

Abstract
Inactivation of tumor suppressor genes via promoter hypermethylation may play an important role in the progression from Barrett's esophagus (BE) to esophageal adenocarcinoma (EA). We have previously shown that acid-induced p16 gene promoter hypermethylation may depend on activation of NADPH oxidase NOX5-S in BAR-T cells and OE33 EA cells. DNA methyltransferase 1 (DNMT1) is known to participate in maintaining established patterns of DNA methylation in dividing cells and may play an important role in the development of cancer. Therefore, we examined whether DNMT1 is involved in acid-induced p16 gene promoter hypermethylation in BAR-T cells. We found that the acid significantly increased p16 gene promoter methylation, decreased p16 mRNA, and increased cell proliferation, effects that may depend on activation of DNMT1 in BAR-T cells. DNMT1 is overexpressed in EA cells FLO and OE33 and EA tissues. Acid treatment upregulated DNMT1 mRNA expression and increased DNMT1 promoter activity. Acid-induced increases in DNMT1 mRNA expression and promoter activity were significantly decreased by knockdown of NOX5-S and NF-κB1 p50. Conversely, overexpression of NOX5-S, p50, or p65 significantly increased DNMT1 promoter activity. Knockdown of NOX5-S significantly decreased the acid-induced increase in luciferase activity in cells transfected with pNFκB-Luc. An NF-κB binding element GGGGTATCCC was identified in the DNMT1 gene promoter. We conclude that the acid-induced increase in p16 gene promoter methylation, downregulation of p16 mRNA, and increase in cell proliferation may depend on activation of DNMT1 in BAR-T cells. Acid-induced DNMT1 expression may depend on sequential activation of NOX5-S and NF-κB1 p50.
Keywords: DNMT1, NOX5-S, NF-κB, esophageal adenocarcinoma, Barrett's esophagus
gastroesophageal reflux disease (GERD) complicated by Barrett's esophagus (BE) is a major risk factor for esophageal adenocarcinoma (EA; Ref. 27). Five to fifteen percent of GERD patients develop BE (9, 22) where esophageal squamous epithelium damaged by reflux esophagitis is replaced by a metaplastic, intestinal-type epithelium. The specialized intestinal metaplasia of BE is associated with nearly a 30- to 125-fold increased risk for the development of EA, with best estimates of cancer incidence of 0.12–0.8% per year, i.e., 1 cancer per 125–860 patients for each year of observation (3, 14, 20, 22, 26, 33, 44). However, the mechanisms of the progression from BE (intestinal metaplasia) to EA are not fully understood.
Many epigenetic alterations and hypermethylation of gene promoters may be involved in this progression (37, 44). For example, hypermethylation of p16 (45) and APC (7, 25) gene promoters are present at a much higher frequency in Barrett's dysplasia and EA than in Barrett's intestinal metaplasia. DNA methylation typically is modified in the CpG-rich areas of the gene promoter region and is mediated by DNA methyltransferases (DNMTs: DNMT1, DNMT3A, and DNMT3B; Ref. 34). DNMT1 contributes to maintaining established patterns of DNA methylation in dividing cells, while DNMT3A and 3B play an important role in establishing novel methylation sites on nascent DNA (15). Recently, DNMTs have been found to be overexpressed in different human cancers, thus supporting a role of these enzymes in the development and maintenance of the neoplastic phenotype (30, 40). Although numerous genes have been shown to be hypermethylated in esophageal cancer (4, 41, 43), the mechanisms of gene promoter hypermethylation have not been revealed yet.
Acid reflux may contribute to the progression from BE to EA since 1) it has been reported that acid exposure induces DNA damage in human BE cell line BAR-T(46); 2) cultured biopsy specimens of intestinal metaplastic cells demonstrate a significant increase in tritiated thymidine uptake when the explants are briefly exposed to acid, suggesting that in Barrett's specimens brief, episodic acid exposure is sufficient to promote tumorigenesis by stimulating cell proliferation (11); and 3) two prospective studies show that proton pump inhibitor treatment significantly reduces the incidence of high-grade dysplasia in BE patients (8, 24).
Reactive oxygen species (ROS) may also play an important role in the development of EA since levels of ROS are increased in BE (32) and EA (10, 36). ROS may cause damage to DNA, RNA, lipids, and proteins, which may result in increased mutation and altered functions of enzyme and proteins (e.g., activation of oncogene products and/or inhibition of tumor suppressor proteins; Refs. 10, 31). Superoxide-generating homologues of phagocytic NADPH oxidase catalytic subunit gp91phox (NOX1, NOX3-NOX5, DUOX1, and DUOX2) and homologues of other subunits (p41phox or NOXO1 and p51phox or NOXA1) have been found in several cell types (1a, 38), which may generate ROS in different cells with distinctive cellular functions related to immunity, signal transduction, and modification of the extracellular matrix. NOX5 has five isoforms: α, β, δ, γ, and NOX5-S (2, 42). NOX5α, β, δ, and γ, have EF-hand motifs at their NH2 terminus (2), whereas NOX5-S does not (6).
We have shown that the NADPH oxidase isoform NOX5-S is present in EA FLO cells (17) and that levels of NOX5-S are significantly increased in BE mucosa with high-grade dysplasia (12). NOX5-S-derived ROS in turn increase p16 gene promoter methylation thereby contributing to the progression from BE to EA in response to acid treatment (18). In this study we show that the acid causes an increase in p16 gene promoter methylation, downregulates p16 mRNA, and increases cell proliferation, effects that may depend on activation of DNMT1 in BAR-T cells. Acid-induced DNMT1 expression may depend on sequential activation of NOX5-S and NF-κB1 p50 in BAR-T cells.
MATERIALS AND METHODS
Cell culture and acid treatment.
Human esophageal squamous HET-1A cells (ATCC, Manassas, VA) were cultured in the bronchial epithelial cell medium (BEGM BulletKit; Cambrex, East Rutherford, NJ) containing a basal medium (BEBM) plus the additives (BEGM SingleQuots) in wells precoated with a mixture of 0.01 mg/ml fibronectin, 0.03 mg/ml vitrogen 100, and FBS.
Human Barrett's cell line BAR-T was derived from esophageal mucosal biopsies of patients with BE (intestinal metaplasia) and immortalized with telomerase as described previously (21). Human Barrett's cell line CP-A was purchased from ATCC. Barrett's cells were cultured in wells precoated with collagen IV (1 μg/cm2; BD Bioscience, Bedford, MA) and in Keratinocyte Medium-2 (Ca2+-free solution; Cambrex, Rockland, ME) supplemented with 1.8 mM CaCl2, 5% FBS, 400 ng/ml hydrocortisone, 20 ng/ml epidermal growth factor, 0.1 nM cholera toxin, 20 μg/ml adenine, 5 μg/ml insulin, 70 μg/ml bovine pituitary extract, and antibiotics.
Human Barrett's adenocarcinoma cell line FLO was derived from human BE adenocarcinoma (19) and generously provided by Dr. David Beer (University of Michigan). FLO cells were cultured in DMEM containing 10% FBS and antibiotics. OE33 EA cell line was purchased from Sigma (St. Louis, MO) and cultured in DMEM containing 10% fetal bovine serum and antibiotics.
