

Abstract
Hypoglossal motoneurons (HNs) control tongue movement and play a role in maintenance of upper airway patency. Defects in these neurons may contribute to the development of sleep apnea and other cranial motor disorders including Rett syndrome (RTT). HNs are modulated by norepinephrine (NE) through α-adrenoceptors. Although postsynaptic mechanisms are known to play a role in this effect, how NE modulates the synaptic transmissions of HNs remains poorly understood. More importantly, the NE system is defective in RTT, while how the defect affects HNs is unknown. Believing that information of NE modulation of HNs may help the understanding of RTT and the design of new therapeutical interventions to motor defects in the disease, we performed these studies in which glycinergic inhibitory postsynaptic currents and intrinsic membrane properties were examined in wild-type and Mecp2−/Y mice, a mouse of model of RTT. We found that activation of α1-adrenoceptor facilitated glycinergic synaptic transmission and excited HNs. These effects were mediated by both pre- and postsynaptic mechanisms. The latter effect involved an inhibition of barium-sensitive G protein-dependent K+ currents. The pre- and postsynaptic modulations of the HNs by α1-adrenoceptors were not only retained in Mecp2-null mice but also markedly enhanced, which appears to be a compensatory mechanism for the deficiencies in NE and GABAergic synaptic transmission. The existence of the endogenous compensatory mechanism is an encouraging finding, as it may allow therapeutical modalities to alleviate motoneuronal defects in RTT.
Keywords: α1-adrenoceptor, hypoglossal motorneurons, IPSCs, norepinephrine, Rett syndrome
hypoglossal motoneurons (HNs) control tongue movements and play a role in mastication, swallowing, sucking, and maintenance of upper airway patency (4, 20, 39). Defects in these neurons may contribute to the development of sleep apnea and other cranial motor disorders including Rett syndrome (RTT). The HNs receive excitatory and inhibitory synaptic inputs (1, 7, 19, 47, 51, 69). The main source of GABAergic and glycinergic inhibitory inputs to the HNs derives from the nucleus of Roller (2, 62), which affect the HNs via activation of postsynaptic GABAA and glycine receptors (2, 5, 52, 62). These inhibitory synaptic inputs control hypoglossal responses to other synaptic inputs, shape the temporospatial pattern of neuronal activity during reflex and rhythmic behaviors, and contribute to the generation of inspiratory motoneuronal synchrony (5, 49, 53).
The strength of these synaptic inhibitions of HNs is modulated by brainstem norepinephrine (NE) systems by α-adrenoceptors (18). The α-adrenoceptors contain two main types including α1 and α2, each comprising at least three subtypes (13, 42). The α1A-, α1B-, and α1C-adrenoceptors are expressed in cranial and spinal motoneurons (14, 34). The α2A- and α2C-receptors are also expressed in motoneurons at relatively low levels (45, 57), while α2B is expressed only in thalamic neurons (44, 45, 57). The β1- and β2- but not β3-adrenoceptors are expressed in the brain (45), although evidence for their expression in motoneurons remains controversial (44, 48, 50). In the brainstem and the spinal cord, activation of α1-adrenoceptor produces a large variety of excitatory postsynaptic effects in motoneurons (52). In addition, there is also compelling evidence suggesting that the α1-adrenoceptor mediates facilitation of GABAergic transmission of neurons in the hippocampus (40), frontal cortex (8), cerebellum (27), nucleus ambiguous (9), and olfactory bulb (43). In the hypoglossal nucleus, the α1-adrenoceptor is involved in NE-mediated depolarization associated with an increase in input resistance, augmentation of spike frequency, and a decrease in afterhyperpolarization (6, 46, 49, 60). In contrast to these postsynaptic effects, how presynaptic α1-adrenoceptors act on synaptic transmission of HNs is poorly understood. Such presynaptic effects are likely to exist, as experimental evidence suggests that α1-adrenoceptor agonists increase glycinergic transmission of motoneurons in the nucleus ambiguus and the spinal cord (3, 9, 20, 24).
The synaptic modulation of HNs may be affected in certain diseases. RTT is a neurodevelopmental disorder caused by disruptions of the MECP2 gene (21, 58). The NE content is deficient in people with RTT and the Mecp2−/Y mice, mouse models of RTT (29, 61, 65). Our laboratory has also demonstrated that dopamine-β-hydroxylase (DBH), the critical enzyme converting dopamine to NE, is significantly reduced without a loss of the DBH-containing neurons in the locus coeruleus (LC; Ref. 73), and such alternations affect the intrinsic membrane properties and CO2 chemosensitivity of the LC neurons (71, 72). Several recent studies have demonstrated defects in the γ-aminobutyric acid (GABA) system in RTT patients and the mouse models of RTT (12, 28, 30, 41, 55, 74). Indeed, knockout of Mecp2 in a subset of forebrain GABAAergic neurons in mice reveals many features of RTT (12). The GABAAergic synaptic transmission in LC neurons and neurons in the ventrolateral medulla is drastically depressed in Mecp2-null mice (12, 30, 41). This as well as the fact that the GABAergic synaptic transmission is augmented by the NE system suggests that the joined disruptions of NE and GABA systems may abrogate, to a large degree, the GABAergic system. Such defects should have profound impact on the inhibitory synaptic transmissions, especially in the motorneuronal system including HNs, as motor defects are characteristic in people with RTT (11, 22, 53). Under the condition of GABAergic defects, the motoneuronal system may rely more on glycinergic system, because the glycinergic system plays an important role in inhibitory synaptic transmission in motoneurons (5, 59, 67), suggesting the necessity to know how the Mecp2 disruption affects the glycinergic system. Therefore, we performed whole cell patch-clamp studies to determine what happens to the glycinergic synaptic currents in Mecp2-null mice and how the modulation of glycinergic synaptic transmission by α1-adrenoceptor is affected.
EXPERIMENTAL PROCEDURES
Animal.
The Mecp2−/Y male mice were produced by cross-breeding the Mecp2+/− females with the WT C57BL/6 males (Jackson Laboratories, Bar Harbor, ME). The offspring were routinely genotyped with a PCR to confirm the absence of Mecp2 gene (30, 71, 73). The Mecp2−/Y males (13–15 days postnatal) were used in the present study as a mouse model of RTT, while their male littermates served as the WT control. Because of the low availability of these mice, nonlittermate, normal C57BL/6 mice (all males) were also used to increase the sample sizes of the WT group. All experimental procedures using animals were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Georgia State University Institutional Animal Care and Use Committee.
Slice preparation.
