

Abstract
Background & Aims
Intestinal epithelial cells aid in mucosal defense by providing a physical barrier against entry of pathogenic bacteria and secreting anti-microbial peptides (AMPs). Autophagy is an important component of immune homeostasis. However, little is known about its role in specific cell types during bacterial infection in vivo. We investigated the role of autophagy in the response of intestinal epithelial and antigen-presenting cells to Salmonella infection in mice.
Methods
We generated mice deficient in Atg16l1 in epithelial cells (Atg16l1f/f x Villin-cre) or CD11c+ cells (Atg16l1f/f x CD11c-cre); these mice were used to assess cell type-specific, anti-bacterial autophagy. All responses were compared to Atg16l1f/f mice (controls). Mice were infected with Salmonella enterica serovar Typhimurium; cecum and small intestine tissues were collected for immunofluorescence, histology, and quantitative reverse transcription PCR analyses of cytokines and AMPs. Modulators of autophagy were screened to evaluate their effects on anti-bacterial responses in human epithelial cells.
Results
Autophagy was induced in small intestine and cecum following infection with S Typhimurium, and required Atg16l1. S Typhimurium colocalized with microtubule-associated protein 1 light chain 3 beta (Map1lc3b or LC3) in the intestinal epithelium of control mice but not in Atg16l1f/f x Villin-cre mice. Atg16l1f/f x Villin-cre mice also had fewer Paneth cells and abnormal granule morphology, leading to reduced expression of AMP. Consistent with these defective immune responses, Atg16l1f/f x Villin-cre mice had increased inflammation and systemic translocation of bacteria compared with control mice. In contrast, we observed few differences between Atg16l1f/f x CD11c-cre and control mice. Trifluoperazine promoted autophagy and bacterial clearance in HeLa cells; these effects were reduced upon knockdown of ATG16L1.
Conclusions
Atg16l1 regulates autophagy in intestinal epithelial cells and is required for bacterial clearance. It is also required to prevent systemic infection of mice with enteric bacteria.
Keywords: mouse model, autophagy, intestinal barrier, mucosa
Introduction
Autophagy is a cellular degradation system for numerous substrates, including long-lived proteins, mitochondria, peroxisomes, and bacteria. The autophagy pathway has been increasingly recognized as an important mechanism for regulating homeostasis in response to cellular and physiological processes, including anti-microbial defense1. The core machinery that mediates bulk autophagic degradation of cytosolic content is highly conserved from yeast to mammals, but specialized functions, cargos, and mechanisms of substrate recognition are rapidly emerging. In many instances, ubiquitinated cargo is recognized by autophagy receptor proteins, such as p62, NBR1, OPTN, or NDP52, which directly interact with ATG8 homologs to facilitate delivery to autophagosomes. This cargo can include intracellular bacteria, suggesting that impaired autophagy could promote bacterial persistence and hyper-inflammation2, 3.
In vivo studies in lower eukaryotes have demonstrated that autophagy is essential for counteracting bacterial infections, including Salmonella enterica serovar Typhimurium, Mycobacterium tuberculosis, Legionella pneumophila, and Listeria monocytogenes4–7. Salmonella is a gram-negative bacteria and is one of the most comprehensively studied pathogens in in vitro antibacterial autophagy assays. Following cell invasion, S Typhimurium localizes to Salmonella-containing vacuoles (SCVs), compartments which serve as protective niches for replication. However, when SCVs are damaged, S Typhimurium can be modified with ubiquitin or diacylglycerol, leading to autophagic encapsulation of the cytosol-exposed bacteria8–10. The host E3 ubiquitin ligase LRSAM1 is actively involved in ubiquitin tagging of cytosolic S Typhimurium11, while the bacterial type 3 secretion effector protein SseL actively deubiquitinates ubiquitin-coated Salmonella12.
Genome-wide association studies have also implicated autophagy-related proteins in human disease, including single-nucleotide polymorphisms (SNPs) in ATG16L1 and IRGM that are associated with risk for inflammatory bowel disease13, 14. ATG16L1 loss-of-function mouse models exhibit several features of dysfunctional immune homeostasis, including impaired Paneth cell function15, 16 and hyper-secretion of IL-1β and IL-18 from macrophages upon engagement by lipopolysaccharide17. These observations are consistent with a role for autophagy in bacterial survival mechanisms, pro-inflammatory signaling, and initiation of adaptive immune responses. However, the consequences of these immunomodulatory effects have yet to be addressed in an in vivo infection model.
To gain insight into the cell-specific role of autophagy in the context of infectious disease in vivo, we established an experimental model to allow investigation of autophagy deficiency in distinct mucosal cell types in bacterial infection. We generated Atg16l1 conditional knockout mice in which Atg16l1 was deleted specifically in intestinal epithelial cells or CD11c+ mononuclear cells, providing a system to distinguish between the roles of autophagy in intestinal epithelium and in CD11c+ mononuclear cells. Using S Typhimurium as a model pathogen, we demonstrate that autophagy is upregulated in the gastrointestinal tract upon infection and provide evidence that S Typhimurium invades intestinal epithelial cells and is engulfed by autophagosomes in vivo. Furthermore, we demonstrate that epithelial Atg16l1 expression is required for microtubule-associated protein 1 light chain 3 beta (Map1lc3b or LC3) colocalization with S Typhimurium. As a result, mice lacking Atg16l1 in epithelial cells are hyper-susceptible to S Typhimurium infection, whereas mice lacking Atg16l1 in CD11c+ immune cells show phenotypes similar to wild-type mice. Moreover, mice lacking epithelial Atg16l1 have fewer Paneth cells and have abnormal granules, leading to impaired AMP production and potentially exacerbating S Typhimurium-induced inflammation. Thus, we conclude that epithelial Atg16l1 expression and LC3 induction are required for host defense against intestinal Salmonella infection in vivo.
