

Abstract
Endothelial cell (EC) dysfunction is implicated in cardiovascular diseases, including diabetes. The decrease in nitric oxide (NO) bioavailability is the hallmark of endothelial dysfunction, and it leads to attenuated vascular relaxation and atherosclerosis followed by a decrease in blood flow. In the heart, decreased coronary blood flow is responsible for insufficient oxygen supply to cardiomyocytes and, subsequently, increases the incidence of cardiac ischemia. In this study we investigate whether and how reactive oxygen species (ROS) in mitochondria contribute to coronary endothelial dysfunction in type 2 diabetic (T2D) mice. T2D was induced in mice by a high-fat diet combined with a single injection of low-dose streptozotocin. ACh-induced vascular relaxation was significantly attenuated in coronary arteries (CAs) from T2D mice compared with controls. The pharmacological approach reveals that NO-dependent, but not hyperpolarization- or prostacyclin-dependent, relaxation was decreased in CAs from T2D mice. Attenuated ACh-induced relaxation in CAs from T2D mice was restored toward control level by treatment with mitoTempol (a mitochondria-specific O2− scavenger). Coronary ECs isolated from T2D mice exhibited a significant increase in mitochondrial ROS concentration and decrease in SOD2 protein expression compared with coronary ECs isolated from control mice. Furthermore, protein ubiquitination of SOD2 was significantly increased in coronary ECs isolated from T2D mice. These results suggest that augmented SOD2 ubiquitination leads to the increase in mitochondrial ROS concentration in coronary ECs from T2D mice and attenuates coronary vascular relaxation in T2D mice.
Keywords: hyperglycemia, diabetic vascular complication, posttranslational modification, ubiquitin-proteasome system
vascular endothelial cells (ECs) play an important role in regulation of vascular tone, vascular permeability, blood cell coagulation, and new vascular formation. The endothelium regulates vascular tone by 1) producing and releasing vasoconstrictors (e.g., endothelin-1, angiotensin II, and thromboxane A2) and vasodilators [e.g., nitric oxide (NO), prostacyclin, and endothelium-derived hyperpolarizing factors] (28, 29, 44, 64, 68) and 2) inducing hyperpolarization of smooth muscle cells (SMCs) via electrical propagation through the gap junctions (10, 26). NO serves as a vasodilator, as well as an inhibitor of aggregation of platelets and infiltration of inflammatory cells (22). The decrease of NO bioavailability, as a result of endothelial dysfunction, causes increased vascular tension and atherosclerotic plaque and leads to the decrease in blood flow. In the heart, decreased coronary blood flow is responsible for insufficient oxygen supply to the cardiomyocytes and increase in the incidence of cardiac ischemia (5, 20).
Ischemic heart diseases are involved in >50% of diabetes-related deaths, and the incidence of cardiac ischemia is higher in diabetic patients than control subjects. Impaired endothelium-dependent vasodilatation due to chronic elevation of blood glucose level has been reported in various diabetic animal models and in diabetic patients (5, 42, 54, 67, 80), and the reduced NO bioavailability is a common feature of EC dysfunction in diabetes (62, 67, 75).
Under physiological conditions, ECs constantly generate reactive oxygen species (ROS), including superoxide anion (O2−), and ROS participate in modulation of essential endothelial functions (9, 78). The potential sources of O2− include mitochondrial electron transport chain (ETC), xanthine oxidase, uncoupled endothelial NO synthases (eNOS), cytochrome P-450 enzymes, and NADPH oxidases (NOXs). ECs express several antioxidant enzymes, such as SOD, catalase, and glutathione peroxidase. The imbalance between ROS production and elimination results in increased oxidative stress and tissue injury and leads to many cardiovascular diseases (51, 87). There is increasing evidence showing that elevated levels of O2− in ECs contribute to development of atherosclerosis (34), vascular complications of diabetes (2, 25, 31, 36, 55, 70), and hypertension (12, 35). In this study we demonstrate that decreased endothelium-dependent vascular relaxation (EDR) in coronary arteries (CAs) from type 2 diabetic (T2D) mice is due to an increase in mitochondrial ROS production in coronary ECs from T2D mice. It is, at least in part, attributed to decreased SOD2 protein expression via augmented ubiquitination of SOD2 in coronary ECs from T2D mice.
MATERIALS AND METHODS
Biological Materials and Reagents
N-nitro-l-arginine methyl ester (l-NAME) was purchased from Cayman Chemical (Ann Arbor, MI); apamin (Apa) and charybdotoxin (ChTx) from AnaSpec (Fremont, CA); streptozotocin (STZ) and anti-SOD1 and anti-SOD2 antibodies from Enzo Life Science (Plymouth Meeting, PA); M199, antibiotic reagents, dispase II, MitoTracker Green FM, and MitoSOX Red from Invitrogen (Carlsbad, CA); anti-ubiquitin (Ub) and anti-actin antibodies and ImmunoCruz from Santa Cruz Biotechnology (Santa Cruz, CA); anti-CD31 and EC growth supplement from BD Biosciences (San Jose, CA); and collagenase II from Worthington Biochemical (Lakewood, NJ). All other chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
Animal Preparation
All investigations conformed to the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, Revised 1985). This study was conducted in accordance with the guidelines established by the Institutional Animal Care and Use Committee at the University of Illinois at Chicago. Our protocols were approved by the Office of Animal Care and Institutional Biosafety Committees at the University of Illinois at Chicago. Six-week-old male C75BL6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). T2D mice were generated by a single injection of STZ (dissolved in citrate buffer, 75 mg/kg ip) and fed a high-fat (60% kcal) diet from the day of STZ injection (27). All data were obtained from mice 12–16 wk after the injection.
Metabolic Characterization
Total cholesterol, HDL, and triglyceride in plasma were measured with a kit from Wako Chemicals USA (Richmond, VA). Plasma insulin level was measured using a kit from ALPCO Diagnostics (Salem, NH). An oral glucose tolerance test was performed as follows: mice were fasted for 6 h; then glucose (2 g/kg body wt) was administrated orally, and plasma glucose concentration was measured at time 0 (before glucose administration) and 15, 30, and 60 min after glucose administration. An insulin tolerance test was performed as follows: mice were fasted for 4 h; then insulin was injected (0.2 U/kg body wt ip), and plasma glucose concentration was measured at time 0 (before insulin injection) and 15, 30, 60, and 120 min after insulin injection. Data were normalized by the glucose level at time 0 and shown as percentage.