All electrophysiological experiments were performed on slices from mice. The mice were anesthetized by inhalation of saturated isoflurane and then decapitated. The brainstem was removed and quickly submerged in the ice-cold oxygenated sucrose-rich artificial cerebrospinal fluid (sucrose aCSF) buffer containing the following (in mM): 200 sucrose, 3 KCl, 2 CaCl2, 2 MgCl2, 26 NaHCO3, 1. 25 NaH2PO4, and 10 d-glucose. The solution was bubbled with 95% O2-5% CO2 (pH 7. 40). The transverse medullary slices (210–250 μM) were made on a Vibratome 1000 (Vibratome, St. Louis, MO). Slices were transferred to normal aCSF in which the sucrose was substituted with 124 mM NaCl and allowed to recover at 33°C for 1 h. Slices were then transferred to a recording chamber and perfused with oxygenated aCSF containing the following (in mM): 124 NaCl, 2.5 KCl, 1.0 NaH2PO4, 1.3 MgSO4, 10-d-glucose, 26 NaHCO3, and 2 CaCl2 (pH 7. 40) with 95% O2-5%CO2 bubbling through it at a rate of 3 ml/min and maintained at 32–35°C (osmolarity ∼310 mosM).
Whole cell patch-clamp recordings.
Whole cell patch-clamp recordings were performed as described previously (30, 32, 33). During the recording, the slice was maintained fully submerged in the recording chamber and perfused with oxygenated aCSF. HNs were visualized by IR-differential interference contrast microscopy using a ×40 water immersion objective (Carol Zeiss). The patch pipettes were pulled from borosilicate glass using the Sutter pipette puller (Model P-97, Novato, CA) and measured 3–5 MΩ when filled with an intracellular patch solution. Tight-seal (>1 GΩ) whole cell recording was obtained from the cell body of HNs. Cells were accepted for further analysis only when their membrane potential was more negative than −60 mV and the action potential amplitude >50 mV. The input resistance of individual neurons was calculated by the ratio of membrane potential at the 400 ms of command current.
For whole cell voltage-clamp experiments, patch pipettes were filled with the internal solution containing the following (in mM): 125 KCl, 10 NaCl, 1 CaCl2, 2 MgCl2, 10 HEPES, 0.5 EGTA, 2 Mg-ATP, 0.3 GTP, and 5 N-(2, 6-dimethylphenylcarbamoylmethyl) triethylammonium bromide (QX314; pH 7.4; ∼310 mosM). The slice was perfused with aCSF with 95% O2-5% CO2, neurons were voltage-clamped at a holding potential of −70 mV, and spontaneous inhibitory postsynaptic currents (sIPSCs) were recorded with an Axopatch 200B amplifier (Molecular Devices, Union City, CA). Signals were filtered at 2 kHz and digitized at 20 kHz. The series resistances (15∼20 mΩ) and holding current were regularly monitored during recording, and neurons were rejected if the resistance changed 20% or holding current altered >100 pA. Concerning the largest currents 200 pA, the maximal space clamp error is 4 mV. To isolate glycine receptor-mediated sIPSCs, 6-cyano-7-nitroquinoxaline-2, 3-dione (CNQX; 20 μM), dl-2-amino-5-phosphonopentanoic acid (dl-AP5; 10 μM) and bicuculline (10 μM) were added to the aCSF. The miniature (m)IPSCs were recorded at a holding potential of −70 mV in the presence of CNQX, dl-AP5, and tetrodotoxin (TTX; 1 μM). The theoretical chloride reversal potential was ∼2.1 mV. For whole cell current-clamp recording, the internal solution contained the following (in mM): 120 potassium gluconate, 20 KCl, 2 MgCl2, 1 CaCl2, 0.5 EGTA, 10 HEPES, 2 Mg ATP, and 0.3 GTP pH 7.4 (300–310 mosM). In some experiments, the G-protein inhibitor guanosine 5′-O-(2-thiodiphosphate) (GDP-β-S; 1 mM) was placed in the recording pipette for both voltage- and current-clamp experiments (15). dl-AP5, CNQX, bicuculline, strychnine, phenylephrine (PE), and TTX and were purchased from Tocris Cookson (Ellisville, MO). All compounds were aliquoted and stored at −20°C as a concentrated solution (×1,000) and diluted into the aCSF immediately before use.
Data analysis.
Signals were further filtered at 1-kHz offline and were analyzed using Clampfit 10.3 software (Molecular Devices) and Mini Analysis software (Synaptosotf, Fort Lee, NJ). The cumulative probability distributions were compared by the Kolmogorov-Smirnov test. Data are expressed as means ± SE. Statistical significance was Student's t-test and Mann-Whitney test. Difference was considered significant when P < 0.05.
RESULTS
Modulation of postsynaptic membrane properties of HNs by postsynaptic α1-adrenoceptors in WT mice.
To determine the postsynaptic effects of α1-adrenoceptor activation, we performed whole cell current-clamp studies in HNs. As described in a previous study in rats (64), the hypoglossal nucleus in mice contained a heterogeneous population of neurons that can be classified into two main subtypes on the base of their firing patterns: neurons with a decrementing or adapting firing pattern (type D) and neurons with an incrementing or accelerating firing pattern (type I). At P8-P15, most of neurons are type I (64). Similarly, most HNs in our current studies belonged to type I in this age group.
Consistent with previous studies of cranial and spinal motoneurons (16, 63, 68), bath application of PE (3∼5 min) produced reversibly depolarization (Fig. 1A) and an increases in spike frequency in the presence of 10 μM bicuculline (Fig. 1, B and C). On average, the membrane potential was −65 ± 2.7 mV (n = 6 cells) before and −57 ± 3.57 mV (n = 6) during PE (40 μM) application, respectively (P < 0.01, Student's t-test; Fig. 1D). The depolarization was associated with an increase in input resistance (144 ± 18.7 MΩ, n = 6, before and 188.6 ± 20 MΩ, n = 6, during PE, respectively, P < 0.01, Student's t-test; Fig. 1E). The action potential frequency in response to the same current injection increased from 22.9 ± 3.2 Hz (n = 6) to 58.8 ± 11 Hz (n = 6) with PE treatment as well (P < 0.05, Student's t-test; Fig. 1F).
Fig. 1.
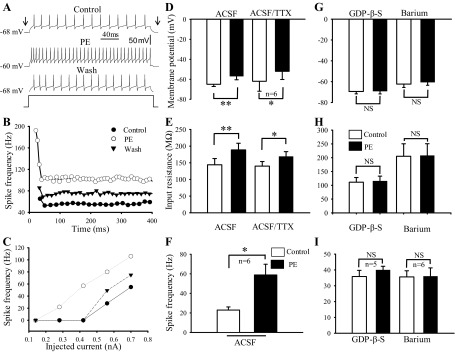
α1-Adrenoceptor agonist augmented postsynaptically hypoglossal motoneuron (HN) excitability in wild-type (WT) mice. A: records of membrane potential, input resistance and spike frequency in response to current injection in an HN in baseline control (top), during phenylephrine (PE; 40 μM) application (middle), and washout PE (bottom). B: frequency-time (f–t) relationship for the same neuron at highest current intensity. C: frequency-current (f–I) relationship for the same neuron. D and E: bar graphs showing the effect of PE on membrane potential (D) and input resistance (E) when perfused with artificial cerebrospinal fluid (aCSF) alone and aCSF plus 1 μM of tetrodotoxin (TTX). F: spike frequency to the current injection under normal aCSF perfusion. G–I: bar graphs showing the effect of PE on membrane potential (G), input resistance (H), and spike frequency (I) under the conditions that electrodes were filled with guanosine 5′-O-(2-thiodiphosphate) (GDP-β-S) and slices were perfused with aCSF plus barium. *P < 0.05; **P < 0.01; NS, not significant; n, number of neurons examined.