Materials and Methods
Western blotting
Cell and tissue extracts were prepared using standard lysis buffer. Following SDS-PAGE, proteins were visualized using ATG16L1, p62, actin, and LC3B antibodies. See Supplementary Materials and Methods for details.
Epithelial cell enrichment
Ileal, cecal and colonic epithelial cells were isolated as previously described18. See Supplementary Materials and Methods for details.
CD11c+ mononuclear cell isolation
Single cell suspensions from spleen and MLN were enriched for CD11c+ mononuclear cells using MACS magnetic beads per manufacturer’s protocol (Miltenyi Biotec), and CD11chigh cells were FACS-sorted using a MoFlo cell sorter. See Supplementary Materials and Methods for details.
Salmonella infection
Bacterial growth and infection were performed as previously described, with slight modification19. The S Typhimurium strain used in this study expresses a dsRed fluorescent protein and is ampicillin resistant20. See Supplementary Materials and Methods for details.
Ex vivo imaging
GFP-LC3 mice were infected with S Typhimurium and after 24 hours were sacrificed for imaging. See Supplementary Materials and Methods for details.
Confocal microscopy and immunofluorescence
Sections were stained with anti-LC3B, anti-Salmonella CSA-1, and DAPI, and digital images were acquired using an A1R confocal microscope (Nikon). See Supplementary Materials and Methods for details.
Bacterial translocation
Tissues were removed aseptically and CFUs were determined by plating dilutions on LB plates supplemented with ampicillin.
Autophagy flux assay
HeLa cells were transfected with siRNA and after 60 hours treated with DMSO, TFP, and/or E64d and Pepstatin A as indicated. After 8 hours, cells were harvested for Western blotting. Similar compound treatments were performed on Atg16l1+/+ and Atg16l1−/− MEFs. See Supplementary Materials and Methods for details.
Bacterial-autophagy compound assay
HeLa cells stably expressing EGFP-LC3 were treated with TFP and incubated at 37°C. After 3 hours, S Typhimurium infections were performed as previously described14, 21. After 20 minutes of infection, cells were incubated with 50 μg/ml gentamycin sulfate and TFP at variable concentrations for 40 minutes. Cells were fixed, stained with Hoechst (Invitrogen), imaged on a Molecular Devices ImageXpress Micro, and analyzed using CellProfiler22. See Supplementary Materials and Methods for details.
Bioluminescent bacterial clearance assay
HeLa cells were infected with S Typhimurium expressing the Photorhabdus luminescens lux operon (Xen26) at 37°C for 30 minutes. After bacteria were removed, 20 μg/mL gentamicin was added, followed by 10 μM compound and incubated at 37°C. Plates were analyzed at indicated time points. See Supplementary Materials and Methods for details.
Results
Atg16l1 conditional knockout mice exhibit defective autophagy in specific cell compartments
Epithelial and mucosal immune cells are crucial for maintaining intestinal homeostasis and protecting against pathogens. Autophagy acts as a mechanism of pathogen clearance and has been implicated in intestinal immunity, but little is known about how this degradative process regulates mucosal immune responses to pathogens in vivo. To address this question, we investigated autophagy in the context of bacterial infection in mice with conditional deletion of Atg16l1, an essential autophagy protein that has also been associated with increased risk of Crohn’s disease in humans14. Viable pups homozygous for the Atg16l1 targeted allele were not observed, consistent with the knock-out-first design of the targeted allele23 (Figure 1A) and the previously described neonatal lethality of mice with a homozygous loss of essential autophagy genes including Atg16l124. However, unlike previous reports in which the coiled-coil domain of Atg16l1 was deleted17, the homozygous KO mouse embryonic fibroblasts (MEFs) isolated from E13 embryos entirely lack Atg16l1 expression. In addition, homozygous KO MEFs lack LC3-I to LC3-II conversion and accumulate p62, suggestive of impaired autophagy (Figure 1B). We next generated mice with conditional targeting of the Atg16l1 locus (Atg16l1f/f), which we bred with cre-expressing mice to enable analysis of cell-specific functions of Atg16l1 in epithelial (Atg16l1f/f x Villin-cre) and CD11c+ (Atg16l1f/f x CD11c-cre) cell compartments (Figure 1A, Supplementary Figure 1).
Figure 1. Systemic and cell-specific deletion of Atg16l1 expression.
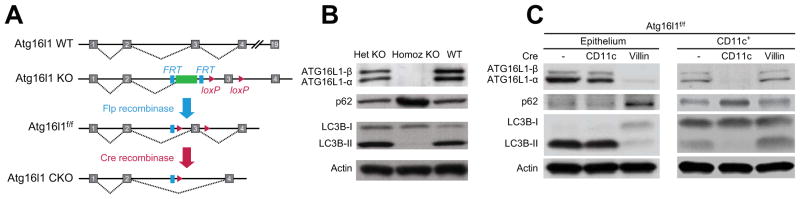
(A) Schematic of Atg16l1 gene targeting as designed by the International Knockout Mouse Consortium. (B) Western blot of MEFs isolated at E13 showing expression of Atg16l1, p62, LC3B, and actin. (C) Atg16l1 conditional knockout (CKO) mice were generated by crossing Atg16l1f/f with Villin-cre or CD11c-cre mice. Western blots demonstrate expression levels of Atg16l1, p62, LC3B, and actin in intestinal epithelial cells (left) and CD11c+ cells (right).