Isometric Tension Measurement of CA Rings
Isometric tension was measured as previously described (58). The heart was isolated and placed in Krebs-Henseleit solution for dissection. Third-order small CAs were cleaned of any adherent connective tissue and cardiomyocytes and cut into 1- to 1.5-mm segments. Rings were mounted in a wire myograph (DMT-USA) with 20-μm wires and set at a resting tension of 0.1 g. All segments were equilibrated for 45 min with intermittent washes every 15 min. After equilibration, each CA ring was contracted by treatment with PGF2α. The degree of ACh-induced vasodilatation was described as a percentage after normalization by PGF2α-induced contraction.
Isolation of Coronary Vascular ECs
Mouse coronary ECs were isolated as previously described (56, 57). Briefly, dissected heart tissues were minced and incubated with M199 containing 1 mg/ml collagenase II and 0.6 U/ml dispase II for 1 h at 37°C. The digested material was filtered through sterile 40-μm nylon mesh and washed in 2% fetal calf serum in M199. Subsequently, the cells were incubated with Dynabeads (Invitrogen), which were prepared as follows: beads coated with sheep anti-rat IgG were incubated with purified rat anti-mouse CD31 monoclonal antibody (1 μg/ml) at 4°C overnight and then washed with PBS containing 0.1% BSA and 2 mM EDTA. The cell suspension was incubated with beads for 1 h at 4°C, and then beads attached to ECs were captured by a Dynal magnet (Invitrogen).
Measurement of Mitochondrial ROS Concentration
Mitochondrial ROS concentration was measured as described previously (41, 57). For detection of mitochondrial ROS, the cells were preloaded with 5 μmol/l MitoSOX Red (an O2− indicator) and 100 nmol/l MitoTracker Green (to visualize the mitochondrial structure) for 30 min. MitoSOX and MitoTracker Green fluorescence from the cells was imaged using a Nikon Eclipse Ti-E inverted fluorescence microscope with a ×60 objective lens. The structure of the mitochondria was determined by MitoTracker Green signal, and the fluorescence intensity of the MitoSOX in the mitochondria was measured. The background intensity was subtracted from the cell intensity.
Western Blot Analysis
After isolation, mouse coronary ECs were lysed and centrifuged at 16,000 g for 10 min at 4°C. Protein samples from ECs were separated through a SDS-polyacrylamide gel and transferred to the membranes. Blots were incubated with a primary antibody [anti-SOD1 (1:1,000 dilution), anti-SOD2 (1:1,000 dilution), anti-Ub (1:500 dilution), or anti-actin (1:4,000 dilution)] and then with a horseradish peroxidase-conjugated secondary antibody. The immunoblots were detected with SuperSignal West Pico reagent (Thermo Fisher Scientific, Rockford, IL). Band intensity was normalized to actin control and expressed in arbitrary units.
Assay of SOD2 mRNA
SOD2 mRNA in coronary ECs from control and T2D mice was measured by real-time PCR. mRNA from mouse coronary ECs was isolated using the RNeasy Plus Micro kit (Qiagen, Chatsworth, CA). Briefly, cells were lysed in Buffer RLT Plus by 10 passages through a 21-gauge needle, and the RNA was purified according to the manufacturer's instructions. cDNA was made by reverse transcription of DNase-free RNA templates using random hexamers and SuperScript III (Invitrogen). The primers for SOD2 and the endogenous reference gene 18S rRNA (18S) are as follows: 5′-GACCTGCCTTACGACTATGG-3′ (forward) and 5′-GACCTTGCTCCTTATTGAAGC-3′ (reverse) for SOD2 and 5′-GTAACCCGTTGAACCCCATT-3′ (forward) and 5′-CCATCCAATCGGTAGTAGCG-3′ (reverse) for 18S. Measurements were made in triplicate, using 1 ng of template per well, with a Bio-Rad Real Time PCR System. The efficiency-correlated cycle threshold (ΔCT) method was used to determine the level, in arbitrary units, of SOD2 mRNA relative to 18S.
High-Glucose and High-Fat Treatment Ex Vivo
Human coronary ECs (Cell Applications, San Diego, CA) were used for the study in Fig. 6. For high-glucose treatment, 20 mmol/l glucose was added to the medium (final glucose concentration 25 mmol/l). In a control group of cells, equimolar mannitol was added to exclude the potential effect of changes in osmolarity (normal glucose, 5 mmol/l). For free fatty acid (FFA) treatment, 300 μmol/l palmitic acid was added to the medium. Control cells were treated with vehicle (BSA) only. Cells were cultured for 48 h and used for the immunoprecipitation (IP) experiment.
Fig. 6.

High-glucose treatment significantly increases ubiquitination of SOD2 protein. A: immunoprecipitation (IP) with anti-SOD2 antibody and immunoblotting (IB) with anti-ubiquitin antibody (Ub) or anti-SOD2 antibodies in human coronary ECs treated with normal glucose (NG, n = 8) or high glucose (HG, n = 9). Values are means ± SE. *P < 0.05 vs. NG. B: IP with anti-SOD2 antibody and IB with anti-Ub or anti-SOD2 antibodies in human coronary ECs treated with 300 μM palmitic acid (free fatty acid, n = 5) or vehicle (BSA, n = 5). Values are means ± SE.
Immunoprecipitation
IP-Ub/immunoblotting-SOD2.
Mouse coronary ECs (from 2–3 hearts/sample) were lysed, and 25 μg of proteins were incubated with ExactaCruz C IP matrix (Santa Cruz Biotechnology), which was prepared as follows: IP matrix (40 μl) was incubated with anti-Ub (2 μg) at 4°C for 2 h and then washed with binding buffer. Lysate with IP matrix was incubated overnight at 4°C. The matrix-bound proteins were collected, and samples were used for immunoblotting.
IP-SOD2/immunoblotting-Ub.
Human coronary ECs were lysed, and 50 μg of proteins were incubated with ExactaCruz B IP matrix, which was prepared as follows: IP matrix (20 μl)was incubated with anti-SOD2 (1 μg/ml) at 4°C for 2 h and then washed with PBS.
Statistical Analysis
Values are means ± SE. Statistical comparison between dose-response curves was made by two-way ANOVA with Bonferroni's correction. Student's t-test for unpaired samples was used to identify significant differences. Differences were considered to be statistically significant when P < 0.05.
RESULTS
Metabolic Characteristics
Blood samples were collected for measurement of metabolic characteristics cholesterol. Body weight and plasma levels of glucose and total cholesterol, HDL, and LDL were significantly higher in T2D than control mice (Table 1). Plasma insulin level was significantly higher in T2D than control mice (Fig. 1A). In addition, T2D mice showed a significant decrease in insulin sensitivity (Fig. 1, B and C).
Table 1.