The PE-induced excitation existed when the perfusate contained 1 μM TTX but was blocked when patch electrodes were filled with 1 mM GDP-β-S. In the presence of TTX, the membrane potential was −62 ± 9.95 mV (n = 6) before and −52 mV ± 8.2 mV (n = 6) during PE application (P < 0.01, Student's t-test; Fig. 1D), while the input resistance was 140 ± 13.7 MΩ (n = 6) in control and 169 ± 14.3 (n = 6) in the presence of PE (P < 0.001, Student's t-test; Fig. 1E). When applied 1 mM GDP-β-S in the recording pipette for >10 min, the membrane potentials were not evidently changed (−69.4 ± 2.3 mV before vs. −69.2 ± 2.7 mV during PE; P = 0.38; Fig. 1G) and neither were the input resistance (112 ± 16.9 MΩ before vs. 115 ± 17.9 MΩ during PE; P = 0.13; Fig. 1H) and the action potential frequency (35.8 ± 3.9 Hz before vs. 40 ± 2.3 Hz during PE; P = 0.12; Fig. 1I) in five HNs (Student's t-test). These results indicate that activation of α1-adrenoceptors augments HN excitability by G protein-dependent postsynaptic mechanisms.
Involvement of G protein-coupled and barium-sensitive K+ channels.
The α1-adrenoceptors are metabotropic and can inhibit G protein-coupled K+ currents including inwardly rectifying K+ (GIRK) channels (10) and TASK channels/the resting leak K+ currents (49). Indeed, previous studies have suggested that at least two ionic mechanisms were involved in PE-induced motor neuron excitation, which are barium sensitive and barium insensitive, in rat facial and HNs (20, 37, 49) (for reviews, see Ref. 52). We have shown that PE depolarized the HNs associated with increased input resistance suggesting barium-sensitive mechanism may be involved in HNs. Thus the effects of PE on membrane potentials, input resistance, and action potential frequency were examined in the presence of 300 μM BaCl2, a concentration known to inhibit selectively the inward rectifier K+ channels (30, 35, 36). Under this condition, the effects of PE on membrane potentials, input resistance and action potential frequency were almost totally eliminated in five HNs (Fig. 1, G–I). The membrane potentials were −62.3 ± 3 mV before PE and −61.5 ± 2.8 mV during PE (P > 0.05, Student t-test; Fig. 1G), the input resistance was 205 ± 45 MΩ before PE and 208 ± 42 MΩ during PE (P = 0.17, Student's t-test; Fig. 1H) and action potential frequency was 35.6 ± 3.9 Hz before PE and 36 ± 5.3 Hz during PE (P = 0.8, Student's t-test; Fig. 1I).
To test whether the barium-sensitive K+ channels is responsible for PE-mediated excitation on HNs, whole cell K+ currents were recorded using a ramp voltage protocol (−150 to 10 mV) at a holding potential of −70 mV in the presence of 20 μM CNQX, 30 μM dl-AP5, 10 μM bicuculline, 1 μM strychnine, and 1 μM TTX. The inward currents were measured at −150 mV before and during PE application in five cells. The currents averaged −0.70 ± 0.05 and −0.45 ± 0.02 nA before and during PE application, respectively (P < 0.01; Fig. 2C). The PE-induced inhibition of whole cell currents was also barium sensitive, which was blocked in the presence of 300 μM BaCl2 (Fig. 2D). Taken together, these results provide further evidence suggesting that G protein-coupled and barium-sensitive K+ channels are involved in the postsynaptic effect of PE in HNs.
Fig. 2.
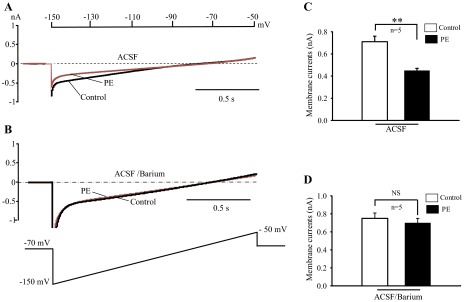
Involvement of barium-sensitive inward K+ currents in the postsynaptic effects of α1-adrenoceptor agonist. A and B: sample traces showing the effect of PE (40 μM) on ramp evoked whole cell currents at holding potential −70 mV in aCSF (A) and aCSF plus barium (B). C and D: bar graphs summarizing PE induced-inhibition on the membrane currents in aCSF (C) and aCSF plus barium (D). All experiments were performed in the present of 20 μM 6-cyano-7-nitroquinoxaline-2, 3-dione (CNQX), 30 μM dl-2-amino-5-phosphonopentanoic acid (dl-AP5), 10 μM bicuculline, 1 μM strychnine, and 1 μM TTX. **P < 0.01; n, number of neurons examined.
Modulation of postsynaptic membrane properties of HNs by postsynaptic α1-adrenoceptors in Mecp2−/Y mice.
Accumulating evidence indicates that the NE system is defective in people with RTT (75, 76) and the mouse models of RTT(29, 56, 65, 73). Such a defect may have an impact on cranial motor neurons, as motor disorders are seen in most Rett patients (11, 22, 53). Thus we performed experiments in Mecp2−/Y mice to elucidate whether the postsynaptic modulation of HN excitability by α1-adrenoceptor agonist is altered in Mecp2−/Y mice. We examined the effect of PE on membrane potentials, input resistance and action potential frequency with current injection in eight neurons. As shown in Fig. 3A, PE application clearly depolarized the HN from an Mecp2−/Y mouse and increased spike frequency to the current injection. On average, the membrane potentials, input resistance, and spike frequency before and during PE application were −71.9 ± 1.72 vs. −56.4 ± 3.3 mV (Fig. 3D; P = 0.001, Student' s t-test), 144.4 ± 17.4 vs. 196 ± 27.9 MΩ (Fig. 3E; P < 0.05, Student's t-test), and 37 ± 4.5 Hz vs. 74.9 ± 8 Hz (Fig. 3F; P < 0.01, Student's t-test), respectively. These results are similar to those obtained from WT (Fig. 1), suggesting that the effects of postsynaptic α1-adrenoceptor activation on HN membrane potentials, input resistance, and spike frequency are retained in Mecp2−/Y mice.
Fig. 3.

Postsynaptic effects of α1-adrenoceptor agonist on HN excitability in Mecp2−/Y mice. A: records of membrane potential, input resistance and spike frequency in response to current injection from an Mecp2−/Y HN in control (top) and during PE (40 μM) application (bottom). B: frequency-time (f–t) relationship for the same neuron for the highest current intensities. C: frequency-current (f–I) relationship for the same neuron. D–F: bar graphs showing the effects of PE on membrane potential (D), input resistance (E), and spike frequency (F) of HNs from Mecp2−/Y mice. *P < 0.05; **P < 0.01; n, number of neurons examined.
