scholar]
- von Itzstein M. The war against influenza: discovery and development of sialidase inhibitors. Nature reviews. Drug discovery. 2007;6:967–974. doi: 10.1038/nrd2400. [DOI] [PubMed] [Google Scholar]
- Wang SY, Su CY, Lin M, Huang SY, Huang WI, Wang CC, Wu YT, Cheng TJ, Yu HM, Ren CT, Wu CY, Wong CH, Cheng YS. HA-pseudotyped retroviral vectors for influenza antagonist screening. J Biomol Screen. 2009;14:294–302. doi: 10.1177/1087057108330786. [DOI] [PubMed] [Google Scholar]
- Ward AC, Azad AA, Macreadie IG. Expression and characterisation of the influenza A virus non-structural protein NS1 in yeast. Archives of virology. 1994;138:299–314. doi: 10.1007/BF01379133. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Watanabe S, Noda T, Fujii Y, Kawaoka Y. Exploitation of nucleic acid packaging signals to generate a novel influenza virus-based vector stably expressing two foreign genes. J Virol. 2003;77:10575–10583. doi: 10.1128/JVI.77.19.10575-10583.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhold EG, Knowles JR. Design and evaluation of a tightly binding fluorescent ligand for influenza A hemagglutinin. J Am Chem Soc. 1992;114:9270–9275. [Google Scholar]
- Ye Q, Krug RM, Tao YJ. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature. 2006;444:1078–1082. doi: 10.1038/nature05379. [DOI] [PubMed] [Google Scholar]
- You L, Cho EJ, Leavitt J, Ma LC, Montelione GT, Anslyn EV, Krug RM, Ellington A, Robertus JD. Synthesis and evaluation of quinoxaline derivatives as potential influenza NS1A protein inhibitors. Bioorganic & medicinal chemistry letters. 2011;21:3007–3011. doi: 10.1016/j.bmcl.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Bartlam M, Lou Z, Chen S, Zhou J, He X, Lv Z, Ge R, Li X, Deng T, Fodor E, Rao Z, Liu Y. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature. 2009;458:909–913. doi: 10.1038/nature07720. [DOI] [PubMed] [Google Scholar]
- Zhang J, Liu T, Tong X, Li G, Yan J, Ye X. Identification of novel virus inhibitors by influenza A virus specific reporter cell based screening. Antiviral Res. 2012;93:48–54. doi: 10.1016/j.antiviral.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]