We validated our targeting strategy by Western blotting isolated epithelial and CD11c+ mononuclear cells (Figure 1C and Supplementary Figure 1D). As expected, wild-type epithelial cells expressed high levels of LC3B-II and barely-detectable levels of the substrate p62, an indicator of autophagic flux. In contrast, epithelial cells from Atg16l1f/f x Villin-cre mice did not express Atg16l1, showed increased p62 expression, and had impaired LC3B-I to LC3B-II conversion (Figure 1C). Epithelial cells from Atg16l1f/f x CD11c-cre mice expressed Atg16l1, p62, and LC3B-I/II at levels comparable to wild-type. In contrast, CD11c+ mononuclear cells from Atg16l1f/f x CD11c-cre mice lacked Atg16l1 expression and showed altered LC3B-I to LC3B-II conversion, consistent with our cell-targeting strategy (Figure 1C). CD11c+ mononuclear cells from Atg16l1f/f x Villin-cre mice showed wild-type levels of Atg16l1, p62, and LC3B-I/II. These data indicate that our conditional knockout models appropriately ablate Atg16l1 expression and show cell-specific disruptions of autophagy.
Autophagy is induced following in vivo S Typhimurium infection
To develop a model of in vivo antibacterial autophagy, we used a protocol in which mice were pre-treated with streptomycin before bacterial infection to improve intestinal colonization19. Mice were then infected with dsRed-tagged S Typhimurium SL1344, a strain known to transverse epithelium and become disseminated to mesenteric lymph nodes (MLN), spleen, and liver where bacteria replicate in host cell SCVs14, 19. To determine whether this pathogen induces autophagy in vivo and could serve as a model to study autoinflammatory and infectious diseases, we examined subcellular localization and expression levels of LC3 in intestinal tissues using confocal microscopy. Analysis of frozen tissue sections revealed that LC3 expression was upregulated in intestinal epithelial and mesenchymal cell compartments of the small intestine (Figure 2A) and cecal patches (Figure 2B) in wild-type mice after S Typhimurium infection. Supporting these data, isolated epithelial cells from ileum and cecum showed up-regulation of LC3B mRNA following S Typhimurium infection (Figure 2C). These data indicate that the epithelium responds to S Typhimurium infection with enhanced expression of autophagy regulators.
Figure 2. LC3 expression is induced following in vivo S Typhimurium infection.

(A–B) Mice were infected with S Typhimurium and tissues were harvested after 24 hours. Terminal ileum (A) and cecum (B) were immunostained with DAPI (blue) and anti-LC3 (green) and imaged by confocal microscopy. Uninfected wild-type tissues are shown on the left. (C) LC3B mRNA levels from isolated wild-type ileal and cecal epithelial cells were determined by qPCR. Numbers shown are relative to streptomycin-only-treated wild-type mice. Bars indicate mean plus S.D. *P ≤ .01. Data are representative of at least 3 independent experiments (n = 9).
Interestingly, Atg16l1f/f x Villin-cre mice demonstrated reduced immunoreactivity for anti-LC3 in the epithelium of the small intestine (Figure 2A) and cecal patches (Figure 2B) after S Typhimurium infection. In contrast, S Typhimurium infection of Atg16l1f/f x CD11c-cre mice resulted in upregulated LC3 in the epithelium, but not in the follicle (Figure 2A and B). Together these data indicate that LC3 expression is induced in an Atg16l1-dependent manner in the intestinal epithelium during host defense against S Typhimurium.
Intracellular S Typhimurium colocalization with LC3 in intestinal epithelial cells requires Atg16l1
We next determined whether LC3 is recruited to S Typhimurium escaping from the SCVs of epithelial cells. Using confocal microscopy of dsRed-tagged Salmonella in GFP-LC3 mice, we found that GFP-LC3 was recruited to autophagosomes containing single Salmonella during invasion of cecal patch follicle-associated epithelium (FAE) (Figure 3A). We also observed recruitment of GFP-LC3 to intracellular compartments containing multiple Salmonella within epithelial cells of the FAE (Figure 3A). In addition, immunostaining with anti-LC3 and anti-Salmonella antibodies demonstrated colocalization of intracellular S Typhimurium and LC3 aggregates in wild-type mice in both the FAE and follicle immune cells (Figure 3B). As predicted by our finding that Atg16l1f/f x Villin-cre mice did not show increased LC3 punctae after infection, suggestive of impaired autophagy (Figure 2A and B), we found that S Typhimurium did not colocalize with LC3 in the FAE of these mice (Figure 3C). However, S Typhimurium present in the follicle of the cecal patch of Atg16l1f/f x Villin-cre mice did colocalize with LC3, suggesting that autophagy is intact outside of the epithelium (Figure 3C). These results suggest that intestinal epithelial cells lacking Atg16l1 are unable to process this pathogen by autophagosomal capture.