Effects of presynaptic α1-adrenoceptor activation on glycinergic synaptic transmission in hypoglossal motoneurons of WT mice.
Inward spontaneous postsynaptic currents were studied in HNs with high concentrations of Cl− (∼140 mM) applied to both pipette and bath solutions at a holding potential of −70 mV in voltage clamp (see experimental procedures). As shown in Fig. 4A, ionotropic glycine receptor-mediated sIPSCs were isolated by selective inhibitions of the GABAA currents with 10–30 μM bicuculline, the non-N-methyl-d-aspartate (NMDA) currents with 20 μM CNQX, and the NMDA currents with 30 μM dl-AP5. The sIPSCs were completely suppressed when strychnine was applied (0.5–1 μM; n = 3; Fig. 4A).
Fig. 4.
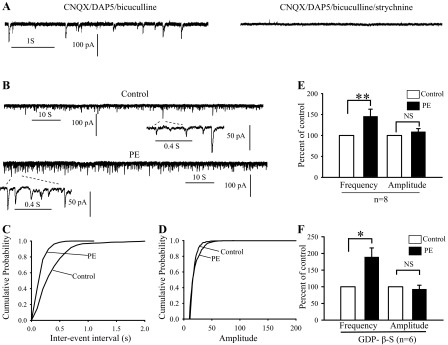
Activation of α1-adrenoceptor facilitated glycinergic synaptic transmission in the WT HNs. A: sample traces showing spontaneous inhibitory postsynaptic currents (sIPSCs) recorded in the presence of 30 μM bicuculline, 20 μM CNQX, and 30 μM d-AP5 (left). sIPSCs were completely abolished when perfused with bicuculline, CNQX, dl-AP5, and 1 μM of strychnine (right). B: sample traces showing sIPSCs recorded under control (top) and during bath application of PE (40 μM; bottom). Note that sIPSCs trace at large time scale obtained from the area indicated by 2 dashed lines. C and D: cumulative distribution of the interevent interval and amplitude curves obtained from the same neuron as in B. PE shifted the interevent interval curve (C) at left, but had no significant effect on the amplitude distribution curve (D). E and F: bar graphs summarizing the effects of PE on sIPSCs frequency and amplitude expressed as percentage of control ± SE in normal recording solution (E) and recording solution added GDP-β-S (F). *P < 0.05; **P < 0.01; n, number of neurons examined.
In the first series of experiments, we examined the effect of the highly selective α1-adrenoceptor agonist PE on sIPSCs of WT neurons. The sIPSCs were recorded every 3–5 min. For control, we recorded sIPSCs for more than 10 min to confirm that the baseline was stable. The effect of PE was then bath applied for 3–5 min, followed by washout recording after another 10 min. For data analysis, only the last 3–5 min of results from each period were analyzed with 200–10,000 events. Only sIPSCs with short rising time (<1 ms) were used for data analysis. The amplitude of sIPSCs was averaged, and frequency was calculated using minianalysis program. The concentration of 40 μM was chosen as PE at 30–50 μM affects both GABAA and glycine receptors mediated-responses in cerebellar and nucleus ambiguous neurons (9, 27). Bath application of PE increased the frequency (146 ± 16.8% of control; P < 0.01, Student's t-test) but had no significant effect on amplitude (109 ± 7.3% of control; P = 0.12, Student's t-test) of sIPSCs in eight HNs (Fig. 4, B–E). Since the α1 receptor is expressed on postsynaptic membranes of several brainstem motoneurons including HNs (20, 52), we applied 1 mM GDP-β-S, a nonhydrolyzable GTP analog and G-protein inhibitor (15), to the pipette solution to block G-protein signaling in the postsynaptic neurons (17, 25, 30, 31, 66). The GDP-β-S was applied for >10 min before PE application. Under this condition, the PE-mediated facilitation on sIPSCs frequency was not reduced at all (189 ± 27.5% of control; P = 0.013, Student t-test) in six cells (Fig. 4F).
Subsequently, we tested whether the PE effect on sIPSCs depends on WT neuronal firing activity. We recorded mIPSCs in the presence of bicuculline, CNQX, dl-AP5, and 1 μM TTX in WT neurons (Fig. 5A). Bath application of PE also increased the frequency (156 ± 14.3% of control; P < 0.001, Student's t-test) but not the amplitude (108.6 ± 4.9% control; P = 0.26, Student's t-test) of mIPSCs in seven cells (Fig. 5D). These results thus indicate that activation of α1-adrenoceptor facilitates glycinergic synaptic inputs in HNs and such an effect is likely to be mediated by enhancing spontaneous glycine release from presynaptic terminals.
Fig. 5.
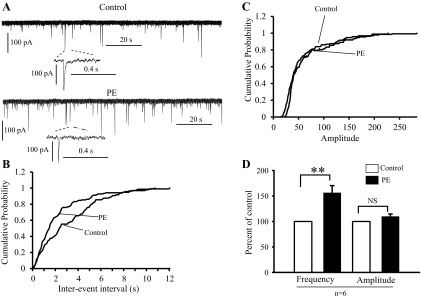
α1-Adrenoceptor agonist facilitated spiking-independent glycinergic synaptic transmission in the WT HNs. A: sample traces showing the effect of PE on mIPSCs recorded in the presence of CNQX, dl-AP5, bicuculline, and 1 μM TTX, under control (top) and during 40 μM PE application (bottom). B and C: cumulative distribution of the interevent interval and amplitude curves obtained from the same neuron as in A. PE shifted the interevent interval curve (B) at left but had no significant effect on the amplitude distribution curve (C). D: bar graphs summarizing the effect of PE on mIPSCs frequency and amplitude expressed as percentage of control ± SE. **P < 0.01; n, number of neurons examined.
Modulation of glycinergic synaptic transmission of HNs in Mecp2−/Y mice.
The effects of PE on glycinergic sIPSCs were examined in the presence of bicuculline, CNQX and d-AP5. Bath application of PE (40 μM) increased the frequency of sIPSCs (Fig. 6, A–D) in eight HNs from 3 Mecp2−/Y mice. Surprisingly, the effect of PE on sIPSC frequency was much stronger in Mecp2−/Y mice than in WT (299.5 ± 47.6 vs. 146 ± 16.8%, respectively; P < 0.01, Mann-Whitney test). Unlike the WT neurons, PE also augmented significantly the amplitude of the glycinergic sIPSCs in Mecp2−/Y mice (by 214.4 ± 44.3 vs. 109 ± 7.3% in WT; P < 0.05, Mann-Whitney test). Although the frequency effect is likely to be mediated by presynaptic mechanisms, the effect of PE on sIPSC amplitude could be both pre- or postsynaptic. For this reason, we employed GDP-β-S to block the postsynaptic α1-receptor-mediated signaling. With GDP-β-S, the frequency and amplitude were 253 ± 39% (P < 0.01, compared with control) and 102 ± 16% (P > 0.05), respectively (n = 5 cells, Student's t-test; Fig. 6E), suggesting that postsynaptic mechanisms are involved in the modulation of sIPSC amplitude.