For acid treatment, BAR-T or CP-A cells were exposed to acidic keratinocyte medium-2 (pH 6.0) or normal keratinocyte medium-2 (pH 7.2, control) for 24 h, and then the culture medium and cells were collected for measurements. FLO cells were exposed to acidic DMEM medium (pH 4.0) or normal medium (pH 7.2) for 1 h and then cultured at pH 7.2 for additional 24 h.
Human esophageal tissues.
Normal esophageal mucosa, Barrett's mucosa, and EA tissues were obtained from patients with EA undergoing esophagogastrectomy. EA patients with preoperative chemoradiotherapy were excluded from the study. The experimental protocols were approved by the Human Research Institutional Review Committee at Rhode Island Hospital.
Construction of pGL3-DNMT1P reporter plasmid.
The DNA fragment containing part of the promoter region (−1751 to 0 from ATG) of DNMT1 gene (GenBank accession no. NM_001130823) was amplified by PCR from human genomic DNA. The primers used were DNMT1P-sense: 5′-GGGGTACCACGGAGTCTC GCTCTGTTG-3′ (the introduced Kpn I is underlined) and DNMT1P-antisense: 5′-CCGCTCGAGATCTCGGAGGCTTCAGCA-3′ (the introduced XhoI is underlined). The obtained cDNA fragment was then cloned into pGL3-basic (Promega) between Kpn I and Xho I.
Small interfering RNA and plasmid transfection.
Twenty-four hours before transfection at 70–80% confluence, cells were trypsinized and diluted 1:5 with fresh medium without antibiotics (1–3 ×105 cells/ml) and transferred to 12-well plates (1 ml/well). Transfection of small interfering RNA (siRNA) was carried out with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Per well, 75 pmol of NOX5-S siRNA, DNMT1 siRNA, NF-κB p50, or control siRNA formulated into liposomes were applied; the final volume was 1.2 ml/well. Transfection efficiencies were determined by fluorescence microscopy after transfection of Block-it fluorescent oligonucleotide (Invitrogen) and were ∼70% at 48 h.
The pCMV-tag5a-NOX5-S plasmid was generously provided to us by Dr. David Lambeth (Emory University School of Medicine, Atlanta, GA). NF-κB in vivo activation reporter plasmid pNF-κB-Luc, which contains five repeats of NF-κB binding element GGGGACTTTCC in the enhancer element of the plasmid, was purchased from Stratagene. pCMV-NF-κB p50 plasmid was purchased from Addgene (plasmid 21965; Ref. 1). pNF-κB p65 plasmid was generously provided to us by Dr. Eugene Chin. For transfection of these plasmids, cells (70% confluence, ∼5 × 106 cells) were transfected with 2 μg above plasmids or control plasmid using Amaxa-Nucleofector-System (Lonza) according to the manufacturer's instructions. Transfection efficiencies were determined by fluorescence microscopy after transfection of pmax-GFP (Lonza) and were ∼80% at 48 h.
Reverse transcription-PCR.
Total RNA was extracted by TRIzol reagent and purified by the total RNA purification system according to the manufacturer's protocol (Invitrogen); 1.5 μg of total RNA were reversely transcribed by using a kit SuperScript First-Strand Synthesis System for RT-PCR according to the manufacturer's protocol (Invitrogen).
Quantitative real-time PCR.
Quantitative real-time PCR was carried out on a Stratagene Mx4000 multiplex quantitative PCR system. The primers used were: DNMT1 F: 5′-GGCGGCTCAAAGATTTGGAAAGAG-3′, DNMT1 R: 5′-CACCGTTCTCCAAGGACAAATC-3′, p16 F: 5′-AAGGTCCCTCAGACATCCC-3′, p16 R: 5′-TGGACATTTACGGTAGTGGG-3′, 18S F: 5′-CGGACAGGATTGACAGATTGATAGC-3′, and 18S R: 5′-TGCCAGAGTCTCGTTCGTTATCG-3′. All reactions were performed in triplicate in a 25-μl total volume containing a 1× concentration of Brilliant SYBR Green QPCR Master Mix (Stratagene). The concentrations of each sense and antisense primer were 100 nM, 1 μl cDNA, and 30 nM reference dyes. Reactions were carried out in a Stratagene Mx4000 multiplex quantitative PCR system for one cycle at 94°C for 5 min; 40 cycles at 94°C for 30 s, 59°C for 30 s, and 72°C for 30 s; one cycle at 94°C for 1 min; and one cycle at 55°C for 30 s. Fluorescence values of SYBR Green I dye, representing the amount of product amplified at that point in the reaction, were recorded in real time at both the annealing step and the extension step of each cycle. The Ct, defined as the point at which the fluorescence signal was statistically significant above background, was calculated for each amplicon in each experimental sample using Stratagene Mx4000 software. This value was then used to determine the relative amount of amplification in each sample by interpolating from the standard curve. The transcript level of each specific gene was normalized to 18S amplification.
Bisulfite conversion of DNA sample.
Bisulfite conversion of cell samples were carried out as described previously (16). Genomic DNA from 104 cells of each different cell lines was extracted and modified using a CpG modification Kit (EZ DNA methylation-Direct Kit; Zymo Research) according to the manufacturer's protocol. Briefly, by bi-sulfite treatment, unmethylated cytosine bases are converted to uracil, whereas methylated cytosines remain unchanged. After treatment, methylated DNA sequence differs from unmethylated DNA, which is used to design methylation-specific primers and probes.
Conventional methylation specific PCR and real-time quantitative methylation.
Specific PCR conventional methylation-specific PCR (MSP) and real-time quantitative methylation-specific PCR were carried out as described previously by us (18). The primers (16) for the bisulfite-converted methylated sequence were p16MF: 5′-TTATTAGAGGGTGGGGCGGATCGC-3′ and p16MR: 5′-GACCCCGAACCGCGACCGTAA-3′. The primers (16) for the bisulfite-converted unmethylated sequence were p16UF: 5′-TTATTAGAGGGTGGGGTGGATTGT-3′ and p16UR: 5′-CAACCCCAAACCACAACCATAA-3′. β-Actin was used as a reference control. Primers for β-actin were β-actin-F: 5′-TGGTGATGGAGGAGGTTTAGTAAGT-3′ and β-actin-R: 5′-AACCAATAAAACCTACTCCTCCCTTAA-3′.
Western blot analysis.
Cells were lysed in a Triton X lysis buffer containing 50 mM Tris-hydrochloride (pH 7.5), 100 mM sodium chloride, 50 mM sodium fluoride, 5 mM EDTA, 1% (vol/vol) Triton X-100, 40 mM β-glycerol phosphate, 40 mM p-nitrophenylphosphate, 200 μM sodium orthovanadate, 100 μM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, and 1 μg/ml aprotinin. The suspension was centrifuged at 15,000 g for 5 min, and the protein concentration in the supernatant was determined. Western blot analysis was done as described previously (5). Primary antibodies used were as follows: IκBα antibody (1:1,000), DNMT1 antibody (1:1,000), and GAPDH antibody (1:2,000).
Luciferase assay.