Figure 3. S Typhimurium colocalization with LC3 in intestinal epithelial cells requires Atg16l1.

(A) GFP-LC3 mice were infected with dsRed-expressing S Typhimurium and cecal tissues were imaged in vivo by confocal microscopy after 24 hours. Colocalization (arrow) of S Typhimurium (red) and GFP-LC3 (green) is shown. LC3-positive vacuoles contained single (upper panels) or multiple (lower panels) Salmonella. (B–C) Wild-type (B) and Atg16l1f/fx Villin-cre (C) mice were infected with S Typhimurium and cecal tissues were harvested after 24 hours. Cecal patches were stained with DAPI (blue), anti-Salmonella (red), and anti-LC3 (green). Colocalization of LC3 and Salmonella shown in yellow (arrows). Upper panels show FAE; bottom panels focus deep within the cecal patch/follicle. Data are representative of at least 3 independent experiments (n ≥ 4).
Mice lacking epithelial Atg16l1 have Paneth cell abnormalities
Paneth cells deficient in essential autophagy proteins exhibit abnormalities in their granules, which are important for secretion of AMPs from this specialized epithelial cell compartment15, 25. As shown in Figure 4A, we found increased LC3 punctae in Paneth cells in wild-type mice following S Typhimurium infection, suggesting that autophagy is triggered here upon infection. Examining the functional effects of Salmonella infection in the terminal ileum, we next confirmed that wild-type mice showed upregulation of AMPs (α-Defensin, Defcr-rs-10, Lysozyme, RegIIIγ, and Cryptidin 1) after S Typhimurium infection (Figure 4B). Consistent with previous reports, expression of Lgr5, an intestinal stem cell gene, remained unaltered (Figure 4B)26. Expression of AMPs in the terminal ileum of Atg16l1f/f x Villin-cre mice were not enhanced after S Typhimurium infection, suggesting that this arm of antibacterial defense is compromised in these mice (Figure 4B). As expected Atg16l1f/f x CD11c-cre mice showed upregulation of these genes to levels comparable to wild-type mice after infection.
Figure 4. Mice lacking epithelial Atg16l1 have Paneth cell abnormalities.

(A) Terminal ileum from wild-type mice was stained with DAPI (blue), anti-Salmonella (red), and anti-LC3 (green) 24 hours after mock or S Typhimurium infection. (B) Gene expression in terminal ileum 72 hours after streptomycin treatment (Strep only) or after 24 hours streptomycin and 48 hours S Typhimurium infection (Infected). Numbers shown are relative to streptomycin-only treated wild-type mice. Bars indicate mean plus S.D. * P ≤ .01. (C) Terminal ileum of uninfected (top) and S Typhimurium-infected (bottom) mice were immunostained with DAPI (blue) and Lysozyme (red) 48 hours post-infection. Dotted lines denote the laser capture zone; arrows indicate presence of lysozyme-positive cells outside this zone. Scale bars = 100 μm. (D) Lysozyme-positive cells per crypt within the laser capture zone were quantified 48 hours post-infection (n=2 mice per group; 25 crypts per mouse). (E) AMP expression by qPCR from laser capture zone shown in C. Numbers shown are relative to streptomycin-only treated wild-type mice. (F) Light microscopy images of Periodic acid-Schiff-stained sections of terminal ileum from uninfected (top) and S Typhimurium-infected (bottom) mice (60X magnification) 48 hours post-infection. Scale bars = 50 μm. Data are representative of at least 2 independent experiments (n ≥ 2).
To address the differences observed in AMP mRNA expression between wild-type and Atg16l1f/f x Villin-cre mice, we next stained two AMPs, Lysozyme and RegIIIγ. Using immunofluorescence (Lysozyme) and immunohistochemistry (RegIIIγ), we observed less AMP staining in Atg16l1f/f x Villin-cre mice compared to wild-type, confirming our qPCR results (Figure 4C and Supplementary Figure 3A–B). We also quantified the number of ileal Paneth cells present with and without S Typhimurium infection via Lysozyme staining (Figure 4C–D). Interestingly, Atg16l1f/f x Villin-cre mice possessed fewer Paneth cells per crypt in the ileum before and after infection, compared to wild-type mice (Figure 4D). A similar trend was observed in jejunal crypts stained with Cryptidin 4 after infection (Supplementary Figure 3C). Ileal crypts were isolated using laser capture microdissection and RNA was harvested for qPCR (Figure 4C, dotted lines). Similar to the data in Figure 4B showing AMP expression in total ileal tissue, significantly lower levels of Lysozyme and Cryptidin mRNA were expressed in isolated crypts from Atg16l1f/f x Villin-cre mice compared to wild-type mice (Figure 4E). Isolated crypt bases showed no Lysozyme induction after infection in wild-type mice; therefore, the approximately 2-fold induction observed in total ileal tissue (Figure 4B) is likely due to the expansion of Lysozyme-positive cells outside the crypt base (Figure 4C, arrows).
Lastly, Periodic acid-Schiff staining showed that Atg16l1f/f x Villin-cre mice have abnormal granules before and after S Typhimurium infection compared to wild-type mice, suggesting an inherent defect in Paneth cells lacking Atg16l1 (Figure 4F). Together these data propose that epithelial Atg16l1 is essential for normal Paneth cell differentiation and morphology.