Fig. 6.
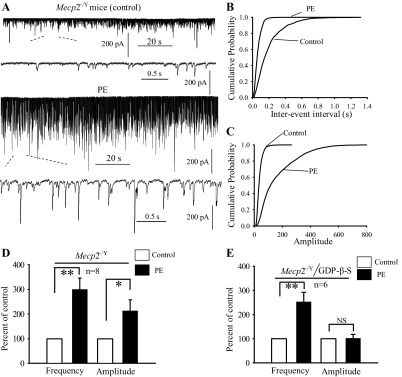
α1-Adrenoceptor agonist facilitated glycinergic synaptic transmission in HNs of Mecp2−/Y mice. A, sample traces showing sIPSCs recorded under control (top) and during PE application (bottom). B and C: cumulative distribution of the interevent interval and amplitude analyzed from the same neuron as in A. PE shifted interevent interval at left (B) and amplitude at right (C). D: bar graph summarizing the effect of PE on sIPSCs frequency and amplitude expressed as percentage of control ± SE in Mecp2−/Y HNs. E: bar graph showing the effect of PE on frequency and amplitude of sIPSCs recorded with electrodes filled with GDP-β-S. *P < 0.05; **P < 0.01; n, number of neurons examined.
The effects of PE on mIPSCs in Mecp2−/Y mice were also tested in five neurons in the presence AMPA, kainite, NMDA, GABAA receptors antagonists, and TTX (1 μM). The frequency and amplitude of mIPSCs were 143 ± 15.9% (P < 0.05, Student's t-test) and 119.6 ± 15.1% of control (P = 0.2, Student's t-test), respectively (Fig. 7, A–D). These were not significantly different from those in the WT neurons (P = 0.29 and 0.9 for amplitude and frequency, respectively (Mann-Whitney test). Thus these results suggest that the modulation of glycinergic synaptic transmission by α1-adrenoceptors is augmented in Mecp2−/Y mice, and the augmentations are spiking dependent involving likely both pre- and postsynaptic mechanisms.
Fig. 7.
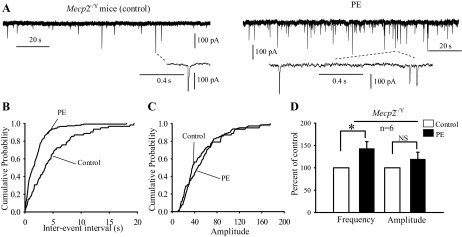
α1-Adrenoceptor facilitated spiking-independent glycinergic transmission in Mecp2−/Y. A: sample traces showing the effect of PE on mIPSCs recorded in the of presence 1 μM TTX, under control (left), and during 40 μM of PE application (right). B and C: cumulative distribution of the interevent interval and amplitude curves from same neuron as in A. Interevent interval (B) but not the amplitude (C) curve was shifted at left. D: bar graphs summarizing the effect of PE on mIPSCs frequency and amplitude expressed as percentage of control ± SE from Mecp2-null mice. *P < 0.05; n, number of neurons examined.
DISCUSSION
The present study has shown that presynaptic α1-adrenoceptor activation facilitates glycinergic transmission and excites HNs, and this presynaptic facilitation is altered in Mecp2-null mice through both pre- and postsynaptic mechanisms. In addition, our data demonstrated that postsynaptic modulation HNs by α1-adrenoceptors involving the G protein-dependent and barium-sensitive K+ channels.
Postsynaptic α1-adrenoceptor modulation of HN excitability in WT mice.
Previous studies have demonstrated that activation of α1-adrenoceptors produces depolarization associated with an increase in input resistance in cranial and spinal motoneurons (16, 20, 37, 49, 52, 63, 68). These excitatory effects are postsynaptic and are not derived from network drive since they are retained in the presence of TTX (52). Consistent with these findings (52), we found that the PE-induced depolarization and increase in input resistance persist in the presence of TTX. Also, the activation of postsynaptic α1-adrenoceptors is critical, as the PE-mediated depolarization, increase in input resistance, and spiking were all blocked when the recording pipette was filled with GDP-β-S. The complete blockade seems to be an overestimate, as it is very difficult to inhibit totally the G protein-dependent signaling by intracellular dialysis of GDP-β-S through the recording pipette. Such an effect seems to be a result of both the GDP-β-S and the glycinergic IPSCs that are augmented by the presynaptic α1-adrenoceptors.
Several mechanisms have been described for PE mediated-excitatory effects in the central nervous system (52). The PE-induced depolarization is due to inhibition of K+ currents in facial motoneurons (37) and Ba2+-sensitive K+ currents in juvenile and young adult HNs (49). In addition, the PE-mediated excitation of HNs also involves the activation of Ba2+-insensitive Na+ currents (49) and inhibition of TASK channels (60). In the present study, we have shown that the PE effects on HN membrane excitability are attributable to the inhibition of K+ currents that are sensitive to GDP-β-S and micromolar concentration of Ba2+, suggesting that the G-protein-coupled and Ba2+-sensitive K+ channels play a role in the postsynaptic effect of PE.
Presynaptic α1-adrenergic modulation of HN glycinergic transmission in WT mice.
We have shown that PE modulates glycinergic sIPSCs in HNs. It has been reported that in several brain regions NE facilitates GABA and glycinergic synaptic transmissions by activation of presynaptic α1-adrenoceptors (3, 9, 23, 26, 27). Indeed, our results indicate that PE augments frequency of the sIPSCs and mIPSCs only, without changing their amplitude. Consistent with these results, the PE-mediated facilitation of sIPSCs was not changed when postsynaptic metabotropic α1-adrenoceptors were blocked by GDP-β-S. Thus our results suggest that activation of presynaptic α1-adrenoceptors involved in the facilitation of an action potential-dependent glycinergic transmission at HNs.
Pre- and postsynaptic modulations of HNs in Mecp2−/Y mice.
RTT is a neurodevelopmental disorder caused by Mecp2 gene mutations (21, 58). Defects in the brainstem NE system have been suggested as an underlying cause for the breathing irregularities of the Mecp2−/Y mice (65, 72). Potential mechanisms for defects in the NE system have been reported, such as inadequate tyrosine hydroxylase and dopamine β-hydroxylase expressions (54, 61, 71), increased neuronal excitability (61, 71, 73), disrupted CO2 chemosensitivity (72), and defect of GABAergic synaptic transmission 3 (30). In the present study, we have found that the PE-dependent facilitation of glycinergic synaptic transmission is enhanced in Mecp2−/Y mice. PE augments both frequency and amplitude of sIPSCs in Mecp2−/Y neurons, both of which are significantly greater than in WT neurons. The enhanced ISPC frequency appears presynaptic, while the augmentation of IPSC amplitude is likely postsynaptic since it was blocked when electrodes were filled with GDP-β-S.