Metabolic characterization
| Control | T2D | |
|---|---|---|
| Body weight, g | 29.0 ± 0.7 | 37.9 ± 1.3* |
| Glucose, mg/dl | 133.1 ± 5.3 | 235.5 ± 24.2* |
| Total cholesterol, mg/dl | 88.9 ± 4.6 | 168.3 ± 14.0* |
| Total triglycerides, mg/dl | 17.3 ± 1.1 | 18.1 ± 1.5 |
| HDL, mg/dl | 40.3 ± 2.6 | 73.3 ± 5.2* |
| LDL, mg/dl | 45.1 ± 2.9 | 91.4 ± 9.9* |
Values are means ± SE; n = 10 in each group. T2D, type 2 diabetic.
P < 0.05 vs. control.
Fig. 1.
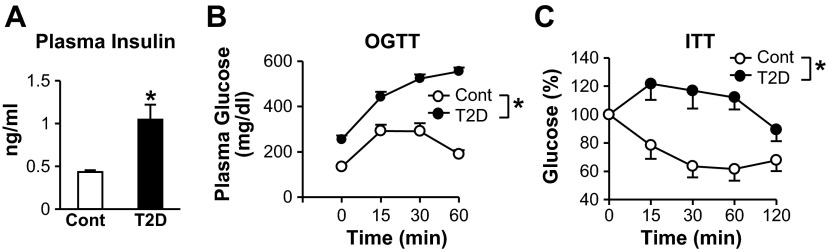
Plasma insulin levels, oral glucose tolerance test (OGTT), and insulin tolerance test (ITT) results. A: insulin concentration in plasma samples from nonfasting type 2 diabetic (T2D, n = 5) and control (Cont, n = 5) mice. B: plasma glucose levels in control (n = 10) and T2D (n = 13) mice fasted for 6 h before the OGTT. C: percent glucose in control (n = 5) and T2D (n = 5) mice fasted for 4 h before the ITT. Values are means ± SE. *P < 0.05 vs. Cont.
Attenuated ACh-Induced Relaxation in CAs From T2D Mice
To investigate endothelial function, ACh-induced vascular relaxation was examined using isolated CA rings from control and T2D mice. As shown in Fig. 2A, endothelium-dependent relaxation (EDR) was significantly decreased in CAs from T2D mice compared with controls, while there was no difference in sodium nitroprusside (SNP)-dependent relaxation between the two groups (Fig. 2B). To identify the molecular mechanism involved in attenuated EDR in diabetic CAs, a series of pharmacological experiments were conducted using the various inhibitors. Pretreatment with l-NAME (a NOS inhibitor, 100 μmol/l) and indomethacin (Indo, a cyclooxygenase inhibitor, 10 μmol/l) attenuated EDR in CAs from control and T2D mice, and the magnitude of the relaxation was not different between control and T2D CAs (Fig. 2C). However, in the presence of the Ca2+-activated K+ channel blockers, Apa and ChTx (100 nmol/l), and Indo, EDR was significantly decreased in CAs from T2D mice compared with controls (Fig. 2D). Pretreatment with all inhibitors (l-NAME, Indo, Apa, and ChTx) diminished EDR in both groups (Fig. 2E). Pretreatment with Indo alone did not affect ACh-induced relaxation in CAs from control and T2D mice (data not shown). These data suggest that an attenuated NO-mediated relaxation contributes to the decrease in ACh-induced relaxation in CAs from T2D mice, while prostacyclin- and endothelium-derived hyperpolarization-dependent relaxation was not altered in CAs from T2D mice.
Fig. 2.

Nitric oxide (NO)-dependent vascular relaxation, but not endothelium-dependent hyperpolarization- or prostacyclin-induced relaxation, is significantly decreased in diabetic coronary arteries (CAs) compared with control. A: dose-response curves of ACh-induced relaxation in CAs from control (n = 11) and T2D (n = 10) mice. B: dose-response curves of sodium nitroprusside (SNP)-induced relaxation in CAs from control (n = 5) and T2D (n = 4) mice. C: ACh-induced vascular response in the presence of N-nitro-l-arginine methyl ester (l-NAME, 100 μmol/l) and indomethacin (Indo, 10 μmol/l) in CAs from control (n = 5) and T2D (n = 4) mice. D: ACh-induced vascular response in the presence of Indo and the Ca2+-activated K+ channel blockers apamin (Apa, 100 nmol/l) and charybdotoxin (ChTx, 100 nmol/l) in CAs from control (n = 5) and T2D (n = 4) mice. E: ACh-induced relaxation in the presence of l-NAME, Indo, and Ca2+-activated K+ channel blockers in CAs from control (n = 4) and T2D (n = 5) mice. Values are means ± SE. *P < 0.05 vs. Cont.
Increased Mitochondrial ROS Concentration in Coronary ECs From T2D Mice
We examined whether mitochondrial ROS concentration was altered in coronary ECs from T2D mice. Coronary ECs were isolated from control and T2D mice and cultured on coverslips. Figure 3, A and B, demonstrates a significant increase of mitochondrial ROS in coronary ECs from T2D mice compared with controls. In addition, the increased ROS in coronary ECs from T2D mice was decreased to the level in the control mice by pretreatment with 10 μmol/l mitoTempol (a mitochondria-specific O2− scavenger).
Fig. 3.
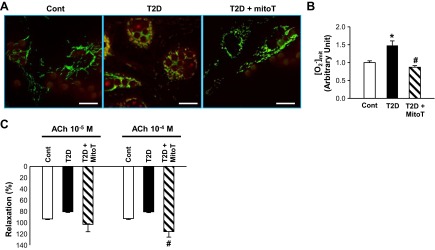
Effect of mitoTempol on mitochondrial reactive oxygen species (ROS) concentration in mouse coronary endothelial cells (ECs) and endothelium-dependent vascular relaxation in diabetes. A: representative images showing mitochondrial ROS concentration in primary cultured coronary ECs isolated from control and T2D mice and ECs from T2D mice treated with mitoTempol (mitoT, a mitochondrial O2− scavenger, 10 μmol/l). Scale bars, 10 μm. B: summarized data showing average of mitochondrial ROS intensity in primary cultured coronary ECs from control (n = 203), T2D (n = 175), and mitoT-treated T2D (n = 177) mice. C: ACh-induced vascular relaxation in control (n = 12), T2D (n = 13), and mitoT-treated (100 μmol/l) T2D (n = 3) mice. Values are means ± SE. *P < 0.05 vs. Cont. #P < 0.05 vs. T2D.
mitoTempol Treatment Restores ACh-Induced Relaxation in Diabetic CA
To determine the role of O2− in decreased EDR in CAs from T2D mice, CAs were pretreated with mitoTempol (100 μmol/l) for 20 min. As shown in Fig. 3C, mitoTempol treatment augmented EDR in CAs from T2D mice toward the level in controls. These data suggest that increased O2− production leads to decreased EDR in CAs from T2D mice.