Mice lacking epithelial Atg16l1 are more susceptible to S Typhimurium infection
Given our findings that S Typhimurium is unable to associate with autophagosomes in Atg16l1-deficient epithelial cells and that Atg16l1f/f x Villin-cre mice have impaired AMP expression following infection, we next assessed the inflammatory response during S Typhimurium infection. After pilot time-course experiments, we chose 48 hours for our analyses, consistent with published reports19. Atg16l1f/f x Villin-cre mice displayed greater cecal inflammation after infection (Figure 5A and B). In addition, Atg16l1f/f x Villin-cre mice had significantly higher levels of the proinflammatory cytokines IL-6 and IL-1β compared to wild-type mice in terminal ileum and cecum (Figure 5C), suggesting these mice are more susceptible to S Typhimurium-induced inflammation. Moreover, epithelial cells isolated from Atg16l1f/f x Villin-cre mice expressed significantly higher levels of the chemokines Cxcl1 and Ccl2 after infection, indicating altered host responses in the absence of epithelial Atg16l1 (Figure 5D). While the absence of Atg16l1 in Villin-expressing cells conferred hyper-susceptibility to this model, Atg16l1f/f x CD11c-cre mice exhibited responses comparable to autophagy-intact wild-type mice (Figure 5A–E).
Figure 5. Mice lacking epithelial Atg16l1 are more susceptible to S Typhimurium infection.

(A) Cecal tissues were harvested from uninfected (top) and S Typhimurium-infected (bottom) mice 48 hours post-infection. Representative H&E-stained sections are shown (20X magnification). (B) Cecal tissue sections were blindly scored for inflammation 48 hours post-infection with S Typhimurium. (C) Cytokine levels in cecum and terminal ileum tissues were assessed via qPCR in S Typhimurium-infected mice 48 hours post-infection. Fold expression shown relative to wild-type. (D) Chemokine levels in isolated ileal epithelial cells were assessed via qPCR in S Typhimurium-infected mice 48 hours post-infection. Fold expression shown relative to wild-type. (E) S Typhimurium translocation to MLN and spleen was quantified 48 hours post-infection. Total CFUs per organ are displayed. In B–E, bars indicate mean plus S.D. * P = .002; ** P ≤.001. Data are representative of at least 3 independent experiments (n ≥ 12).
To exclude the possibility that other cell compartments known to assist in bacterial clearance were altered and contributing to this phenotype, we quantified immune populations by flow cytometry in the intestinal, MLN, and splenic compartments. No differences in absolute cell numbers were detected in immune cell subsets prior to infection, suggesting the observed phenotype is due largely to epithelial defects in the absence of Atg16l1 (Supplementary Figure 2A and B). Importantly, significantly greater numbers of CD11c+ mononuclear cells were present in the MLN of Atg16l1f/f x Villin-cre mice after infection, possibly due to the inability of the Atg16l1-deficient epithelial cells to control the acute infection (Supplementary Figure 2C). Dendritic cells and macrophages migrate from intestinal tissues to the MLN to present antigens and engage additional immune populations to help mount a response against uncontrolled pathogens. Indeed, when bacterial translocation was assessed in spleen and MLN, Atg16l1f/f x Villin-cre mice showed significantly greater numbers of Salmonella colony-forming units (CFU), suggesting that these mice were unable to control dissemination of the intestinal pathogen (Figure 5E). Taken together, these data demonstrate that Atg16l1 in intestinal epithelial cells is required for in vivo antibacterial defense in this model.
Identification of ATG16L1-dependent antibacterial chemical modulators
Our results establish a critical role for Atg16l1 in innate immunity against Salmonella in vivo, with our key observations complementing previous reports in cultured cell lines21. Therefore, cell culture infection models may be powerful systems for screening chemical compounds that modulate autophagy and can expedite our understanding of their modes of action in cellular pathways. Based on our observations that intestinal epithelial cells are important for antibacterial autophagy, we assembled a collection of 80 compounds known to regulate autophagy by diverse mechanisms (probe kit) for testing in an epithelial cell line. After confirming the effects of probe kit compounds on EGFP-LC3 puncta formation (Supplementary Figure 4), a subset of scoring compounds known to activate autophagy by different mechanisms were tested for their effects on bacterial survival using a gentamicin protection assay in S Typhimurium-infected HeLa cells (Figure 6A). Trifluoperazine (TFP), an FDA-approved phenothiazine, enhanced bacterial clearance in infected HeLa cells (Figure 6A and B), but did not alter the number of metabolically active HeLa cells or bacterial growth in cell-free cultures (Supplementary Figure 5A), suggesting that TFP activates antibacterial mechanisms in HeLa cells. TFP was previously validated as an mTOR-independent autophagy modulator that promotes the degradation of long-lived protein and polyglutamine aggregates in H4 cells27. Consistent with these reports, TFP treatment increased LC3-II levels in a concentration-dependent manner in HeLa cells, suggesting TFP activates autophagy (Figure 6C). Furthermore, co-administration of TFP and the lysosomal inhibitors E64d and Pepstatin A increased LC3-II levels to a much greater extent than treatment with lysosomal inhibitors alone, demonstrating that TFP induces autophagic flux and is a true activator of autophagy28 (Figure 6C). These TFP-dependent effects were reduced upon knockdown of ATG16L1 expression, providing further support that this compound’s effects are autophagy-specific (Figure 6C). Interestingly, autophagic flux in HeLa cells with very low ATG16L1 protein expression was strongly impaired, but reached levels similar to ATG16L1-proficient HeLa cells upon treatment with higher concentrations (5–10 μM) of TFP (Figure 6C). However, while TFP increased autophagic flux in wild-type MEFs, LC3-II was undetectable in untreated and TFP-treated Atg16l1−/− MEFs (Supplementary Figure 5B). Thus, TFP-induced autophagy is strictly ATG16L1-dependent, but low levels of ATG16L1 protein appear sufficient to mediate TFP-induced autophagic flux.