The 0.1 μg renilla and 1.0 μg reporter plasmids pNF-κB-Luc or pGL3-DNMT1P in combination with NOX5 siRNA, p50 siRNA, control siRNA, pcDNA3.1, or NOX5-S plasmid were transiently transfected in duplicate with Lipofectamine 2000 (Invitrogen) according to manufacturer's instruction. Twenty-four hours after transfection, cells were washed, lysed, and sequentially evaluated for luciferase activity using the Dual-Luciferase Reporter Assay System (Promega). For acid treatment, BAR-T cells were treated with acidic medium (pH 6.0, 24 h) 12 h after transfection and then the cells are collected for measurements. Luciferase activity was measured for 10 s after a 2-s delay using a BD Monolight 3010 luminometer (BD Biosciences, San Jose, CA). Variation in transfection efficiency was normalized by dividing the construct luciferase activity by the corresponding renilla luciferase activity.
Chromatin immunoprecipitation assay.
Chromatin immunoprecipitation (ChIP) assay was performed using the ChIP assay kit (Upstate, Charlottesville, VA) following manufacturer's protocol. Briefly, BAR-T cells grown in plastic dishes for 2 days (∼1 × 106 cells) were treated with acidic medium, pH 4.0, for 1 h and then treated with 1% formaldehyde for 10 min to cross-link NF-κB p50 to DNA. After removal of the formaldehyde, the cells were washed with ice cold phosphate-buffered saline containing 0.1% EDTA and protease inhibitors (1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, and 1% protease inhibitor mixture) and gently scraped into a conical tube, centrifuged for 5 min at 700 g at 4°C. Pelleted cells were resuspended in 400 μl of lysis buffer [10 mM HEPES, pH 7.9, 60 mM KCl, 0.5% (vol/vol) Nonidet P-40] with protease inhibitors and incubated on ice for 10 min. Nuclei were recovered by centrifugation at 1,000 g for 10 min and resuspended in 400 μl of SDS lysis buffer (1% SDS, 10 mM EDTA, and 50 mM Tris·HCl pH 8.1) containing protease inhibitors. The mixture was incubated on ice for 10 min, and the lysate was sonicated eight times for 10 s each time on ice to shear the genomic DNA to lengths of 0.2–1 kb. The debris was removed by centrifugation, and the supernatant was then diluted 10× with ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris·HCl, 16.7 mM NaCl, and protease inhibitors pH 8.0). Five-hundred microliters of the diluted lysate were kept for input control. The chromatin solution was precleared with salmon sperm DNA/protein A-agarose for 1 h at 4°C. Anti-NF-κB p50 (Upstate) antibody was added, respectively, to the supernatant fraction and incubated overnight at 4°C with rotation. The mixture was then incubated with 60 μl of salmon sperm DNA/protein A-agarose slurry for 1 h at 4°C with rotation. Normal rabbit IgG (Upstate) or c-Myc antibody (SeroTec, Raleigh, NC) was used instead of the specific antibody for the negative control. The protein A-agarose-antibody-histone complex was pelleted by gentle centrifugation (1,000 g at 4°C for 1 min). The pellet was washed sequentially (3–5 min per wash) on a rotating platform with 1 ml each of low salt washing buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris·HCl, and 150 mM NaCl pH 8.0), high salt washing buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris·HCl, and 500 mM NaCl pH 8.0), LiCl washing buffer (0.25 M LiCl, 1% Nonidet P-40, 1% sodium deoxycholate, 1 mM EDTA, and 10 mM Tris·HCl pH 8.0), and 1× TE buffer (10 mM Tris·HCl and 1 mM EDTA pH 8.0). After the final wash, the pellet was eluted by two 15-min incubations with 250 μl of freshly made elution buffer (1% SDS and 50 mM NaHCO3). Two fractions of elutes were combined, and 20 μl of 5 M NaCl were added to the supernatant. Cross-linking was reversed by heating at 65°C for 4 h, followed by the addition of 10 μl of 0.5 mM EDTA, 20 μl of 1 M Tris·HCl pH 6.5, and 2 μl of 10 mg/ml proteinase K. The sample was incubated at 45°C for 2 h, and DNA was then extracted by phenol chloroform extraction followed by ethanol precipitation. The DNA pellet was resuspended in 50 μl of H2O, and 5 μl were used for PCR analysis. PCR was carried with the primer pairs that targeted the −1147 to −1019 region of the human DNMT1 promoter (sense 5′-ACCTCAGCCTCCCAAGTA-3′and antisense 5′-ATCGCTTGAGGTTAGGAGTT-3′) at 94°C for 5 min, 94°C for 30 s, 62°C for 30 s, and 72°C for 30 s for 35 cycles followed by a 7-min extension at 72°C. Results were visualized in 2% agarose gels stained with ethidium bromide.
Gel mobility shift assay.
Gel shift assay was performed using the gel shift assay kit (Licor, NE) following the manufacturer's protocol. Oligonucleotides derived from human DNMT1 promoter (−1055 to −1075) were synthesized and labeled with IRDye 700 by Integrated DNA Technologies (IDT). Gel shift assay was performed by incubating 5 μg of HeLa nuclear extract with 1 μl of 50 nM IRDye 700-labeled probe in a 20-μl reaction buffer containing 10 mM Tris pH 7.5, 0.05 mg/ml poly (dI-dC)_poly (dI-dC), 4% glycerol, 0.5 mM EDTA, 0.1 M KCl, 0.5 mM phenylmethylsulfonyl fluoride, and 0.5 mM dithiothreitol, for 20 min at room temperature. For competition experiments and supershift assay with normal rabbit IgG (control) or rabbit NF-κB p50 antibody, the competing unlabeled probes or antibodies were preincubated for 20 min at room temperature with the nuclear extracts before the addition of the labeled probes. The wild-type competitor DNMT1-WT (−1055 to −1075; 5′-GAGATGGGGTATCCCTATGT-3′) or mutant competitor DNMT1-MUT (−1055 to −1075; 5′-GAGATGAGGTATCAATATGT-3′) was used. The DNA-protein complexes were resolved on a 5% nondenaturing polyacrylamide gel for 2 h at 100 V in 0.5× TBE buffer. After electrophoresis, the gel was imaged immediately by Licor Odyssey imager system.
[3H]thymidine incorporation.
For acid treatment, BAR-T cells were exposed to acidic keratinocyte medium-2 (pH 6.0) or normal keratinocyte medium-2 (control) for 24 h. For the H2O2 treatment, BAR-T cells were treated with (H2O2 10−11 M) 24 h. For siRNA transfection, after introducing with DNMT1 siRNA, NF-κB p50 siRNA, or control siRNA, cells were treated without or with acid and then incubated with methyl-[3H]thymidine (0.05 μCi/ml) for 4 h. After being washed three times with PBS to remove unincorporated radioactivity, cells were collected and homogenized with a lysis buffer containing (pH 7.4): 50 mM HEPES, 50 mM NaCl, 1% Triton X-100, 1% Nonidet P-40, 0.1 mM phenylmethylsulfonyl fluoride, and 1 mM dithiothreitol. Methyl-[3H]thymidine uptake was measured in a scintillation counter. The level of protein in the homogenates was also determined and the level of methyl-[3H]thymidine incorporation was normalized to protein content.
Materials.