Decreased SOD2 Protein Expression in Coronary ECs from T2D Mice
To determine which subtypes of SOD protein expression were altered, we used coronary ECs freshly isolated from control and T2D mice. As shown in Fig. 4, SOD2 protein expression was significantly decreased in coronary ECs from T2D mice compared with controls [1.03 ± 0.07 (n = 8) and 0.66 ± 0.12 (n = 9) in control and T2D, respectively, P = 0.02], whereas SOD1 protein expression level was not changed in coronary ECs from T2D mice.
Fig. 4.

SOD2 protein expression is significantly decreased in coronary ECs isolated from T2D mice compared with controls. Top: Western blots showing SOD1 (A) and SOD2 (B) and actin protein levels. Actin was used as a loading control. Bottom: SOD1 and SOD2 protein levels normalized by actin. Values are means ± SE; n = 4 Cont and 4 T2D (A) and n = 8 Cont and 9 T2D (B). *P < 0.05 vs. Cont.
Protein Ubiquitination of SOD2
To identify the mechanism whereby SOD2 protein expression was decreased in coronary ECs from T2D mice, we first measured and compared SOD2 mRNA level in coronary ECs isolated from control and T2D mice. Figure 5A demonstrates no difference of SOD2 mRNA expression between coronary ECs from control and T2D mice. Next, we determined the level of SOD2 protein ubiquitination in coronary ECs isolated from control and T2D mice. Coronary ECs from T2D mice exhibit significant increase in the level of ubiquitinated SOD2 protein compared with coronary ECs from controls (Fig. 5B). These data suggest that increased mitochondrial O2− in coronary ECs from T2D mice may be due to decreased SOD2 protein expression via the increase in ubiquitination of SOD2 protein in coronary ECs from T2D mice.
Fig. 5.

Ubiquitination of SOD2 protein is significantly increased in coronary ECs from T2D mice. A: SOD2 mRNA expression level in coronary ECs isolated from control and T2D mice detected by real-time PCR. B: immunoprecipitation (IP) with anti-ubiquitin (Ub) antibody and immunoblotting (IB) with anti-SOD2 antibody in coronary ECs from control and T2D mice. Values are means ± SE; n = 5 Cont and 5 T2D. *P < 0.05 vs. Cont.
High-Glucose Treatment Increases Protein Ubiquitination of SOD2
We used human coronary ECs to examine the effect of high-glucose or high-FFA treatment on ubiquitination of SOD2 protein. At 48 h after treatment, cells were lysed and used for the IP study. High-glucose treatment significantly increased the level of ubiquitinated SOD2 protein compared with control (1.03 ± 0.06 and 2.00 ± 0.52 with normal and high glucose, respectively, P < 0.05; Fig. 6A), whereas FFA treatment did not affect the level of SOD2 protein ubiquitination (Fig. 6B). These data suggest that increased mitochondrial O2− in coronary ECs from T2D mice may be due to decreased SOD2 protein expression via the increase in ubiquitination of SOD2 protein by hyperglycemia.
DISCUSSION
EC dysfunction is the leading cause of cardiovascular complications in diabetes (5, 19, 73, 80, 88); however, the mechanisms that cause EC dysfunction were varied between the different organs and different animal models that were used in the experiments. Our T2D mice exhibit hyperglycemia, hyperinsulinemia, and hyperlipidemia with significant increase in their body weight (Table 1, Fig. 1); this model is very close to human T2D induced by a Western diet (27, 39, 49, 53). Coronary heart disease is the common cause of death in T2D patients, and it is mainly induced by coronary atherosclerosis, cardiac ischemia, and, in some cases, by coronary vasospasm. Although coronary EC dysfunction is implicated in these vascular complications, only one report demonstrated attenuation of coronary EC function in patients with T2D and used human samples to show detailed molecular mechanisms (4). Several reports demonstrate coronary EC dysfunction in db/db (genetically derived T2D) mice (6, 13, 30, 66) and in Otsuka Long-Evans Tokushima Fatty rats (a spontaneous model of T2D) (40). EDR was attenuated in db/db mice mainly due to the decrease in NO production (6, 13), while there was no change in NO production and NO-mediated vascular relaxation between Otsuka Long-Evans Tokushima Fatty and control rats (40). In our T2D mice, we found that NO-dependent relaxation was significantly attenuated compared with controls (Fig. 2). Furthermore, scavenging mitochondrial O2− by mitoTempol restored attenuated ACh-induced relaxation in CAs of T2D mice (Fig. 3C). O2− is a ROS that reacts quickly with NO in the cells and forms peroxynitrite, which no longer stimulates cGMP production in smooth muscle cells. Our data imply that NO bioavailability was decreased by excess O2− production in coronary ECs from T2D mice.
Increased ROS (including O2−) and its maladaptive effects on vascular function have been demonstrated in patients with T2D (11, 18, 24). Augmented ROS production in the vessels has also been shown in T2D mice (8, 21, 38, 74, 82) and rats (32, 70–72), but none of these studies examined coronary ECs. We demonstrated, for the first time, that mitochondrial ROS concentration was significantly increased in coronary ECs from T2D mice compared with controls and that the increased ROS concentration in coronary ECs from T2D mice was restored toward the level in controls by treatment with mitoTempol (Fig. 3, A and B). This direct evidence suggests that attenuated ACh-induced relaxation in diabetic CAs is caused by the increased O2− in coronary ECs, but not SMCs, which is why SNP-induced vascular relaxation was not altered in diabetic CAs.
There are various sources of O2− generation in ECs, including NOX, the mitochondrial ETC, xanthine oxidase, uncoupled eNOS, and cytochrome P-450 (51). High-glucose treatment enhances mitochondrial O2− production by alteration of mitochondrial complex III activity in rat renal proximal tubular cells (65) or by phosphorylation of p47phox (interaction with NOX1) in rat aortic vascular SMCs (52). Treatment with oxidized LDL increases the activity of complex I in mitochondria of human umbilical vein ECs (7). In femoral arteries of type 1 diabetic rats, an increase in gp91phox (also known as NOX2) is followed by excess O2− production (23). NOX1 knockout mice do not develop endothelial dysfunction in the aorta in type 1 diabetic mice (86). NOX inhibitor restores the level of O2− toward the control in coronary SMCs of T2D mice (30). Excess mitochondrial O2− production and impaired mitochondrial antioxidant defense result in increased mitochondrial O2−. Mitochondrial O2− generated by the ETC is largely released to the matrix at complex I and the intermembrane space at complex III. SOD2 (also known as MnSOD) in the matrix and SOD1 (also known as Cu/ZnSOD) in the intermembrane space catalyze the dismutation of O2− to H2O2. H2O2 is detoxified to H2O in the matrix by catalase, the thioredoxin-thioredoxin peroxidase system, or the glutathione-glutathione peroxidase system. Figure 4 demonstrates that SOD2, but not SOD1, was significantly decreased in coronary ECs isolated from T2D mice compared with controls, suggesting that the decrease in SOD2 may contribute to the increase of mitochondrial ROS in coronary ECs from T2D mice. Our observation is in line with other investigators' data using other tissue samples in T2D (3, 46, 61, 63), while some data show the increase in SOD2 protein expression (17, 33, 59) or no change (69, 77). Possible explanations for these different outcomes might be that their model may not increase and/or change the level of mitochondrial O2− that was not directly measured in their study.