Figure 6. Trifluoperazine induces antibacterial autophagy in epithelial cells.

(A) Left: HeLa cells infected with Salmonella were incubated with indicated compound and gentamycin for 16 hours and CFUs were quantified. Right: Percent viability of HeLa cells 16 hours after compound treatment was assessed by luminescence measurement following addition of an equal volume of CellTiter-Glo to cells in media. Error bars represent mean plus S.D. of triplicate samples. (B) HeLa cells infected with S Typhimurium were incubated with 10 μM TFP and 20 μg/ml gentamicin for 16 h and CFUs were quantified. Bars indicate means plus S.D. of three independent experiments in triplicate. (C) HeLa cells transfected with indicated siRNA were treated for 8 hours with TFP in the presence or absence of lysosomal inhibitors. Western blot demonstrates expression levels of Atg16l1, actin, and LC3B. (D) HeLa cells stably expressing EGFP-LC3 were treated with TFP for 3 hours. Cells were then infected with S Typhimurium, followed by incubation with gentamycin sulfate and TFP. Numbers of EGFP-LC3-positive bacteria were calculated as a percentage of total bacteria. (E) Bioluminescent Salmonella-infected HeLa cells were treated with TFP. Intracellular bacteria were quantified by luminescence 24 hours post-infection. Error bars represent means plus S.D. from two independent experiments in quadruplicate. (F) Intracellular bioluminescent Salmonella were quantified by luminescence at indicated times following infection and treatment with 10 μM TFP. Error bars represent means plus S.D. from two independent experiments in quadruplicate.
To determine whether TFP-induced antibacterial activity is due to enhanced encapsulation of S Typhimurium into autophagosomes, EGFP-LC3 HeLa cells were infected with dsRed-expressing S Typhimurium. We observed a dose-dependent increase in the co-localization of EGFP-LC3-labeled autophagosomes and dsRed-S Typhimurium one hour post-infection (Figure 6D). To study the dose-dependent and kinetic effects of TFP on bacterial replication and survival, we monitored bacterial luminescence in TFP-treated HeLa cells infected with S Typhimurium expressing the Photorhabdus luminescens lux operon. These studies indicated that TFP suppresses bacterial growth, and possibly survival, in a dose- and time-dependent manner (Figure 6E and F). Taken together, these results demonstrate that TFP promotes anti-Salmonella autophagy by enhancing capture of Salmonella in autophagosomes and suppressing bacterial replication in an ATG16L1-dependent manner.
Discussion
Little is known about the functionality and efficacy of autophagic control of bacterial survival and spread in vivo in mice. Using Salmonella as a model system for invasive bacterial infections in mice, we demonstrated that intestinal epithelial cells can trap intracellular Salmonella in autophagosomes in vivo. Furthermore, loss of expression of the essential autophagy protein Atg16l1 in intestinal epithelial cells abolished autophagic engulfment of S Typhimurium, resulting in hyper-inflammation and systemic bacterial translocation in infected mice. In addition, Atg16l1f/f x Villin-cre mice showed significantly more CD11c+ mononuclear cells in the MLN after S Typhimurium infection, consistent with the severe inflammation and bacterial translocation observed in these mice due to impaired bacterial clearance by the epithelium.
A recent paper by Benjamin et al. similarly reported increased epithelial autophagy after Salmonella infection, and demonstrated that this process requires epithelial MyD88 and Atg5 expression29. Our study extends these findings by using a new conditional model to demonstrate that epithelial Atg16l1 is necessary for host defense against intestinal pathogens and by assessing the role of autophagy in CD11c-expressing antigen-presenting cells during Salmonella infection. Interestingly, ablation of Atg16l1 expression in CD11c+ mononuclear cells resulted in enhanced proinflammatory cytokine expression compared to wild-type mice, suggesting that autophagy regulates antigen-presenting cell responses during acute infection. Indeed, previous studies have demonstrated hyper-activation of caspase-1 and subsequent excessive generation and secretion of bioactive IL-1β and IL-18 in Atg16l1-deficient macrophages17. Nevertheless, Atg16l1 deficiency in CD11c+ mononuclear cells did not significantly impact susceptibility to Salmonella, suggesting that epithelial cell Atg16l1 is critical for the acute antibacterial response in our model. Autophagy is required for antigen presentation and Atg16l1 recently has been shown to modulate immunologic synapse stability, highlighting that autophagy is important in adaptive immunity and likely plays a role in other systems30, 31. As autophagy has also been implicated in regulating cross-presentation, oral administration of a less virulent Salmonella strain may help assess physiological responses over time, potentially uncovering a role for Atg16l1 in CD11c+ mononuclear cells during chronic immune responses. Moreover, determining the outcomes of concurrent Atg16l1 deficiency in epithelial and antigen-presenting cell compartments may offer insight into the compounding effects of defective autophagy in multiple cell types during antibacterial defense.