[3H]thymidine was purchased from PerkinElmer (Waltham, MA). Human NOX5 siRNA was purchased from Ambion DNMT1 antibody, IκBα antibody, NF-κB p50 antibody, DNMT1 siRNA, and NF-κB p50 siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); TRIzol reagent, total RNA purification system, and RNA reverse transcription kit were purchased from Invitrogen, Bisulfite Conversion CpG modification kit was from Zymo Research. Hydrocortisone, epidermal growth factor, cholera toxin, adenine, insulin, bovine pituitary extract, Triton X-100, phenylmethylsulfonyl-fluoride, and other reagents were purchased from Sigma.
Statistical analysis.
Data are expressed as means ± SE. Statistical differences between two groups were determined by Student's t-test. Differences among multiple groups were tested using ANOVA and checked for significance using Fisher's protected least significant difference test.
RESULTS
DNMT1 is involved in acid-induced p16 hypermethylation in BAR-T cells.
Figure 1, A and B, shows that DNMT1 siRNA significantly decreased DNMT1 protein expression, suggesting that DNMT1 siRNA effectively knocked down DNMT1. Consistent with our previous finding (18), acid treatment significantly increased p16 gene promoter methylation (Fig. 1C) and decreased p16 mRNA expression (Fig. 1D). In addition, knockdown of DNMT1 with DNMT1 siRNA blocked acid-induced p16 gene promoter hypermethylation (Fig. 1C) and p16 mRNA downregulation (Fig. 1D) in BAR-T cells. Acid treatment significantly increased thymidine incorporation, an increase that was significantly decreased by knockdown of DNMT1 (Fig. 1E). These data suggest that DNMT1 may mediate acid-induced increase in p16 gene promoter methylation in BAR-T cells, which in turn downregulates p16 mRNA and increases cell proliferation.
Fig. 1.
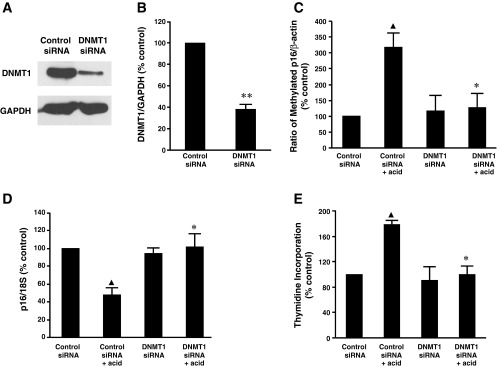
Role of DNA methyltransferase 1 (DNMT1) in acid-induced methylation of p16 gene promoter and cell proliferation in BAR-T cells. Typical image of 3 experiments (A) and summarized data (B) show that DNMT1 small interfering (si)RNA significantly decreased DNMT1 protein expression, suggesting that DNMT1 siRNA effectively knocks down DNMT1. C: acid treatment significantly increased p16 gene promoter methylation, an increase that was significantly reduced by knockdown of DNMT1 with DNMT1 siRNA, indicating that DNMT1 may mediate acid-induced increase in p16 promoter methylation in BAR-T cells. D: knockdown of DNMT1 expression reversed acid-induced decrease in p16 mRNA expression in BAR-T cells. E: knockdown of DNMT1 significantly decreased acid-induced increase in thymidine incorporation in BAR-T cells. Data suggest that acid-induced increase in p16 gene promoter methylation and cell proliferation may depend on activation of DNMT1 (n = 3). ANOVA: ▲P < 0.01, compared with control siRNA group; *P < 0.05, compared with control siRNA + acid group. **P < 0.01 by t-test.
Next, we examined DNMT1 mRNA levels in different cell lines and tissues by real-time PCR. We found that DNMT1 mRNA levels were significantly higher in the EA cell lines FLO and OE33 than the normal esophageal squamous epithelial cell line HET-1A or Barrett's cell lines BAR-T or CP-A (Fig. 2A). DNMT1 mRNA levels were higher in BAR-T and CP-A cells than those in HET-1A cells, but the increase did not reach the statistical significance. Similarly, DNMT1 mRNA levels were significantly increased in EA tissues (Fig. 2B), when compared with normal esophageal mucosa or Barrett's mucosa. DNMT1 mRNA levels were higher in Barrett's mucosa than those in normal esophageal mucosa, but the increase did not reach the statistical significance (P = 0.0992). These data suggest that DNMT1 might be important in development of EA.
Fig. 2.

DNMT1 expression in different cell lines and tissues. A: real-time RT-PCR showed that DNMT1 mRNA levels were significantly higher in esophageal adenocarcinoma (EA) cell lines FLO and OE33 than normal esophageal squamous epithelial cell line HET-1A or Barrett's cell lines BAR-T or CP-A (n = 3–10). DNMT1 mRNA levels were higher in BAR-T and CP-A cells than those in HET-1A cells, but the increase did not reach the statistical significance. B: DNMT1 mRNA levels were significantly increased in EA tissues, when compared with normal esophageal mucosa or Barrett's mucosa (n = 3–12). DNMT1 mRNA levels were higher in Barrett's mucosa than those in normal esophageal mucosa, but the increase did not reach the statistical significance. These data suggest that DNMT1 might be important in development of EA. ANOVA: *P < 0.01, #P < 0.02, compared with HET-1A cells, BAR-T cells, or CP-A cells; **P < 0.01, compared with normal esophageal mucosa (NESO) or Barrett's mucosa (BE).
Role of NOX5-S in acid-induced DNMT1 expression.
We found that acid treatment significantly increased DNMT1 mRNA expression in BAR-T, CP-A, and FLO cells (Fig. 3, A–C). Knockdown of NOX5-S with NOX5-S siRNA, which has been shown by us to effectively knockdown NOX5 (12), blocked the acid-induced increase in DNMT1 mRNA expression (Fig. 3, A–C), suggesting that NOX5-S may mediate acid-induced DNMT1 expression. To further confirm this result, we constructed a DNMT1 reporter plasmid pGL3-DNMT1P. Acid treatment significantly increased DNMT1 promoter activity, an increase that was significantly reduced by knockdown of NOX5-S (Fig. 3D). Overexpression of NOX5-S significantly increased DNMT1 promoter activity (Fig. 3E). These data suggest that acid-induced increase in DNMT1 promoter activity may depend on activation of NOX5-S.
Fig. 3.
