

Abstract
The development of insulin resistance has been associated with impaired mitochondrial fatty acid oxidation (FAO), but the exact relationship between FAO capacity and glucose metabolism continues to be debated. To address this controversy, patients with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) deficiency underwent an oral glucose tolerance test (OGTT) and measurement of energy expenditure, body composition, and plasma metabolites. Compared with controls, patients with LCHAD deficiency had a trend toward higher total body fat and extramyocellular lipid deposition but similar levels of intramyocelluar and intrahepatic lipids. Resting energy expenditure was similar between the groups, but respiratory quotient was higher and total energy expenditure was lower in LCHAD-deficient patients compared with controls. High-molecular-weight (HMW) adiponectin levels were lower and plasma long-chain acylcarnitines were higher among LCHAD-deficient patients. Fasting and post-OGTT levels of glucose, insulin, and ghrelin, along with estimates of insulin sensitivity, were the same between the groups. Despite decreased capacity for FAO, lower total energy expenditure and plasma HMW adiponectin, and increased plasma acylcarnitines, LCHAD-deficient patients exhibited normal glucose tolerance. These data suggest that inhibition of the FAO pathway in humans is not sufficient to induce insulin resistance.
Keywords: long-chain 3-hydroxy acyl-coenzyme A dehydrogenase deficiency, acylcarnitines
several lines of evidence have linked decreased mitochondrial fatty acid oxidation (FAO) with the development of insulin resistance. Reduced FAO has been associated with the subsequent accumulation of cytosolic lipid by-products such as diacylglycerols (DAGs), ceramides, and triglycerides in skeletal muscle and liver, also known as ectopic fat (5, 24, 32, 36). In turn, these cytosolic lipid intermediates have been shown to activate serine stress kinases and signaling pathways that impair normal insulin signaling and are the basis of the lipotoxicity hypothesis for insulin resistance (32, 38). Indeed, increasing FAO and reducing ectopic fat are thought to be mechanisms that contribute to improved insulin sensitivity by various agents or genetic manipulations (1, 6, 7, 17, 37, 55).
In contrast, others have proposed that increased FAO without a concomitant increase in tricarboxylic acid cycle function results in mitochondrial dysfunction and the subsequent development of insulin resistance. Some studies have shown that blocking fatty acid entry into mitochondria can alleviate high-fat-diet-induced insulin resistance, suggesting that enhanced FAO and subsequent mitochondrial stress is the cause of insulin resistance (22, 23). Impaired mitochondrial function and subsequent production of partially oxidized substrates, such as intermediate metabolites from FAO in the form of fatty acylcarnitines (23, 30, 34), and increased reactive oxygen species (16, 25), have been associated with insulin resistance. These metabolites may serve as biomarkers of mitochondrial function or may be involved as potential mediators of insulin resistance (2). The question, therefore, is whether decreased or increased FAO is sufficient to induce insulin resistance.
Following the first description of inherited deficiencies of FAO in humans in the 1980s, ∼22 different defects have been identified (40). Patients with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) deficiency have a dramatically reduced ability to oxidize fatty acids and typically present with hypoketotic hypoglycemia in infancy, accumulate long-chain acylcarnitine and long-chain hydroxyacylcarnitine species in their blood, and have exercise intolerance later in life (41, 42, 52, 53). Treatment for these disorders consists of limiting long-chain fats in the diet and providing alternative fuel sources such as carbohydrates and protein (49). In practice, subjects with LCHAD deficiency consume very high carbohydrate diets and eat frequently to avoid fasting (13, 14). Some, but not all, patients become overweight. Although this constellation of features would typically be expected to increase the risk of impaired glucose clearance, to date no individual with LCHAD deficiency and glucose intolerance has been reported in the literature.
Because subjects with these rare disorders could potentially inform us about the relationship between reduced FAO and insulin sensitivity, we measured energy expenditure and body composition and performed oral glucose tolerance tests in patients with LCHAD deficiency compared with age-, sex-, and body mass index (BMI)-matched control subjects. We hypothesized that impaired ability to oxidize fat would not be sufficient to induce insulin resistance among subjects with a long-chain FAO disorder.