Contrary to our expectation, SOD2 mRNA expression was not changed in coronary ECs from T2D mice compared with controls (Fig. 5A). The level of protein expression is regulated by translational modification (e.g., RNA stability) and posttranslational modification (e.g., protein stability). The Ub-proteasome system is the major intracellular proteolytic system responsible for degradation of large amounts of proteins. Dysregulation of the Ub-proteasome system has been implicated in the development of cardiovascular diseases (76, 81), and T2D is no exception (15, 16, 43, 60). Although eNOS is not ubiquitinated (47), Stangl and Stangl (76) reported that the inhibition of proteasome increases eNOS protein expression via regulation of its transcription factors, and subsequently augments EDR (84). SOD1 and SOD2 protein expression levels are regulated by ubiquitination (45, 85), and SOD2 protein ubiquitination is enhanced by oxidized LDL treatment in human aortic ECs (79). We demonstrated that coronary ECs from T2D mice significantly increased the ubiquitination of SOD2 protein (Fig. 5B). In addition, high-glucose, but not high-fat, treatment ex vivo significantly increased SOD2 ubiquitination (Fig. 6).
What is the upstream mechanism in which the ubiquitination of SOD2 protein is increased in T2D mice? The potential mechanism might be, at least in part, regulation of the ubiquitination-deubiquitination system by ROS. There is increasing evidence that the activity of deubiquitination enzymes, which dissociate Ub from the substrate protein and prevent protein degradation, is negatively regulated by ROS (14, 48, 50). Excess ROS production also inhibits the assembly of 26S proteasomes, which results in a decrease of protein ubiquitination (1, 37, 83). Further experiments are required to identify how mitochondrial ROS affects mitochondrial protein ubiquitination.
Taken together, these data suggest that increased mitochondrial ROS generation in coronary ECs from T2D mice is due to decreased SOD2 protein expression as a result of ubiquitination. The increased mitochondrial ROS generation is partially responsible for impaired EDR in diabetic CAs by decreasing NO bioavailability.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant HL-115578 (to A. Makino).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.-E.C., A.B., A.D., M.H., and A.M. performed the experiments; Y.-E.C., A.B., A.D., and A.M. analyzed the data; Y.-E.C., A.B., A.D., and A.M. prepared the figures; Y.-E.C. drafted the manuscript; Y.-E.C., A.B., A.D., M.H., and A.M. approved the final version of the manuscript; A.M. is responsible for conception and design of the research; A.M. interpreted the results of the experiments; A.M. edited and revised the manuscript.
ACKNOWLEDGMENTS
A. Makino is the recipient of the New Investigator Award from the Cell and Molecular Physiology section of the American Physiological Society.
REFERENCES
- 1.Aiken CT, Kaake RM, Wang X, Huang L. Oxidative stress-mediated regulation of proteasome complexes. Mol Cell Proteomics 10: R110.006924, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alp NJ, Mussa S, Khoo J, Cai S, Guzik T, Jefferson A, Goh N, Rockett KA, Channon KM. Tetrahydrobiopterin-dependent preservation of nitric oxide-mediated endothelial function in diabetes by targeted transgenic GTP-cyclohydrolase I overexpression. J Clin Invest 112: 725–735, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bauer S, Wanninger J, Neumeier M, Wurm S, Weigert J, Kopp A, Bala M, Schaffler A, Aslanidis C, Buechler C. Elevated free fatty acids and impaired adiponectin bioactivity contribute to reduced SOD2 protein in monocytes of type 2 diabetes patients. Exp Mol Pathol 90: 101–106, 2011 [DOI] [PubMed] [Google Scholar]
- 4.Beleznai T, Feher A, Spielvogel D, Lansman SL, Bagi Z. Arginase 1 contributes to diminished coronary arteriolar dilation in patients with diabetes. Am J Physiol Heart Circ Physiol 300: H777–H783, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belin de Chantemele EJ, Stepp DW. Influence of obesity and metabolic dysfunction on the endothelial control in the coronary circulation. J Mol Cell Cardiol 52: 840–847, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belmadani S, Palen DI, Gonzalez-Villalobos RA, Boulares HA, Matrougui K. Elevated epidermal growth factor receptor phosphorylation induces resistance artery dysfunction in diabetic db/db mice. Diabetes 57: 1629–1637, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ceaser EK, Ramachandran A, Levonen AL, Darley-Usmar VM. Oxidized low-density lipoprotein and 15-deoxy-Δ12,14-PGJ2 increase mitochondrial complex I activity in endothelial cells. Am J Physiol Heart Circ Physiol 285: H2298–H2308, 2003 [DOI] [PubMed] [Google Scholar]
- 8.Cheang WS, Wong WT, Tian XY, Yang Q, Lee HK, He GW, Yao X, Huang Y. Endothelial nitric oxide synthase enhancer reduces oxidative stress and restores endothelial function in db/db mice. Cardiovasc Res 92: 267–275, 2011 [DOI] [PubMed] [Google Scholar]
- 9.Chen AF, Chen DD, Daiber A, Faraci FM, Li H, Rembold CM, Laher I. Free radical biology of the cardiovascular system. Clin Sci (Lond) 123: 73–91, 2012 [DOI] [PubMed] [Google Scholar]
- 10.Chen G, Suzuki H, Weston AH. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br J Pharmacol 95: 1165–1174, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen LL, Yu F, Zeng TS, Liao YF, Li YM, Ding HC. Effects of gliclazide on endothelial function in patients with newly diagnosed type 2 diabetes. Eur J Pharmacol 659: 296–301, 2011 [DOI] [PubMed] [Google Scholar]
- 12.Chen X, Touyz RM, Park JB, Schiffrin EL. Antioxidant effects of vitamins C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke-prone SHR. Hypertension 38: 606–611, 2001 [DOI] [PubMed] [Google Scholar]