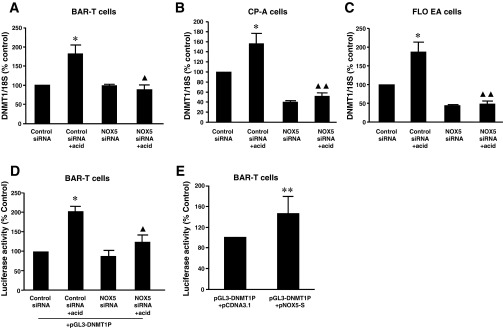
Effect of NOX5-S on acid-induced DNMT1 expression. A: in BAR-T cells, acid treatment significantly increased DNMT1 mRNA expression. Knockdown of NOX5-S with NOX5-S siRNA blocked acid-induced increase in DNMT1 mRNA expression. B: in CP-A cells, acid treatment significantly increased DNMT1 mRNA expression. Knockdown of NOX5-S with NOX5-S siRNA blocked acid-induced increase in DNMT1 mRNA expression. C: in FLO cells, acid treatment significantly increased DNMT1 mRNA expression. Knockdown of NOX5-S with NOX5-S siRNA blocked acid-induced increase in DNMT1 mRNA expression. D: a reporter plasmid of DNMT1 (pGL3-DNMT1P) was generated by ligating a DNMT1 promoter fragment (−1751 to 0 from ATG) into the pGL3-basic vector. Acid treatment significantly increased luciferase activity, an increase that was significantly reduced by knockdown of NOX5-S. E: overexpression of NOX5-S significantly increased luciferase activity in BAR-T cells. These data suggest that acid-induced increase in DNMT1 expression and promoter activity may depend on activation of NOX5-S (n = 3). ANOVA: *P < 0.001, compared with control siRNA group; ▲P < 0.05, ▲▲P < 0.001, compared with control siRNA + acid group. **P < 0.05 by t-test.
Role of NF-κB in acid-induced DNMT1 expression.
To test whether NF-κB plays a role in NOX5-S-mediated DNMT1 expression, we determined whether acid activates NF-κB. Figure 4, A and B, shows that pulsed acid treatment significantly decreased the expression of IκBα, suggesting acid-induced activation of NF-κB. To confirm this conclusion, we transfected BAR-T cells with NF-κB cis-reporter plasmid pNF-κB-Luc, which contains five repeats of NF-κB binding element GGGGACTTTCC in the enhancer element of the plasmid. Acid treatment significantly increased the luciferase activity, an increase that was significantly decreased by knockdown of NOX5-S (Fig. 4C), suggesting that acid-induced activation of NF-κB may depend on activation of NOX5-S in BAR-T cells.
Fig. 4.

Acid-induced activation of NF-κB in BAR-T cells. A typical example of Western blot analysis (A) and summarized data (B) showed that acid treatment significantly decreased the expression of IκBα, when compared with the control group. Data suggest that acid treatment may activate NF-κB in BAR-T cells. C: acid treatment significantly increased the luciferase activity in BAR-T cells transfected with NF-κB cis-reporter plasmid pNF-κB-Luc, which contains 5 repeats of NF-κB binding element GGGGACTTTCC in the enhancer element of the plasmid. NOX5 siRNA significantly decreased acid-induced increase in luciferase activity in BAR-T cells. These data suggest that acid-induced activation of NF-κB may depend on activation of NOX5-S in BAR-T cells (n = 3). ANOVA: *P = 0.001, compared with control siRNA group; **P < 0.05, compared with control siRNA + acid group. #P < 0.001 by t-test.
Next, we examined DNMT1 mRNA level in the presence or absence of SN50, a cell permeable inhibitor of NF-κB, or p50 siRNA. We found that acid-induced DNMT1 mRNA expression was significantly decreased by SN-50 in BAR-T cells (Fig. 5A) or by knockdown of NF-κB1 p50 with p50 siRNA in BAR-T cells (Fig. 5B), CP-A cells (Fig. 5C), and FLO EA cells (Fig. 5D). Similarly, acid-induced increase in DNMT1 promoter activity was significantly decreased by knockdown of NF-κB1 p50 with p50 siRNA in BAR-T cells (Fig. 5E). These data suggest that NF-κB1 p50 may contribute to acid-induced DNMT1 expression. p50 siRNA has been shown by us to effectively knockdown p50 (17). Overexpression of NF-κB p50 significantly increased DNMT1 promoter activity in BAR-T cells (Fig. 5F). In addition, overexpression of p65 significantly increased DNMT1 mRNA (Fig. 6A) and protein expression (Fig. 6, B and C) in BAR-T cells. These data suggest that acid-induced DNMT1 expression may depend on activation of NF-κB1 p50.
Fig. 5.
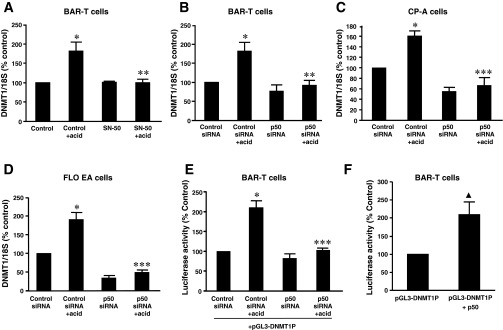
Role of NF-κB in acid-induced DNMT1 expression. A: SN50, a cell permeable inhibitor of NF-κB significantly decreased acid-induced DNMT1 mRNA expression. B: in BAR-T cells, knockdown of NF-κB1 p50 with p50 siRNA significantly decreased acid-induced DNMT1 mRNA expression. C: in CP-A cells, knockdown of NF-κB1 p50 with p50 siRNA significantly decreased acid-induced DNMT1 mRNA expression. D: in FLO EA cells, knockdown of NF-κB1 p50 with p50 siRNA significantly decreased acid-induced DNMT1 mRNA expression. E: acid-induced increase in DNMT1 promoter activity was significantly decreased by knockdown of NF-κB1 p50 with p50 siRNA in BAR-T cells. F: in BAR-T cells, overexpression of NF-κB p50 significantly increased DNMT1 promoter activity (n = 3). ANOVA: *P < 0.001, compared with control or control siRNA group; **P < 0.05, ***P < 0.001, compared with control + acid group or control siRNA + acid group. ▲P < 0.05 by t-test.
Fig. 6.
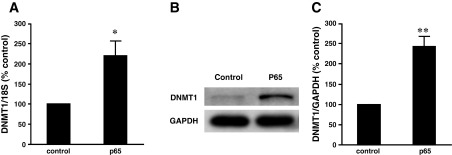
Effect of overexpression of p65 on DNMT1 expression in BAR-T cells. A: overexpression of P65 significantly increased DNMT1 mRNA expression measured by real-time PCR. A typical example of Western blot analysis (B) and summarized data (C) show that overexpression of P65 significantly increased DNMT1 protein expression in BAR-T cells. Data suggest that NF-κB may mediate DNMT1 expression in BAR-T cells (n = 3). *P < 0.001, **P < 0.05 by t-test.
We also examined whether NF-κB participates in acid-induced p16 gene promoter hypermethylation in BAR-T cells. We found that knockdown of p50 significantly downregulated acid-induced increase in p16 gene promoter methylation in BAR-T cells (Fig. 7A) and almost abolished acid-induced decrease in p16 mRNA expression (Fig. 7B), suggesting that acid-induced p16 methylation may depend on activation of NF-κB1 p50. Furthermore, overexpression of p65 significantly increased p16 gene promoter methylation in BAR-T cells, an increase that was significantly decreased by knockdown of DNMT1 by DNMT1 siRNA (Fig. 7C). The data suggest that NF-κB-mediated increase in p16 gene promoter methylation may depend on DNMT1 activation.
Fig. 7.