HNs neurons receive both GABAergic and glycinergic synaptic inputs (7, 38, 47, 69). A defect in GABAergic transmission has been reported in ventrolateral medulla of Mecp2−/Y mice (12, 30, 41). Similar findings have also been made in hippocampal (70) and LC neurons (26). Interestingly, the glycinergic system in ventrolateral medulla remains normal in Mecp2−/Y mice (41). Data from the present study suggest that the adrenoceptor-mediated facilitation of glycinergic transmission indeed is stronger in Mecp2−/Y mice than in the WT. Our data suggest that both pre- and postsynaptic mechanisms are involved in the facilitation of glycinergic synaptic inputs in HNs.
It is possible that the enhanced glycinergic transmission by presynaptic α1-adrenoceptors serves as a compensatory mechanism. GABAergic synaptic currents are dysfunctional in several types of neurons of the Mecp2−/Y mice (12, 30, 41). Thus enhanced glycinergic receptor activity may reflect compensation for reduced GABAergic activity. The Mecp2−/Y mice show a marked deficiency in NE content and function (29, 61, 65). For instance, the NE content is lower in Mecp2−/Y mice than the wild type in brainstem (61, 65) and medulla (65). It was hypothesized that breathing disturbances in Mecp2−/Y mice may be due to a deficiency in noradrenergic modulation of the respiratory network (65). The increased sensitivity of presynaptic glycinergic receptors to α1-adrenoceptor agonist may allow the Mecp2−/Y HNs to maintain inhibitory synaptic inputs with limited NE in the presynaptic terminals. Therefore, the augmentation of glycinergic transmission by α1-adrenoceptors may be a compensatory mechanism in the Mecp2−/Y HNs. We speculate that when the Mecp2 knockout affects the body, the system may try to respond to minimize the defect. Thus our findings open up the possibility for the development of pharmacotherapies to increase the glycinergic transmission by α1-adrenoceptor agonists for the treatment of motor disorders of RTT.
In conclusion, activation of α1-adrenoceptor facilitates glycinergic synaptic transmission and excites HNs. These effects are mediated by pre- and postsynaptic mechanisms. The latter effect is likely to be mediated by an inhibition of the G protein-coupled K+ channels. The pre- and postsynaptic modulations of HNs by α1-adrenoceptors are not only retained in Mecp2−/Y mice but also markedly enhanced, which may be a compensation mechanism for the deficiencies in NE modulatory system and GABAergic synaptic transmission. These findings are encouraging, as they suggest that endogenous compensation mechanisms, and others that may exist as well, for an alleviation of the defects in the central nervous system caused by Mecp2 gene disruption provides a new hope for therapeutical interventions to RTT.
GRANTS
This work was supported by National Institute of Neurological Disorders and Stroke Grant 1R01-NS-073875 and the International Rett Syndrome Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: X.-T.J. and C.J. conception and design of research; X.-T.J., N.C., W.Z., and Z.W. performed experiments; X.-T.J. and X.J. analyzed data; X.-T.J. and C.J. interpreted results of experiments; X.-T.J. prepared figures; X.-T.J. drafted manuscript; X.-T.J., Z.W., and C.J. edited and revised manuscript; X.-T.J., Z.W., and C.J. approved final version of manuscript.
REFERENCES
- 1.Aldes LD. The enkephalinergic innervation of the genioglossus musculature in the rat: implications for the respiratory control of the tongue. Brain Res 780: 67–73, 1998 [DOI] [PubMed] [Google Scholar]
- 2.Aldes LD, Chronister RB, Shelton C, 3rd, Haycock JW, Marco LA, Wong DL. Catecholamine innervation of the rat hypoglossal nucleus. Brain Res Bull 21: 305–312, 1988 [DOI] [PubMed] [Google Scholar]
- 3.Baba H, Shimoji K, Yoshimura M. Norepinephrine facilitates inhibitory transmission in substantia gelatinosa of adult rat spinal cord (part 1): effects on axon terminals of GABAergic and glycinergic neurons. Anesthesiology 92: 473–484, 2000 [DOI] [PubMed] [Google Scholar]
- 4.Bartlett D, Jr, Leiter JC, Knuth SL. Control and actions of the genioglossus muscle. Prog Clin Biol Res 345: 99–107; discussion 108, 1990 [PubMed] [Google Scholar]
- 5.Berger AJ. Development of synaptic transmission to respiratory motoneurons. Respir Physiol Neurobiol 179: 34–42, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berger AJ, Bayliss DA, Bellingham MC, Umemiya M, Viana F. Postnatal development of hypoglossal motoneuron intrinsic properties. Adv Exp Med Biol 381: 63–71, 1995 [DOI] [PubMed] [Google Scholar]
- 7.Berger AJ, Isaacson JS. Modulation of motoneuron N-methyl-d-aspartate receptors by the inhibitory neurotransmitter glycine. J Physiol (Paris) 93: 23–27, 1999 [DOI] [PubMed] [Google Scholar]
- 8.Bergles DE, Doze VA, Madison DV, Smith SJ. Excitatory actions of norepinephrine on multiple classes of hippocampal CA1 interneurons. J Neurosci 16: 572–585, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boychuk CR, Bateman RJ, Philbin KE, Mendelowitz D. α1-Adrenergic receptors facilitate inhibitory neurotransmission to cardiac vagal neurons in the nucleus ambiguus. Neuroscience 193: 154–161, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bunemann M, Meyer T, Pott L, Hosey M. Novel inhibition of gbetagamma-activated potassium currents induced by M(2) muscarinic receptors via a pertussis toxin-insensitive pathway. J Biol Chem 275: 12537–12545, 2000 [DOI] [PubMed] [Google Scholar]
- 11.Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron 56: 422–437, 2007 [DOI] [PubMed] [Google Scholar]
- 12.Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JL, Noebels JL, Rosenmund C, Zoghbi HY. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468: 263–269, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Docherty JR. Subtypes of functional alpha1- and alpha2-adrenoceptors. Eur J Pharmacol 361: 1–15, 1998 [DOI] [PubMed] [Google Scholar]
- 14.Domyancic AV, Morilak DA. Distribution of alpha1A adrenergic receptor mRNA in the rat brain visualized by in situ hybridization. J Comp Neurol 386: 358–378, 1997 [DOI] [PubMed] [Google Scholar]