Interestingly, LC3 upregulation has been reported in duodenal and ileal Paneth cells of Crohn’s disease patients, similar to our observed LC3 upregulation in Paneth cells following S Typhimurium infection32. Moreover, abnormal granule morphology and impaired secretory functions of Paneth cells have been described in patients with inflammatory bowel disease harboring the disease-associated SNP in ATG16L133, 34. We show that epithelial Atg16l1 deficiency, unlike other loss-of-function mutations in the autophagy pathway, leads to fewer numbers of Paneth cells. In addition, we demonstrate that Paneth cell granule morphology is abnormal in mice lacking epithelial Atg16l1 both prior to and after S Typhimurium infection. Likewise, previous reports have shown impaired granule exocytosis in Paneth cells deficient in Atg5, Atg7, or Atg16l1 expression, leading to defective AMP secretion25. We propose that the impaired AMP expression observed in Atg16l1f/f x Villin-cre mice may be due solely to fewer numbers of Paneth cells, or may be further compounded by defective exocytosis due to granule abnormalities. Either scenario proposes that Atg16l1f/f x Villin-cre mice have compromised epithelial antibacterial responses in the intestine, consistent with their hyper-susceptibility to S Typhimurium infection.
We and others have demonstrated that autophagy regulates pathogen-specific immune responses in a variety of cell compartments during bacterial infection. M. tuberculosis infects macrophages and initiates ubiquitin-mediated autophagy by permeabilizing M. tuberculosis-containing phagosomal membranes35. Interestingly, using mice lacking Atg5 expression in macrophages, several studies have shown that macrophage autophagy is important for protection from M. tuberculosis by suppressing bacterial growth, inhibiting proinflammatory cytokine secretion, and regulating T cell polarization35, 36. In addition, during Shigella flexneri or S Typhimurium infection of intestinal epithelial cells, amino acid starvation occurs, which inhibits the kinase mTOR, an autophagy inhibitor37. This cellular defense mechanism initiates autophagy to aid in bacterial handling; however, S Typhimurium have developed a mechanism to evade autophagy by normalizing amino acid levels37. Together with our observations supporting a role for epithelial autophagy during acute S Typhimurium infection, these data propose that both pathogen- and cell compartment-specific autophagy responses are important during in vivo infections. In addition, the opportunistically invasive commensal Enterococcus faecalis has recently been shown to induce autophagosome formation in intestinal epithelial cells, highlighting a broad role for autophagy in bacterial sensing29.
There is a compelling need for new therapeutic approaches to modulate specific pathways important in autoinflammatory and infectious diseases. We have demonstrated that autophagy is important for antibacterial effects in intestinal epithelial cells in vivo, and, by phenotypic small molecule screening, we demonstrated that modulators of autophagy, including TFP, can enhance antibacterial defenses. In addition to TFP, several other phenothiazines suppress bacterial growth, and it would therefore be interesting to determine whether a common mechanism of action exists for this class of compounds in antibacterial autophagy, which may have broader implications in infections and autoinflammatory diseases.
Supplementary Material
Acknowledgments
Natalia Nedelsky for editorial assistance (DK043351).
Grant support: This work was supported by the following grants: DK097485, DK092405, and DK060049 (R.J.X.); The Leona M. and Harry B. Helmsley Charitable Trust (S.L.S., R.J.X.); DK043351 (R.J.X., H.C.R.); AI093588, DK068181, and DK033506 (H.C.R.). DK082427 (H.N.S.); CCFA Genetics Initiative (H.W.V., R.J.X., T.S.S.); AI054483 (H.W.V.).
Abbreviations used in this paper
- AMP
anti-microbial peptide
- SCV
Salmonella-containing vacuole
- SNP
single-nucleotide polymorphism
- MEF
mouse embryonic fibroblast
- MLN
mesenteric lymph nodes
- FAE
follicle-associated epithelium
- CFU
colony-forming units
- TFP
Trifluoperazine
Footnotes
Conflicts of interest: The authors report no conflict of interest.
Author contributions: K.L.C. and P.K. contributed equally to this manuscript. J.S. and K.K.P. contributed equally to this manuscript. K.L.C., P.K., A.B.C., L.N.A., H.N.S., A.F.S, H.C.R., and R.J.X. designed experiments; K.L.C., P.K, J.S., K.K.P., A.B.C., O.H.Y., H.B.J., M.Z., E.J.V., J.M.P., I.A.L., E.M., L.N.A., and D.M. performed experiments; K.L.C., P.K, J.S., K.K.P., A.B.C., O.H.Y., H.B.J, E.J.V, J.M.P., G.G., I.A.L, E.M., L.N.A., A.K.B., A.F.S., T.S.S., H.C.R., and R.J.X. analyzed data; K.L.C., P.K., and R.J.X. wrote the manuscript; J.S., K.K.P., A.B.C., H.B.J., M.Z., E.J.V., J.M.P., H.N.S., A.K.B., T.S.S., S.Y.S., S.L.S., H.W.V., A.F.S., and H.C.R. participated in editing the manuscript and provided conceptual advice.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Author names in bold designate shared co-first authorship.