- 13.Choi SK, Galan M, Kassan M, Partyka M, Trebak M, Matrougui K. Poly(ADP-ribose) polymerase 1 inhibition improves coronary arteriole function in type 2 diabetes mellitus. Hypertension 59: 1060–1068, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clague MJ. Biochemistry: oxidation controls the DUB step. Nature 497: 49–50, 2013 [DOI] [PubMed] [Google Scholar]
- 15.Costes S, Huang CJ, Gurlo T, Daval M, Matveyenko AV, Rizza RA, Butler AE, Butler PC. Beta-cell dysfunctional ERAD/ubiquitin/proteasome system in type 2 diabetes mediated by islet amyloid polypeptide-induced UCH-L1 deficiency. Diabetes 60: 227–238, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costes S, Vandewalle B, Tourrel-Cuzin C, Broca C, Linck N, Bertrand G, Kerr-Conte J, Portha B, Pattou F, Bockaert J, Dalle S. Degradation of cAMP-responsive element-binding protein by the ubiquitin-proteasome pathway contributes to glucotoxicity in beta-cells and human pancreatic islets. Diabetes 58: 1105–1115, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Assis AM, Rech A, Longoni A, Rotta LN, Denardin CC, Pasquali MA, Souza DO, Perry ML, Moreira JC. ω-3-Polyunsaturated fatty acids prevent lipoperoxidation, modulate antioxidant enzymes, and reduce lipid content but do not alter glycogen metabolism in the livers of diabetic rats fed on a high fat thermolyzed diet. Mol Cell Biochem 361: 151–160, 2012 [DOI] [PubMed] [Google Scholar]
- 18.De Mattia G, Laurenti O, Fava D. Diabetic endothelial dysfunction: effect of free radical scavenging in type 2 diabetic patients. J Diabetes Complications 17: 30–35, 2003 [DOI] [PubMed] [Google Scholar]
- 19.De Vriese AS, Verbeuren TJ, Van de Voorde J, Lameire NH, Vanhoutte PM. Endothelial dysfunction in diabetes. Br J Pharmacol 130: 963–974, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deussen A, Ohanyan V, Jannasch A, Yin L, Chilian W. Mechanisms of metabolic coronary flow regulation. J Mol Cell Cardiol 52: 794–801, 2012 [DOI] [PubMed] [Google Scholar]
- 21.Dong YF, Liu L, Kataoka K, Nakamura T, Fukuda M, Tokutomi Y, Nako H, Ogawa H, Kim-Mitsuyama S. Aliskiren prevents cardiovascular complications and pancreatic injury in a mouse model of obesity and type 2 diabetes. Diabetologia 53: 180–191, 2010 [DOI] [PubMed] [Google Scholar]
- 22.Dusting GJ. Nitric oxide in coronary artery disease: roles in atherosclerosis, myocardial reperfusion and heart failure. EXS 76: 33–55, 1996 [DOI] [PubMed] [Google Scholar]
- 23.Ebrahimian TG, Heymes C, You D, Blanc-Brude O, Mees B, Waeckel L, Duriez M, Vilar J, Brandes RP, Levy BI, Shah AM, Silvestre JS. NADPH oxidase-derived overproduction of reactive oxygen species impairs postischemic neovascularization in mice with type 1 diabetes. Am J Pathol 169: 719–728, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ergul A, Johansen JS, Stromhaug C, Harris AK, Hutchinson J, Tawfik A, Rahimi A, Rhim E, Wells B, Caldwell RW, Anstadt MP. Vascular dysfunction of venous bypass conduits is mediated by reactive oxygen species in diabetes: role of endothelin-1. J Pharmacol Exp Ther 313: 70–77, 2005 [DOI] [PubMed] [Google Scholar]
- 25.Fatehi-Hassanabad Z, Chan CB, Furman BL. Reactive oxygen species and endothelial function in diabetes. Eur J Pharmacol 636: 8–17, 2010 [DOI] [PubMed] [Google Scholar]
- 26.Feletou M, Vanhoutte PM. Endothelium-derived hyperpolarizing factor: where are we now? Arterioscler Thromb Vasc Biol 26: 1215–1225, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Fricovsky ES, Suarez J, Ihm SH, Scott BT, Suarez-Ramirez JA, Banerjee I, Torres-Gonzalez M, Wang H, Ellrott I, Maya-Ramos L, Villarreal F, Dillmann WH. Excess protein O-GlcNAcylation and the progression of diabetic cardiomyopathy. Am J Physiol Regul Integr Comp Physiol 303: R689–R699, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Furchgott RF, Vanhoutte PM. Endothelium-derived relaxing and contracting factors. FASEB J 3: 2007–2018, 1989 [PubMed] [Google Scholar]
- 29.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288: 373–376, 1980 [DOI] [PubMed] [Google Scholar]
- 30.Gao X, Belmadani S, Picchi A, Xu X, Potter BJ, Tewari-Singh N, Capobianco S, Chilian WM, Zhang C. Tumor necrosis factor-α induces endothelial dysfunction in Leprdb mice. Circulation 115: 245–254, 2007 [DOI] [PubMed] [Google Scholar]
- 31.Gryglewski RJ, Palmer RM, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 320: 454–456, 1986 [DOI] [PubMed] [Google Scholar]
- 32.Gupte S, Labinskyy N, Gupte R, Csiszar A, Ungvari Z, Edwards JG. Role of NAD(P)H oxidase in superoxide generation and endothelial dysfunction in Goto-Kakizaki (GK) rats as a model of nonobese NIDDM. PLos One 5: e11800, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamed S, Brenner B, Aharon A, Daoud D, Roguin A. Nitric oxide and superoxide dismutase modulate endothelial progenitor cell function in type 2 diabetes mellitus. Cardiovasc Diabetol 8: 56, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hathaway CA, Heistad DD, Piegors DJ, Miller FJ., Jr Regression of atherosclerosis in monkeys reduces vascular superoxide levels. Circ Res 90: 277–283, 2002 [DOI] [PubMed] [Google Scholar]
- 35.Higashi Y, Sasaki S, Nakagawa K, Matsuura H, Oshima T, Chayama K. Endothelial function and oxidative stress in renovascular hypertension. N Engl J Med 346: 1954–1962, 2002 [DOI] [PubMed] [Google Scholar]
- 36.Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, Skatchkov M, Thaiss F, Stahl RA, Warnholtz A, Meinertz T, Griendling K, Harrison DG, Forstermann U, Munzel T. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res 88: E14–E22, 2001 [DOI] [PubMed] [Google Scholar]
- 37.Huang Q, Wang H, Perry SW, Figueiredo-Pereira ME. Negative regulation of 26S proteasome stability via calpain-mediated cleavage of Rpn10 subunit upon mitochondrial dysfunction in neurons. J Biol Chem 288: 12161–12174, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hwang J, Kleinhenz DJ, Rupnow HL, Campbell AG, Thule PM, Sutliff RL, Hart CM. The PPARγ ligand, rosiglitazone, reduces vascular oxidative stress and NADPH oxidase expression in diabetic mice. Vasc Pharmacol 46: 456–462, 2007 [DOI] [PubMed] [Google Scholar]