METHODS
Patients with a FAO disorder were recruited by announcements posted on family support networks and physician referral. The diagnosis of a FAO disorder was confirmed by reviewing medical records and evidence of two of the following four diagnostic criteria: 1) history of rhabdomyolysis with absence of urinary ketones, 2) abnormal acylcarnitine profile suggestive of a FAO disorder, 3) one or two known disease-causing mutations, or 4) decreased enzymatic activity in cultured fibroblasts (4, 39). Based on these criteria, initially 12 patients were identified and studied, consisting of 2 subjects with defects in the carnitine palmitoyltransferase (CPT)-2 enzyme, 1 subject with a mild adolescent-onset LCHAD deficiency, and 9 subjects with infant-onset LCHAD deficiency. Because the patients with CPT2 and mild LCHAD introduced minor enzymatic and phenotypic heterogeneity, it was decided to include only those patients with classical LCHAD defects in this analysis. Control subjects were recruited who were the same gender and had a similar age and BMI for a one-to-one matching design from the Portland area through the Oregon Health & Science University (OHSU) research recruitment webpage and referrals. The groups were matched for age, gender, and BMI. This study was approved by the OHSU institutional review board (IRB no. 817) and is listed on clinical trials.gov (NCT00654004). All subjects and their guardians gave written informed consent after the study had been explained to them.
Patients with LCHAD deficiency and control subjects were admitted to the OHSU Clinical and Translational Research Center for completion of all study procedures except for the doubly labeled water (DLW) sampling. Ingestion of the DLW and subsequent collection of urine samples for analysis took place after returning home to control for the change in ground water isotope enrichment.
Resting energy expenditure.
Resting energy expenditure was measured after an overnight fast by indirect calorimetry using a Sensor Medics Vmax 29N encore. Participants lay supine in a room with ambient temperature and low lighting for 30 min. The Plexiglas canopy was then placed over their head and chest, and gas exchange was measured for 45 min. Urine was collected for 24 h and analyzed for total urinary urea nitrogen concentration. Substrate oxidation was estimated using resting V̇o2 (l/min) inspired, V̇co2 (l/min) expired, and 24-h urea excretion (g/min) (18).
Total energy expenditure.
Total energy expenditure was measured with DLW. Baseline urine samples were collected immediately before and three times within a 6-h period following labeled water consumption. Two final urine samples were collected 7 days later about 1 h apart. Samples were analyzed for H2 and 18O enrichment by isotope ratio mass spectrometry as previously described (Laboratory of Schoeller, University of Wisconsin-Madison, Madison, WI). Total energy expenditure was calculated from the difference in elimination curves of H2 and 18O enrichment in urine assuming a respiratory quotient of 0.85 (43).
Body composition.
Weight and height were measured in light clothing without shoes after an overnight 10-h fast. Body composition was measured by dual-energy X-ray absorptiometry (Hologic QDR 4500 Densitometer; Hologic, Bedford, MA) to determine total and regional fat and fat-free body mass.
Magnetic resonance imaging and spectroscopy.
All magnetic resonance images (MRI) and spectroscopy (MRS) were obtained at the OHSU Advanced Imaging Research Center using a Siemens Magnetom Tim Trio 3 Tesla (Siemens Medical Solutions, Malvern, PA, and Erlangen, Germany) whole body system following an overnight fast. MRI of the abdomen for visceral and subcutaneous fat was obtained as previously described (21). A single slice at the level on the umbilicus was used for analysis and calculation of the visceral fat and subcutaneous fat areas. Liver (intrahepatic lipid, or IHL) and muscle (extramyocellular, or EMCL, and intramyocellular, or IMCL) lipid deposition were measured using image-guided, 1H-localized MRS following high-resolution T-weighted spin-echo image (51). Volumes of interest within muscle were centered over the midsoleus muscles (12–15 cm3) with minimal contribution from gastrocnemius. Similarly, volumes of interest in liver were located away from major vascular structures, typically within the right lobe, with size varying from 18 to 27 cm3. Localized proton spectra within muscle were collected using a PRESS sequence with the following parameters: repetition time = 5 s, time to echo = 40 ms, 1,024 data points over 2,000 kHz spectral width. Liver spectra were collected using the same sequence and parameters except for a method of signal averaging to correct for liver motion due to patient respiration.
Oral glucose tolerance test.
Following completion of the indirect calorimetry and continuing in a fasting state, an antecubital intravenous catheter was placed for blood sampling. Two fasting blood samples were drawn 15 min apart to account for pulsatile insulin secretion. Following this, subjects were given a standard flavored glucose to consume orally over 10 min (Glucola: 75 g for ≥16 yr; 50 g for ≤16 yr). Postprandial blood samples were obtained via the intravenous catheter at 30, 60, 90, and 120 min following glucose ingestion.
Blood chemistries.