- 15.Eckstein F, Cassel D, Levkovitz H, Lowe M, Selinger Z. Guanosine-O(2-thiodiphosphate): an inhibitor of adenylate cyclase stimulation by guanine nucleotides and fluoride ions. J Biol Chem 254: 9829–9834, 1979 [PubMed] [Google Scholar]
- 16.Elliott P, Wallis DI. Serotonin and l-norepinephrine as mediators of altered excitability in neonatal rat motoneurons studied in vitro. Neuroscience 47: 533–544, 1992 [DOI] [PubMed] [Google Scholar]
- 17.Flavin HJ, Jin XT, Daw NW. 2R,4R-4-Aminopyrrolidine-2,4-dicarboxylate (APDC) attenuates cortical EPSPs. Brain Res 873: 212–217, 2000 [DOI] [PubMed] [Google Scholar]
- 18.Funk GD, Smith JC, Feldman JL. Development of thyrotropin-releasing hormone and norepinephrine potentiation of inspiratory-related hypoglossal motoneuron discharge in neonatal and juvenile mice in vitro. J Neurophysiol 72: 2538–2541, 1994 [DOI] [PubMed] [Google Scholar]
- 19.Funk GD, Smith JC, Feldman JL. Generation and transmission of respiratory oscillations in medullary slices: role of excitatory amino acids. J Neurophysiol 70: 1497–1515, 1993 [DOI] [PubMed] [Google Scholar]
- 20.Funk GD, Zwicker JD, Selvaratnam R, Robinson DM. Noradrenergic modulation of hypoglossal motoneuron excitability: developmental and putative state-dependent mechanisms. Arch Ital Biol 149: 426–453, 2011 [DOI] [PubMed] [Google Scholar]
- 21.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet 27: 322–326, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol 14: 471–479, 1983 [DOI] [PubMed] [Google Scholar]
- 23.Han SK, Chong W, Li LH, Lee IS, Murase K, Ryu PD. Noradrenaline excites and inhibits GABAergic transmission in parvocellular neurons of rat hypothalamic paraventricular nucleus. J Neurophysiol 87: 2287–2296, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Hayar A, Feltz P, Piguet P. Adrenergic responses in silent and putative inhibitory pacemaker-like neurons of the rat rostral ventrolateral medulla in vitro. Neuroscience 77: 199–217, 1997 [DOI] [PubMed] [Google Scholar]
- 25.Hayar A, Heyward PM, Heinbockel T, Shipley MT, Ennis M. Direct excitation of mitral cells via activation of alpha1-noradrenergic receptors in rat olfactory bulb slices. J Neurophysiol 86: 2173–2182, 2001 [DOI] [PubMed] [Google Scholar]
- 26.Herold S, Hecker C, Deitmer JW, Brockhaus J. alpha1-Adrenergic modulation of synaptic input to Purkinje neurons in rat cerebellar brain slices. J Neurosci Res 82: 571–579, 2005 [DOI] [PubMed] [Google Scholar]
- 27.Hirono M, Obata K. Alpha-adrenoceptive dual modulation of inhibitory GABAergic inputs to Purkinje cells in the mouse cerebellum. J Neurophysiol 95: 700–708, 2006 [DOI] [PubMed] [Google Scholar]
- 28.Hogart A, Nagarajan RP, Patzel KA, Yasui DH, Lasalle JM. 15q11–13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum Mol Genet 16: 691–703, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ide S, Itoh M, Goto Y. Defect in normal developmental increase of the brain biogenic amine concentrations in the mecp2-null mouse. Neurosci Lett 386: 14–17, 2005 [DOI] [PubMed] [Google Scholar]
- 30.Jin X, Cui N, Zhong W, Jin XT, Jiang C. GABAergic synaptic inputs of locus coeruleus neurons in wild-type and Mecp2-null mice. Am J Physiol Cell Physiol 304: C844–C857, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin XT, Beaver CJ, Ji Q, Daw NW. Effect of the group I metabotropic glutamate agonist DHPG on the visual cortex. J Neurophysiol 86: 1622–1631, 2001 [DOI] [PubMed] [Google Scholar]
- 32.Jin XT, Pare JF, Raju DV, Smith Y. Localization and function of pre- and postsynaptic kainate receptors in the rat globus pallidus. Eur J Neurosci 23: 374–386, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Jin XT, Pare JF, Smith Y. Differential localization and function of GABA transporters, GAT-1 and GAT-3, in the rat globus pallidus. Eur J Neurosci 33: 1504–1518, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones LS, Gauger LL, Davis JN. Anatomy of brain alpha 1-adrenergic receptors: in vitro autoradiography with [125I]-heat. J Comp Neurol 231: 190–208, 1985 [DOI] [PubMed] [Google Scholar]
- 35.Knot HJ, Zimmermann PA, Nelson MT. Extracellular K(+)-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J Physiol 419–430, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi M, Takei H, Yamamoto K, Hatanaka H, Koshikawa N. Kinetics of GABAB autoreceptor-mediated suppression of GABA release in rat insular cortex. J Neurophysiol 107: 1431–1442, 2012 [DOI] [PubMed] [Google Scholar]
- 37.Larkman PM, Kelly JS. Ionic mechanisms mediating 5-hydroxytryptamine- and noradrenaline-evoked depolarization of adult rat facial motoneurones. J Physiol 456: 473–490, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li YQ, Takada M, Kaneko T, Mizuno N. Distribution of GABAergic and glycinergic premotor neurons projecting to the facial and hypoglossal nuclei in the rat. J Comp Neurol 378: 283–294, 1997 [DOI] [PubMed] [Google Scholar]
- 39.Lowe AA. The neural regulation of tongue movements. Prog Neurobiol 15: 295–344, 1980 [DOI] [PubMed] [Google Scholar]
- 40.Madison DV, Nicoll RA. Norepinephrine decreases synaptic inhibition in the rat hippocampus. Brain Res 442: 131–138, 1988 [DOI] [PubMed] [Google Scholar]
- 41.Medrihan L, Tantalaki E, Aramuni G, Sargsyan V, Dudanova I, Missler M, Zhang W. Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J Neurophysiol 99: 112–121, 2008 [DOI] [PubMed] [Google Scholar]
- 42.Minneman KP, Esbenshade TA. Alpha 1-adrenergic receptor subtypes. Annu Rev Pharmacol Toxicol 34: 117–133, 1994 [DOI] [PubMed] [Google Scholar]
- 43.Nai Q, Dong HW, Hayar A, Linster C, Ennis M. Noradrenergic regulation of GABAergic inhibition of main olfactory bulb mitral cells varies as a function of concentration and receptor subtype. J Neurophysiol 101: 2472–2484, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nicholas AP, Hokfelt T, Pieribone VA. The distribution and significance of CNS adrenoceptors examined with in situ hybridization. Trends Pharmacol Sci 17: 245–255, 1996 [DOI] [PubMed] [Google Scholar]
- 45.Nicholas AP, Pieribone VA, Hokfelt T. Cellular localization of messenger RNA for beta-1 and beta-2 adrenergic receptors in rat brain: an in situ hybridization study. Neuroscience 56: 1023–1039, 1993 [DOI] [PubMed] [Google Scholar]