- 39.Islam MS, Loots du T. Experimental rodent models of type 2 diabetes: a review. Methods Find Exp Clin Pharmacol 31: 249–261, 2009 [DOI] [PubMed] [Google Scholar]
- 40.Kajikuri J, Watanabe Y, Ito Y, Ito R, Yamamoto T, Itoh T. Characteristic changes in coronary artery at the early hyperglycaemic stage in a rat type 2 diabetes model and the effects of pravastatin. Br J Pharmacol 158: 621–632, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalyanaraman B, Darley-Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ, 2nd, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med 52: 1–6, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kamata K, Miyata N, Abiru T, Kasuya Y. Functional changes in vascular smooth muscle and endothelium of arteries during diabetes mellitus. Life Sci 50: 1379–1387, 1992 [DOI] [PubMed] [Google Scholar]
- 43.Kaniuk NA, Kiraly M, Bates H, Vranic M, Volchuk A, Brumell JH. Ubiquitinated-protein aggregates form in pancreatic beta-cells during diabetes-induced oxidative stress and are regulated by autophagy. Diabetes 56: 930–939, 2007 [DOI] [PubMed] [Google Scholar]
- 44.Karwatowska-Prokopczuk E, Wennmalm A. Endothelium-derived constricting factor(s): the last novelty—endothelin. Clin Physiol 10: 113–121, 1990 [DOI] [PubMed] [Google Scholar]
- 45.Kim MS, Ramakrishna S, Lim KH, Kim JH, Baek KH. Protein stability of mitochondrial superoxide dismutase SOD2 is regulated by USP36. J Cell Biochem 112: 498–508, 2011 [DOI] [PubMed] [Google Scholar]
- 46.Kim MY, Lim JH, Youn HH, Hong YA, Yang KS, Park HS, Chung S, Koh SH, Shin SJ, Choi BS, Kim HW, Kim YS, Lee JH, Chang YS, Park CW. Resveratrol prevents renal lipotoxicity and inhibits mesangial cell glucotoxicity in a manner dependent on the AMPK-SIRT1-PGC1α axis in db/db mice. Diabetologia 56: 204–217, 2013 [DOI] [PubMed] [Google Scholar]
- 47.Kone BC, Kuncewicz T, Zhang W, Yu ZY. Protein interactions with nitric oxide synthases: controlling the right time, the right place, and the right amount of nitric oxide. Am J Physiol Renal Physiol 285: F178–F190, 2003 [DOI] [PubMed] [Google Scholar]
- 48.Kulathu Y, Garcia FJ, Mevissen TE, Busch M, Arnaudo N, Carroll KS, Barford D, Komander D. Regulation of A20 and other OTU deubiquitinases by reversible oxidation. Nat Commun 4: 1569, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kusakabe T, Tanioka H, Ebihara K, Hirata M, Miyamoto L, Miyanaga F, Hige H, Aotani D, Fujisawa T, Masuzaki H, Hosoda K, Nakao K. Beneficial effects of leptin on glycaemic and lipid control in a mouse model of type 2 diabetes with increased adiposity induced by streptozotocin and a high-fat diet. Diabetologia 52: 675–683, 2009 [DOI] [PubMed] [Google Scholar]
- 50.Lee JG, Baek K, Soetandyo N, Ye Y. Reversible inactivation of deubiquitinases by reactive oxygen species in vitro and in cells. Nat Commun 4: 1568, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol 287: R1014–R1030, 2004 [DOI] [PubMed] [Google Scholar]
- 52.Liu S, Ma X, Gong M, Shi L, Lincoln T, Wang S. Glucose down-regulation of cGMP-dependent protein kinase I expression in vascular smooth muscle cells involves NAD(P)H oxidase-derived reactive oxygen species. Free Radic Biol Med 42: 852–863, 2007 [DOI] [PubMed] [Google Scholar]
- 53.Luo J, Quan J, Tsai J, Hobensack CK, Sullivan C, Hector R, Reaven GM. Nongenetic mouse models of non-insulin-dependent diabetes mellitus. Metabolism 47: 663–668, 1998 [DOI] [PubMed] [Google Scholar]
- 54.Luscher TF, Tanner FC, Tschudi MR, Noll G. Endothelial dysfunction in coronary artery disease. Annu Rev Med 44: 395–418, 1993 [DOI] [PubMed] [Google Scholar]
- 55.Mackenzie RM, Salt IP, Miller WH, Logan A, Ibrahim HA, Degasperi A, Dymott JA, Hamilton CA, Murphy MP, Delles C, Dominiczak AF. Mitochondrial reactive oxygen species enhance AMP-activated protein kinase activation in the endothelium of patients with coronary artery disease and diabetes. Clin Sci (Lond) 124: 403–411, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Makino A, Platoshyn O, Suarez J, Yuan JX, Dillmann WH. Downregulation of connexin40 is associated with coronary endothelial cell dysfunction in streptozotocin-induced diabetic mice. Am J Physiol Cell Physiol 295: C221–C230, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Makino A, Scott BT, Dillmann WH. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia 53: 1783–1794, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Makino A, Wang H, Scott BT, Yuan JX, Dillmann WH. Thyroid hormone receptor-α and vascular function. Am J Physiol Cell Physiol 302: C1346–C1352, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Manavalan JS, Cremers S, Dempster DW, Zhou H, Dworakowski E, Kode A, Kousteni S, Rubin MR. Circulating osteogenic precursor cells in type 2 diabetes mellitus. J Clin Endocrinol Metab 97: 3240–3250, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marfella R, D'Amico M, Di Filippo C, Siniscalchi M, Sasso FC, Ferraraccio F, Rossi F, Paolisso G. The possible role of the ubiquitin proteasome system in the development of atherosclerosis in diabetes. Cardiovasc Diabetol 6: 35, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marrotte EJ, Chen DD, Hakim JS, Chen AF. Manganese superoxide dismutase expression in endothelial progenitor cells accelerates wound healing in diabetic mice. J Clin Invest 120: 4207–4219, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Masha A, Dinatale S, Allasia S, Martina V. Role of the decreased nitric oxide bioavailability in the vascular complications of diabetes mellitus. Curr Pharm Biotechnol 12: 1354–1363, 2011 [DOI] [PubMed] [Google Scholar]