Role of NF-κB in acid-induced methylation of p16 gene promoter in BAR-T cells. A: knockdown of P50 significantly decreased acid-induced increase in p16 promoter methylation in BAR-T cells, indicating that NF-κB may mediate acid-induced increase in p16 promoter methylation in BAR-T cells. B: knockdown of P50 reversed acid-induced decrease in p16 mRNA level in BAR-T cells, indicating that NF-κB may participate in acid-induced decrease in p16 mRNA in BAR-T cells. C: knockdown of DNMT1 significantly decreased p16 promoter hypermethylation induced by overexpression of p65, a subunit of NF-κB, indicating that P65-induced p16 promoter methylation may depend on activation of DNMT1 (n = 3). ANOVA: *P < 0.05, compared with control siRNA group; **P < 0.05, compared with control siRNA + acid group or control siRNA + p65 group.
After analyzing the genomic DNA sequence of DNMT1, we found one possible NF-κB binding element in the DNMT1 promoter GGGGTATCCC (positions −1069 to −1060). To determine whether NF-κB binds to DNMT1 genomic DNA, we performed ChIP assay. DNMT1 DNA was detectable in the immunoprecipitated chromatin samples of BAR-T cell lysate by PCR using a pair of primers targeting the −1146 to −1018 (position from ATG) region of the DNMT1 promoter (Fig. 8). This pair of primers covers the possible NF-κB-binding site as described above. The data suggest that NF-κB may bind to the DNMT1 promoter. To further confirm this result, we did gel mobility supershift assay. In the gel shift assay, one prominent complex was detectable with IRdye 700-labeled DNMT1 oligonucleotide DNMT1-WT, containing the NF-κB binding site GGGGTATCCC (Fig. 9A). Competition experiments with a high concentration of unlabeled (cold) DNMT1-WT significantly reduced binding. The addition of the mutant DNMT1 oligonucleotide DNMT1-MUT had no apparent effect on binding of the DNMT1-WT oligonucleotide (Fig. 9A). In the supershift assay, a supershifted band was detected with a NF-κB p50 antibody but not with immunoglobulin G (Fig. 9B). These data suggest that NF-κB binds to the site GGGGTATCCC on the DNMT1 promoter region.
Fig. 8.

Typical example of 3 experiments shows that DNMT1 DNA was detectable in the immunoprecipitated chromatin sample of BAR-T cells by using an antibody against NF-κB1 p50, suggesting that NF-κB1 p50 binds to DNMT1 promoter. Positive, genomic DNA used as a positive control; rabbit IgG and c-Myc antibody were used as negative controls.
Fig. 9.
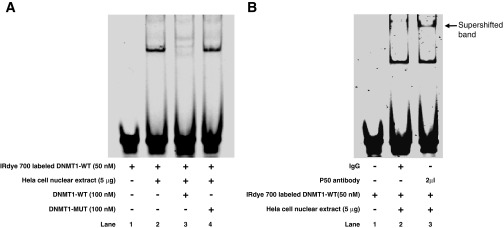
A: in a gel mobility shift assay, a double-stranded IRdye 700-labeled oligonucleotide containing the sequence GAGATGGGGTATCCCTATGT (DNMT1-WT) and HeLa nuclear extract provided by the kit were used. One prominent complex was detected (lane 2). Competition experiments with unlabeled DNMT1-WT oligonucleotide significantly reduced binding (lane 3); however, addition of the mutant oligonucleotide GAGATGAGGTATCAATATGT (DNMT1-MUT, lane 4) had no effect on binding. These data suggest that NF-κB binds to the site GGGGTATCCC at the DNMT1 promoter. B: in a supershift assay with a NF-κB1 P50 antibody, the supershifted band was observed with NF-κB1 P50 antibody but not with immunoglobulin G, suggesting that NF-κB1 p50 binds to the binding element at DNMT1 promoter.
DISCUSSION
GERD complicated by BE is a major risk factor for EA (27). However, the mechanisms of the progression from BE (intestinal metaplasia) to EA are not fully understood. Many epigenetic alterations and hypermethylation of gene promoters may be involved in this progression (37, 44). We have shown that acid reflux present in patients with BE may activate NADPH oxidase NOX5-S and produce high levels of ROS. NOX5-S-derived ROS in turn increase p16 gene promoter methylation, downregulate p16 expression, and increase cell proliferation, thereby contributing to the progression from BE to EA (18).
DNA methylation typically is modified in the CpG rich areas of the gene promoter region and is mediated by DNA methyltransferases (34). DNMT1 is the major methyltransferase responsible for methylating DNA and is a therapeutic target for chemotherapy and chemoprevention (28–29, 39). Therefore, we hypothesize that NOX5-S mediates acid-induced p16 gene promoter methylation via activation of DNMT1. To test our hypothesis, we first examined whether DNMT1 participates in acid-induced p16 gene promoter hypermethylation and cell proliferation in BAR-T cells. We found that acid-induced increase in p16 gene promoter methylation was significantly reduced by knockdown of DNMT1 in BAR-T cells. In addition, knockdown of DNMT1 reversed acid-induced decrease in p16 mRNA expression and significantly decreased acid-induced increase in cell proliferation. These data suggest that DNMT1 may mediate acid-induced increase in p16 gene promoter methylation and decrease in p16 mRNA expression in BAR-T cells, thereby increasing cell proliferation.
We also found that DNMT1 expression was significantly increased in EA cells and tissues and that acid treatment may increase DNMT1 expression. Acid-induced DNMT1 expression may depend on activation of NOX5-S since 1) knockdown of NOX5-S blocked acid-induced upregulation of DNMT1 and significantly decreased DNMT1 promoter activity; and 2) overexpression of NOX5-S significantly increased DNMT1 promoter activity.
We found that acid treatment significantly decreased IκBα protein levels and increased luciferase activity in cells transfected with NF-κB reporter plasmid pNFκB-Luc, suggesting that acid may activate NF-κB. We also found that knockdown of NOX5-S significantly decreased the acid-induced increase in luciferase activity in cells transfected with pNFκB-Luc, suggesting that activation of NF-κB may depend on activation of NOX5-S. This result is consistent with the literature showing that the p47phox subunit of NADPH oxidases participates in the activation of RelA in endothelial cells (13).
Next, we examined the role of NF-κB in acid-induced DNMT1 expression. NF-κB is thought to be a family of Rel domain-containing proteins, including RelA (also called p65), RelB, c-Rel, NF-κB1 (p105/p50), and NF-κB2 (p100/p52). p105 and p100 are larger precursor proteins containing IκB (an inhibitor of κB)-like ankyrin repeat sequences in their carboxyl termini. Because of their IκB-like ankyrin repeat sequences, these precursors are retained in the cytoplasm and require proteolytic processing to generate their mature DNA-binding proteins, p50 and p52, respectively (23). In the cytoplasm NF-κB is in an inactive state, and its activity is regulated by at least two pathways. In the first pathway, a heterotrimer composed of p50, p65, and IκB is degraded in a ubiquitin-dependent reaction, leading to the translocation of the p65-p50 dimers to the nucleus (23). In the second pathway, the dimers consisting of p100 and Rel B undergo proteolytic removal of the IκB-like COOH-terminal domain of p100, allowing Rel B-p52 dimers to translocate to nucleus, where NF-κB activates gene transcription (23). p50 plays an important role in lymphoid organogenesis and inflammation, whereas p52 is mainly involved in lymphoid organogenesis (35). Therefore, we focused on the role of p50 in acid-induced DNMT1 expression.