- 1.Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–56. doi: 10.1146/annurev-biochem-052709-094552. [DOI] [PubMed] [Google Scholar]
- 2.Kuballa P, Nolte WM, Castoreno AB, et al. Autophagy and the immune system. Annu Rev Immunol. 2012;30:611–46. doi: 10.1146/annurev-immunol-020711-074948. [DOI] [PubMed] [Google Scholar]
- 3.Shi CS, Shenderov K, Huang NN, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–63. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yano T, Mita S, Ohmori H, et al. Autophagic control of listeria through intracellular innate immune recognition in drosophila. Nat Immunol. 2008;9:908–16. doi: 10.1038/ni.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim JJ, Lee HM, Shin DM, et al. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe. 2012;11:457–68. doi: 10.1016/j.chom.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 6.Jia K, Thomas C, Akbar M, et al. Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling-regulated pathogen resistance. Proc Natl Acad Sci U S A. 2009;106:14564–9. doi: 10.1073/pnas.0813319106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tung SM, Unal C, Ley A, et al. Loss of Dictyostelium ATG9 results in a pleiotropic phenotype affecting growth, development, phagocytosis and clearance and replication of Legionella pneumophila. Cell Microbiol. 2010;12:765–80. doi: 10.1111/j.1462-5822.2010.01432.x. [DOI] [PubMed] [Google Scholar]
- 8.Birmingham CL, Smith AC, Bakowski MA, et al. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem. 2006;281:11374–83. doi: 10.1074/jbc.M509157200. [DOI] [PubMed] [Google Scholar]
- 9.Shahnazari S, Yen WL, Birmingham CL, et al. A diacylglycerol-dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell Host Microbe. 2010;8:137–46. doi: 10.1016/j.chom.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Randow F. How cells deploy ubiquitin and autophagy to defend their cytosol from bacterial invasion. Autophagy. 2011;7:304–9. doi: 10.4161/auto.7.3.14539. [DOI] [PubMed] [Google Scholar]
- 11.Huett A, Heath RJ, Begun J, et al. The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin-dependent autophagy of intracellular Salmonella Typhimurium. Cell Host Microbe. 2012;12:778–90. doi: 10.1016/j.chom.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mesquita FS, Thomas M, Sachse M, et al. The Salmonella deubiquitinase SseL inhibits selective autophagy of cytosolic aggregates. PLoS Pathog. 2012;8:e1002743. doi: 10.1371/journal.ppat.1002743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–11. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 14.Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cadwell K, Liu JY, Brown SL, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–63. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cadwell K, Patel KK, Maloney NS, et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–45. doi: 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–8. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 18.Sidhu M, Cotoner CA, Guleng B, et al. Small molecule tyrosine kinase inhibitors for the treatment of intestinal inflammation. Inflamm Bowel Dis. 2011;17:2416–26. doi: 10.1002/ibd.21646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barthel M, Hapfelmeier S, Quintanilla-Martinez L, et al. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect Immun. 2003;71:2839–58. doi: 10.1128/IAI.71.5.2839-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Niess JH, Brand S, Gu X, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307:254–8. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]
- 21.Kuballa P, Huett A, Rioux JD, et al. Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS One. 2008;3:e3391. doi: 10.1371/journal.pone.0003391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carpenter AE, Jones TR, Lamprecht MR, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skarnes WC, Rosen B, West AP, et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474:337–42. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–30. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cadwell K, Patel KK, Komatsu M, et al. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy. 2009;5:250–2. doi: 10.4161/auto.5.2.7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez Rodriguez NR, Eloi MD, Huynh A, et al. Expansion of Paneth cell population in response to enteric Salmonella enterica serovar Typhimurium infection. Infect Immun. 2012;80:266–75. doi: 10.1128/IAI.05638-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Yu J, Pan H, et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci U S A. 2007;104:19023–8. doi: 10.1073/pnas.0709695104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–26. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benjamin JL, Sumpter R, Jr, Levine B, et al. Intestinal Epithelial Autophagy Is Essential for Host Defense against Invasive Bacteria. Cell Host Microbe. 2013;13:723–34. doi: 10.1016/j.chom.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee HK, Mattei LM, Steinberg BE, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 32:227–39. doi: 10.1016/j.immuni.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wildenberg ME, Vos AC, Wolfkamp SC, et al. Autophagy attenuates the adaptive immune response by destabilizing the immunologic synapse. Gastroenterology. 142:1493–503. e6. doi: 10.1053/j.gastro.2012.02.034. [DOI] [PubMed] [Google Scholar]
- 32.Thachil E, Hugot JP, Arbeille B, et al. Abnormal activation of autophagy-induced crinophagy in Paneth cells from patients with Crohn’s disease. Gastroenterology. 142:1097–1099. e4. doi: 10.1053/j.gastro.2012.01.031. [DOI] [PubMed] [Google Scholar]
- 33.Simms LA, Doecke JD, Walsh MD, et al. Reduced alpha-defensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn’s disease. Gut. 2008;57:903–10. doi: 10.1136/gut.2007.142588. [DOI] [PubMed] [Google Scholar]
- 34.Wehkamp J, Salzman NH, Porter E, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci U S A. 2005;102:18129–34. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. 2012;150:803–15. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Castillo EF, Dekonenko A, Arko-Mensah J, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A. 2012;109:E3168–76. doi: 10.1073/pnas.1210500109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tattoli I, Sorbara MT, Vuckovic D, et al. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe. 2012;11:563–75. doi: 10.1016/j.chom.2012.04.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.