- 63.Moien-Afshari F, Ghosh S, Elmi S, Rahman MM, Sallam N, Khazaei M, Kieffer TJ, Brownsey RW, Laher I. Exercise restores endothelial function independently of weight loss or hyperglycaemic status in db/db mice. Diabetologia 51: 1327–1337, 2008 [DOI] [PubMed] [Google Scholar]
- 64.Moncada S. Nitric oxide in the vasculature: physiology and pathophysiology. Ann NY Acad Sci 811: 60–69, 1997 [DOI] [PubMed] [Google Scholar]
- 65.Munusamy S, MacMillan-Crow LA. Mitochondrial superoxide plays a crucial role in the development of mitochondrial dysfunction during high glucose exposure in rat renal proximal tubular cells. Free Radic Biol Med 46: 1149–1157, 2009 [DOI] [PubMed] [Google Scholar]
- 66.Park Y, Capobianco S, Gao X, Falck JR, Dellsperger KC, Zhang C. Role of EDHF in type 2 diabetes-induced endothelial dysfunction. Am J Physiol Heart Circ Physiol 295: H1982–H1988, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Potenza MA, Gagliardi S, Nacci C, Carratu MR, Montagnani M. Endothelial dysfunction in diabetes: from mechanisms to therapeutic targets. Curr Med Chem 16: 94–112, 2009 [DOI] [PubMed] [Google Scholar]
- 68.Rubanyi GM. Endothelium-derived relaxing and contracting factors. J Cell Biochem 46: 27–36, 1991 [DOI] [PubMed] [Google Scholar]
- 69.Santl Letonja M, Letonja M, Ikolajevic-Starcevic JN, Petrovic D. Association of manganese superoxide dismutase and glutathione S-transferases genotypes with carotid atherosclerosis in patients with diabetes mellitus type 2. Int Angiol 31: 33–41, 2012 [PubMed] [Google Scholar]
- 70.Sartoretto JL, Oliveira MA, Nigro D, Carvalho MH, Tostes RC, Fortes ZB. Constrictor responses to noradrenaline, hemodynamic profile, and superoxide levels measured by hydroethidine oxidation in diabetic rats. Biol Pharm Bull 30: 1938–1942, 2007 [DOI] [PubMed] [Google Scholar]
- 71.Sena CM, Nunes E, Louro T, Proenca T, Fernandes R, Boarder MR, Seica RM. Effects of α-lipoic acid on endothelial function in aged diabetic and high-fat fed rats. Br J Pharmacol 153: 894–906, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Serpillon S, Floyd BC, Gupte RS, George S, Kozicky M, Neito V, Recchia F, Stanley W, Wolin MS, Gupte SA. Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose-6-phosphate dehydrogenase-derived NADPH. Am J Physiol Heart Circ Physiol 297: H153–H162, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sharma A, Bernatchez PN, de Haan JB. Targeting endothelial dysfunction in vascular complications associated with diabetes. Int J Vasc Med 2012: 750126, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shen M, Sun D, Li W, Liu B, Wang S, Zhang Z, Cao F. The synergistic effect of valsartan and LAF237 [(S)-1-[(3-hydroxy-1-adamantyl)ammo]acetyl-2-cyanopyrrolidine] on vascular oxidative stress and inflammation in type 2 diabetic mice. Exp Diabetes Res 2012: 146194, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shi Y, Vanhoutte PM. Reactive oxygen-derived free radicals are key to the endothelial dysfunction of diabetes. J Diabetes 1: 151–162, 2009 [DOI] [PubMed] [Google Scholar]
- 76.Stangl K, Stangl V. The ubiquitin-proteasome pathway and endothelial (dys)function. Cardiovasc Res 85: 281–290, 2010 [DOI] [PubMed] [Google Scholar]
- 77.Suge R, Shimazu T, Hasegawa H, Inoue I, Hayashibe H, Nagasaka H, Araki N, Katayama S, Nomura M, Watanabe S. Cerebral antioxidant enzyme increase associated with learning deficit in type 2 diabetes rats. Brain Res 1481: 97–106, 2012 [DOI] [PubMed] [Google Scholar]
- 78.Suvorava T, Kojda G. Reactive oxygen species as cardiovascular mediators: lessons from endothelial-specific protein overexpression mouse models. Biochim Biophys Acta 1787: 802–810, 2009 [DOI] [PubMed] [Google Scholar]
- 79.Takabe W, Li R, Ai L, Yu F, Berliner JA, Hsiai TK. Oxidized low-density lipoprotein-activated c-Jun NH2-terminal kinase regulates manganese superoxide dismutase ubiquitination: implication for mitochondrial redox status and apoptosis. Arterioscler Thromb Vasc Biol 30: 436–441, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tousoulis D, Kampoli AM, Stefanadis C. Diabetes mellitus and vascular endothelial dysfunction: current perspectives. Curr Vasc Pharmacol 10: 19–32, 2012 [DOI] [PubMed] [Google Scholar]
- 81.Willis MS, Townley-Tilson WH, Kang EY, Homeister JW, Patterson C. Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ Res 106: 463–478, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wong WT, Tian XY, Xu A, Ng CF, Lee HK, Chen ZY, Au CL, Yao X, Huang Y. Angiotensin II type 1 receptor-dependent oxidative stress mediates endothelial dysfunction in type 2 diabetic mice. Antioxid Redox Signal 13: 757–768, 2010 [DOI] [PubMed] [Google Scholar]
- 83.Xu J, Wang S, Zhang M, Wang Q, Asfa S, Zou MH. Tyrosine nitration of PA700 links proteasome activation to endothelial dysfunction in mouse models with cardiovascular risk factors. PLos One 7: e29649, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 84.Xu J, Wu Y, Song P, Zhang M, Wang S, Zou MH. Proteasome-dependent degradation of guanosine 5′-triphosphate cyclohydrolase I causes tetrahydrobiopterin deficiency in diabetes mellitus. Circulation 116: 944–953, 2007 [DOI] [PubMed] [Google Scholar]
- 85.Ying Z, Wang H, Fan H, Zhu X, Zhou J, Fei E, Wang G. Gp78, an ER associated E3, promotes SOD1 and ataxin-3 degradation. Hum Mol Genet 18: 4268–4281, 2009 [DOI] [PubMed] [Google Scholar]
- 86.Youn JY, Gao L, Cai H. The p47phox- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia 55: 2069–2079, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang DX, Gutterman DD. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am J Physiol Heart Circ Physiol 292: H2023–H2031, 2007 [DOI] [PubMed] [Google Scholar]
- 88.Zhang H, Dellsperger KC, Zhang C. The link between metabolic abnormalities and endothelial dysfunction in type 2 diabetes: an update. Basic Res Cardiol 107: 237, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]