We found that inhibition of NF-κB by SN50 or knockdown of NF-κB p50 by p50 siRNA significantly decreased DNMT1 expression in response to acid treatment in BAR-T cells. In addition, knockdown of p50 significantly decreased the acid-induced increase in DNMT1 promoter activity and overexpression of p50 increased DNMT1 promoter activity. Overexpression of p65 significantly increased DNMT1 mRNA and protein expression. These data suggest that NF-κB1 p50 and p65 heterodimers may be responsible for acid-induced DNMT1 expression in BAR-T cells. Furthermore, NF-κB p50 may be involved in acid-induced increase in p16 promoter methylation and decrease in p16 mRNA expression since 1) acid-induced increase in p16 gene promoter methylation was significantly reduced by knockdown of p50; 2) acid-induced decrease in p16 mRNA expression was reversed by knockdown of p50; and 3) overexpression of p65 significantly increased p16 gene promoter methylation in BAR-T cells. p16 gene promoter hypermethylation induced by overexpression of p65 was inhibited by downregulation of DNMT1, suggesting that DNMT1 may mediate NF-κB-dependent p16 gene promoter hypermethylation.
We found that NF-κB may bind to one possible NF-κB binding element in the DNMT1 promoter, GGGGTATCCC (positions −1069 to −1060) since 1) DNMT1 DNA was detectable in the immunoprecipitated chromatin samples of BAR-T cell lysate; 2) in the gel shift assay, one prominent complex was detectable with IRdye 700-labeled DNMT1 oligonucleotide, containing the NF-κB binding site GGGGTATCCC; 3) competition experiments with a high concentration of unlabeled (cold) DNMT1 oligonucleotide significantly reduced binding, whereas addition of the mutant DNMT1 oligonucleotide had no apparent effect on binding; and 4) in the supershift assay, a supershifted band was detected with a NF-κB p50 antibody but not with immunoglobulin G.
We conclude that acid-induced p16 gene promoter hypermethylation, downregulation of p16 mRNA, and increase in cell proliferation may depend on sequential activation of NOX5-S, NF-κB1 p50, and DNMT1 (Fig. 10). It is possible that acid reflux present in patients with BE may activate NOX5-S. High levels of ROS derived from NOX5-S may activate NF-κB1 p50 and upregulate DNMT1, which in turn induces p16 gene promoter hypermethylation and downregulates p16 mRNA, thereby contributing to the progression from BE to EA.
Fig. 10.

Signal transduction pathway of acid-induced downregulation of p16 and increase in cell proliferation.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-080703.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.H., D.L., and W.C. conception and design of research; J.H. and D.L. performed experiments; J.H. and W.C. analyzed data; J.H., D.L., and W.C. interpreted results of experiments; J.H., D.L., and W.C. prepared figures; J.H. and W.C. drafted manuscript; J.W., R.S., and W.C. edited and revised manuscript; W.C. approved final version of manuscript.
REFERENCES
- 1.Ballard DW, Dixon EP, Peffer NJ, Bogerd H, Doerre S, Stein B, Greene WC. The 65-kDa subunit of human NF-κ B functions as a potent transcriptional activator and a target for v-Rel-mediated repression. Proc Natl Acad Sci USA 89: 1875–1879, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1a.Banfi B, Maturana A, Jaconi S, Arnaudeau S, Laforge T, Sinha B, Ligeti E, Demaurex N, Krause KH. A mammalian H+ channel generated through alternative splicing of the NADPH oxidase homolog NOH-1. Science 287: 138–142, 2000 [DOI] [PubMed] [Google Scholar]
- 2.Banfi B, Molnar G, Maturana A, Steger K, Hegedus B, Demaurex N, Krause KH. A Ca(2+)-activated NADPH oxidase in testis, spleen, and lymph nodes. J Biol Chem 276: 37594–37601, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Bhat S, Coleman HG, Yousef F, Johnston BT, McManus DT, Gavin AT, Murray LJ. Risk of malignant progression in Barrett's esophagus patients: results from a large population-based study. J Natl Cancer Inst 103: 1049–1057, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brabender J, Arbab D, Huan X, Vallbohmer D, Grimminger P, Ling F, Neiss S, Bollschweiler E, Schneider PM, Holscher AH, Metzger R. Death-associated protein kinase (DAPK) promoter methylation and response to neoadjuvant radiochemotherapy in esophageal cancer. Ann Surg Oncol 16: 1378–1383, 2009 [DOI] [PubMed] [Google Scholar]
- 5.Cao W, Sohn UD, Bitar KN, Behar J, Biancani P, Harnett KM. MAPK mediates PKC-dependent contraction of cat esophageal and lower esophageal sphincter circular smooth muscle. Am J Physiol Gastrointest Liver Physiol 285: G86–G95, 2003 [DOI] [PubMed] [Google Scholar]
- 6.Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 269: 131–140, 2001 [DOI] [PubMed] [Google Scholar]
- 7.Eads CA, Lord RV, Kurumboor SK, Wickramasinghe K, Skinner ML, Long TI, Peters JH, DeMeester TR, Danenberg KD, Danenberg PV, Laird PW, Skinner KA. Fields of aberrant CpG island hypermethylation in Barrett's esophagus and associated adenocarcinoma. Cancer Res 60: 5021–5026, 2000 [PubMed] [Google Scholar]
- 8.El-Serag HB, Aguirre TV, Davis S, Kuebeler M, Bhattacharyya A, Sampliner RE. Proton pump inhibitors are associated with reduced incidence of dysplasia in Barrett's esophagus. Am J Gastroenterol 99: 1877–1883, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Falk GW. Barrett's esophagus. Gastroenterology 122: 1569–1591, 2002 [DOI] [PubMed] [Google Scholar]
- 10.Farhadi A, Fields J, Banan A, Keshavarzian A. Reactive oxygen species: are they involved in the pathogenesis of GERD, Barrett's esophagus, and the latter's progression toward esophageal cancer? Am J Gastroenterol 97: 22–26, 2002 [DOI] [PubMed] [Google Scholar]
- 11.Fitzgerald RC, Omary MB, Triadafilopoulos G. Dynamic effects of acid on Barrett's esophagus. An ex vivo proliferation and differentiation model. J Clin Invest 98: 2120–2128, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu X, Beer DG, Behar J, Wands J, Lambeth D, Cao W. cAMP-response element-binding protein mediates acid-induced NADPH oxidase NOX5-S expression in Barrett esophageal adenocarcinoma cells. J Biol Chem 281: 20368–20382, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Gu Y, Xu YC, Wu RF, Nwariaku FE, Souza RF, Flores SC, Terada LS. p47phox participates in activation of RelA in endothelial cells. J Biol Chem 278: 17210–17217, 2003 [DOI] [PubMed] [Google Scholar]
- 14.Haggitt RC. Barrett's esophagus, dysplasia, adenocarcinoma. Hum Pathol 25: 982–993, 1994 [DOI] [PubMed] [Google Scholar]