- 46.Nunez-Abades PA, Morillo AM, Pasaro R. Brainstem connections of the rat ventral respiratory subgroups: afferent projections. J Auton Nerv Syst 42: 99–118, 1993 [DOI] [PubMed] [Google Scholar]
- 47.O'Brien JA, Isaacson JS, Berger AJ. NMDA and non-NMDA receptors are co-localized at excitatory synapses of rat hypoglossal motoneurons. Neurosci Lett 227: 5–8, 1997 [DOI] [PubMed] [Google Scholar]
- 48.Palacios JM, Kuhar MJ. Beta-adrenergic-receptor localization by light microscopic autoradiography. Science 208: 1378–1380, 1980 [DOI] [PubMed] [Google Scholar]
- 49.Parkis MA, Bayliss DA, Berger AJ. Actions of norepinephrine on rat hypoglossal motoneurons. J Neurophysiol 74: 1911–1919, 1995 [DOI] [PubMed] [Google Scholar]
- 50.Rainbow TC, Parsons B, Wolfe BB. Quantitative autoradiography of beta 1- and beta 2-adrenergic receptors in rat brain. Proc Natl Acad Sci USA 81: 1585–1589, 1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rekling JC. Interaction between thyrotropin-releasing hormone (TRH) and NMDA-receptor-mediated responses in hypoglossal motoneurones. Brain Res 578: 289–296, 1992 [DOI] [PubMed] [Google Scholar]
- 52.Rekling JC, Funk GD, Bayliss DA, Dong XW, Feldman JL. Synaptic control of motoneuronal excitability. Physiol Rev 80: 767–852, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rett A. [On a unusual brain atrophy syndrome in hyperammonemia in childhood]. Wien Med Wochenschr 116: 723–726, 1966 [PubMed] [Google Scholar]
- 54.Roux JC, Panayotis N, Dura E, Villard L. Progressive noradrenergic deficits in the locus coeruleus of Mecp2 deficient mice. J Neurosci Res 88: 1500–1509, 2010 [DOI] [PubMed] [Google Scholar]
- 55.Samaco RC, Hogart A, LaSalle JM. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet 14: 483–492, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Samaco RC, Mandel-Brehm C, Chao HT, Ward CS, Fyffe-Maricich SL, Ren J, Hyland K, Thaller C, Maricich SM, Humphreys P, Greer JJ, Percy A, Glaze DG, Zoghbi HY, Neul JL. Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proc Natl Acad Sci USA 106: 21966–21971, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scheinin M, Lomasney JW, Hayden-Hixson DM, Schambra UB, Caron MG, Lefkowitz RJ, Fremeau RT., Jr Distribution of alpha 2-adrenergic receptor subtype gene expression in rat brain. Brain Res Mol Brain Res 21: 133–149, 1994 [DOI] [PubMed] [Google Scholar]
- 58.Shahbazian MD, Zoghbi HY. Rett syndrome and MeCP2: linking epigenetics and neuronal function. Am J Hum Genet 71: 1259–1272, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Singer JH, Berger AJ. Development of inhibitory synaptic transmission to motoneurons. Brain Res Bull 53: 553–560, 2000 [DOI] [PubMed] [Google Scholar]
- 60.Talley EM, Lei Q, Sirois JE, Bayliss DA. TASK-1, a two-pore domain K+ channel, is modulated by multiple neurotransmitters in motoneurons. Neuron 25: 399–410, 2000 [DOI] [PubMed] [Google Scholar]
- 61.Taneja P, Ogier M, Brooks-Harris G, Schmid DA, Katz DM, Nelson SB. Pathophysiology of locus ceruleus neurons in a mouse model of Rett syndrome. J Neurosci 29: 12187–12195, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van Brederode JF, Yanagawa Y, Berger AJ. GAD67-GFP+ neurons in the Nucleus of Roller: a possible source of inhibitory input to hypoglossal motoneurons. I. Morphology and firing properties. J Neurophysiol 105: 235–248, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.VanderMaelen CP, Aghajanian GK. Intracellular studies showing modulation of facial motoneurone excitability by serotonin. Nature 287: 346–347, 1980 [DOI] [PubMed] [Google Scholar]
- 64.Viana F, Bayliss DA, Berger AJ. Repetitive firing properties of developing rat brainstem motoneurones. J Physiol 486: 745–761, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Viemari JC, Maussion G, Bevengut M, Burnet H, Pequignot JM, Nepote V, Pachnis V, Simonneau M, Hilaire G. Ret deficiency in mice impairs the development of A5 and A6 neurons and the functional maturation of the respiratory rhythm. Eur J Neurosci 22: 2403–2412, 2005 [DOI] [PubMed] [Google Scholar]
- 66.Wang XF, Daw NW, Jin X. The effect of ACPD on the responses to NMDA and AMPA varies with layer in slices of rat visual cortex. Brain Res 812: 186–192, 1998 [DOI] [PubMed] [Google Scholar]
- 67.Werman R, Davidoff RA, Aprison MH. Inhibition of motoneurones by iontophoresis of glycine. Nature 214: 681–683, 1967 [DOI] [PubMed] [Google Scholar]
- 68.White SR, Fung SJ, Barnes CD. Norepinephrine effects on spinal motoneurons. Prog Brain Res 88: 343–350, 1991 [DOI] [PubMed] [Google Scholar]
- 69.Yang CC, Chan JY, Chan SH. Excitatory innervation of caudal hypoglossal nucleus from nucleus reticularis gigantocellularis in the rat. Neuroscience 65: 365–374, 1995 [DOI] [PubMed] [Google Scholar]
- 70.Zhang L, He J, Jugloff DG, Eubanks JH. The MeCP2-null mouse hippocampus displays altered basal inhibitory rhythms and is prone to hyperexcitability. Hippocampus 18: 294–309, 2008 [DOI] [PubMed] [Google Scholar]
- 71.Zhang X, Cui N, Wu Z, Su J, Tadepalli JS, Sekizar S, Jiang C. Intrinsic membrane properties of locus coeruleus neurons in Mecp2-null mice. Am J Physiol Cell Physiol 298: C635–C646, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang X, Su J, Cui N, Gai H, Wu Z, Jiang C. The disruption of central CO2 chemosensitivity in a mouse model of Rett syndrome. Am J Physiol Cell Physiol 301: C729–C738, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang X, Su J, Rojas A, Jiang C. Pontine norepinephrine defects in Mecp2-null mice involve deficient expression of dopamine beta-hydroxylase but not a loss of catecholaminergic neurons. Biochem Biophys Res Commun 394: 285–290, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang ZW, Zak JD, Liu H. MeCP2 is required for normal development of GABAergic circuits in the thalamus. J Neurophysiol 103: 2470–2481, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zoghbi HY, Milstien S, Butler IJ, Smith EO, Kaufman S, Glaze DG, Percy AK. Cerebrospinal fluid biogenic amines and biopterin in Rett syndrome. Ann Neurol 25: 56–60, 1989 [DOI] [PubMed] [Google Scholar]
- 76.Zoghbi HY, Percy AK, Glaze DG, Butler IJ, Riccardi VM. Reduction of biogenic amine levels in the Rett syndrome. N Engl J Med 313: 921–924, 1985 [DOI] [PubMed] [Google Scholar]