- 15.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 349: 2042–2054, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 93: 9821–9826, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hong J, Behar J, Wands J, Resnick M, Wang LJ, DeLellis RA, Lambeth D, Souza RF, Spechler SJ, Cao W. Role of a novel bile acid receptor TGR5 in the development of oesophageal adenocarcinoma. Gut 59: 170–180, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong J, Resnick M, Behar J, Wang LJ, Wands J, DeLellis RA, Souza RF, Spechler SJ, Cao W. Acid-induced p16 hypermethylation contributes to development of esophageal adenocarcinoma via activation of NADPH oxidase NOX5-S. Am J Physiol Gastrointest Liver Physiol 299: G697–G706, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes SJ, Nambu Y, Soldes OS, Hamstra D, Rehemtulla A, Iannettoni MD, Orringer MB, Beer DG. Fas/APO-1 (CD95) is not translocated to the cell membrane in esophageal adenocarcinoma. Cancer Res 57: 5571–5578, 1997 [PubMed] [Google Scholar]
- 20.Hvid-Jensen F, Pedersen L, Drewes AM, Sorensen HT, Funch-Jensen P. Incidence of adenocarcinoma among patients with Barrett's esophagus. N Engl J Med 365: 1375–1383, 2011 [DOI] [PubMed] [Google Scholar]
- 21.Jaiswal KR, Morales CP, Feagins LA, Gandia KG, Zhang X, Zhang HY, Hormi-Carver K, Shen Y, Elder F, Ramirez RD, Sarosi GA, Jr, Spechler SJ, Souza RF. Characterization of telomerase-immortalized, non-neoplastic, human Barrett's cell line (BAR-T). Dis Esophagus 20: 256–264, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Kahrilas PJ. The problems with surveillance of Barrett's esophagus. N Engl J Med 365: 1437–1438, 2011 [DOI] [PubMed] [Google Scholar]
- 23.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer 2: 301–310, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Kastelein F, Spaander MC, Steyerberg EW, Biermann K, Valkhoff VE, Kuipers EJ, Bruno MJ. Proton pump inhibitors reduce the risk of neoplastic progression in patients with Barrett's esophagus. Clin Gastroenterol Hepatol 11: 382–388, 2013 [DOI] [PubMed] [Google Scholar]
- 25.Kawakami K, Brabender J, Lord RV, Groshen S, Greenwald BD, Krasna MJ, Yin J, Fleisher AS, Abraham JM, Beer DG, Sidransky D, Huss HT, Demeester TR, Eads C, Laird PW, Ilson DH, Kelsen DP, Harpole D, Moore MB, Danenberg KD, Danenberg PV, Meltzer SJ. Hypermethylated APC DNA in plasma and prognosis of patients with esophageal adenocarcinoma. J Natl Cancer Inst 92: 1805–1811, 2000 [DOI] [PubMed] [Google Scholar]
- 26.Kim R, Weissfeld JL, Reynolds JC, Kuller LH. Etiology of Barrett's metaplasia and esophageal adenocarcinoma. Cancer Epidemiol Biomarkers Prev 6: 369–377, 1997 [PubMed] [Google Scholar]
- 27.Lagergren J, Bergstrom R, Lindgren A, Nyren O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med 340: 825–831, 1999 [DOI] [PubMed] [Google Scholar]
- 28.Li A, Omura N, Hong SM, Goggins M. Pancreatic cancer DNMT1 expression and sensitivity to DNMT1 inhibitors. Cancer Biol Ther 2010. February 25 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin RK, Hsieh YS, Lin P, Hsu HS, Chen CY, Tang YA, Lee CF, Wang YC. The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J Clin Invest 120: 521–532, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mizuno S, Chijiwa T, Okamura T, Akashi K, Fukumaki Y, Niho Y, Sasaki H. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood 97: 1172–1179, 2001 [DOI] [PubMed] [Google Scholar]
- 31.Ohshima H, Tatemichi M, Sawa T. Chemical basis of inflammation-induced carcinogenesis. Arch Biochem Biophys 417: 3–11, 2003 [DOI] [PubMed] [Google Scholar]
- 32.Olyaee M, Sontag S, Salman W, Schnell T, Mobarhan S, Eiznhamer D, Keshavarzian A. Mucosal reactive oxygen species production in oesophagitis and Barrett's oesophagus. Gut 37: 168–173, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet 381: 400–412, 2013 [DOI] [PubMed] [Google Scholar]
- 34.Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW, Baylin SB, Vogelstein B. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 416: 552–556, 2002 [DOI] [PubMed] [Google Scholar]
- 35.Shih VF, Tsui R, Caldwell A, Hoffmann A. A single NFkappaB system for both canonical and non-canonical signaling. Cell Res 21: 86–102, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sihvo EI, Ruohtula T, Auvinen MI, Koivistoinen A, Harjula AL, Salo JA. Simultaneous progression of oxidative stress and angiogenesis in malignant transformation of Barrett esophagus. J Thorac Cardiovasc Surg 126: 1952–1957, 2003 [DOI] [PubMed] [Google Scholar]
- 37.Souza RF, Morales CP, Spechler SJ. Review article: a conceptual approach to understanding the molecular mechanisms of cancer development in Barrett's oesophagus. Aliment Pharmacol Ther 15: 1087–1100, 2001 [DOI] [PubMed] [Google Scholar]
- 38.Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature 401: 79–82, 1999 [DOI] [PubMed] [Google Scholar]
- 39.Tikoo K, Ali IY, Gupta J, Gupta C. 5-Azacytidine prevents cisplatin induced nephrotoxicity and potentiates anticancer activity of cisplatin by involving inhibition of metallothionein, pAKT and DNMT1 expression in chemical induced cancer rats. Toxicol Lett 191: 158–166, 2009 [DOI] [PubMed] [Google Scholar]
- 40.Toyota M, Kopecky KJ, Toyota MO, Jair KW, Willman CL, Issa JP. Methylation profiling in acute myeloid leukemia. Blood 97: 2823–2829, 2001 [DOI] [PubMed] [Google Scholar]
- 41.Tsunoda S, Smith E, De Young NJ, Wang X, Tian ZQ, Liu JF, Jamieson GG, Drew PA. Methylation of CLDN6, FBN2, RBP1, RBP4, TFPI2, and TMEFF2 in esophageal squamous cell carcinoma. Oncol Rep 21: 1067–1073, 2009 [DOI] [PubMed] [Google Scholar]
- 42.Vignais PV. The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci 59: 1428–1459, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang JS, Guo M, Montgomery EA, Thompson RE, Cosby H, Hicks L, Wang S, Herman JG, Canto MI. DNA promoter hypermethylation of p16 and APC predicts neoplastic progression in Barrett's esophagus. Am J Gastroenterol 104: 2153–2160, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wild CP, Hardie LJ. Reflux, Barrett's oesophagus and adenocarcinoma: burning questions. Nat Rev Cancer 3: 676–684, 2003 [DOI] [PubMed] [Google Scholar]
- 45.Wong DJ, Barrett MT, Stoger R, Emond MJ, Reid BJ. p16INK4a promoter is hypermethylated at a high frequency in esophageal adenocarcinomas. Cancer Res 57: 2619–2622, 1997 [PubMed] [Google Scholar]
- 46.Zhang HY, Hormi-Carver K, Zhang X, Spechler SJ, Souza RF. In benign Barrett's epithelial cells, acid exposure generates reactive oxygen species that cause DNA double-strand breaks. Cancer Res 69: 9083–9089, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]