Concentrations of glucose were measured by a colorimetric enzyme assay (Raichem Glucose Color Reagent; Cliniqa, San Marcos, CA). Insulin concentrations were measured by radioimmunoassay (EMD Millipore, Billerica, MA). Phenylmethanesulfonyl fluoride and hydrochloric acid were added to plasma at the time of collection to prevent ghrelin degradation. These samples were assayed in duplicate using a ghrelin sandwich assay to measure both des- and acyl-ghrelin as previously described (27). Tetrahydrolipostatin was added to EDTA plasma at the time of collection to inhibit lipase hydrolysis of free fatty acids ex vivo. Free fatty acid concentrations were measured by a commercially available enzymatic colorimetric kit (Wako Chemicals, Richmond, VA). Plasma was analyzed for acylcarnitines by electrospray tandem mass spectrometry at the Biochemical Genetics Laboratory, Mayo Clinic (46). Plasma was analyzed for amino acids by high-performance liquid chromatography using an amino acid analyzer at the Biochemical Genetics laboratory of the Knight Diagnostic Laboratories (Portland, OR). Protease inhibitors were added to fasting serum to prevent adiponectin degradation. Adiponectin and high-molecular-weight (HMW) adiponectin were measured by ELISA (EMD Millipore). Fasting leptin concentrations were measured by radioimmunoassay (EMD Millipore) (9).
Data analysis.
Insulin sensitivity was estimated by the homeostatic model assessment using the fasting glucose and insulin levels (glucose × insulin/405) (29) and the insulin sensitivity index (ISI) of Matsuda and Defronzo [10,000/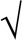 (fasting glucose × fasting insulin) × (mean glucose × mean insulin) during the oral glucose tolerance test] (28). Insulin secretion capacity was estimated using the insulinogenic index, the area under the curve (0–120 min) of insulin/area under the curve for glucose (31).
(fasting glucose × fasting insulin) × (mean glucose × mean insulin) during the oral glucose tolerance test] (28). Insulin secretion capacity was estimated using the insulinogenic index, the area under the curve (0–120 min) of insulin/area under the curve for glucose (31).
Differences in body composition, lipid deposition, energy expenditure, substrate oxidation, fasting leptin, and adiponectin between LCHAD-deficient patients and matched control subjects were determined by paired t-test using Prism 4.0 (Graphpad, La Jolla, CA). Because the data were matched one-to-one, it is necessary in the analysis to take into account the matching. The appropriate analysis, the paired t-test, will be more efficient than the unmatched analysis if there is moderate positive correlation between the paired observations. Although this method is more commonly seen when pairs of observations are obtained on the same subject, it is also widely used for more general matched pairs (10, 20). Differences between groups in plasma glucose, insulin, ghrelin, and acylcarnitines over time were compared using a paired repeated-measures ANOVA (Prism 4.0). If there was a significant difference between groups, a Bonferroni post hoc test was used for analysis of difference between individual time points. For all analysis, P < 0.05 was considered statistically significant.
RESULTS
The patients with LCHAD deficiency ranged in age from 7 to 17 yr (Table 1), and their BMIs ranged from 15.3 to 28.8 kg/m2. No participant met criteria for obesity in an adult. Patients with LCHAD deficiency tended to have a higher percent fat mass and lower percent fat-free mass than the control subjects (Fig. 1A). This difference in body composition appears to be due to both a slight increase in fat mass and decrease in lean mass, as suggested by similar fat mass and lean mass indexed to height between groups (Fig. 1B). The fat content was distributed equally between the trunk and extremities in the subjects, and visceral adipose tissue or subcutaneous adipose tissue areas were not different between groups (Table 2). There was no difference in IHL content between patients with LCHAD deficiency and control subjects (Fig. 1, C and D). LCHAD-deficient patients exhibited a trend for more EMCL deposition but no difference in IMCL in the soleus muscle compared with control subjects (Fig. 1, C and E).
Table 1.
Participant characteristics
| LCHAD-Deficient Patients |
Matched Controls |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutation | Age, yr | Gender | Ht, cm | Ht Z-score | Wt, kg | BMI, kg/m2 | BMI Z-score | BMI, percentile | Age, yr | Gender | Ht, cm | Ht Z-score | Wt, kg | BMI, kg/m2 | BMI Z-score | BMI, percentile |
| c. 1528G>C/c.1528G>C | 7 | M | 140.2 | 3.3 | 32.7 | 16.64 | 0.61 | 75 | 9 | M | 137 | 0.5 | 35.5 | 18.91 | 1.05 | 87 |
| c. 1528G>C/c.2102A>G | 7 | M | 135.9 | 2.5 | 35.5 | 19.22 | 1.62 | 95 | 8 | M | 130.7 | 0.4 | 26.1 | 15.28 | −0.49 | 37 |
| c.1528G>C/c.1132C>T | 8 | M | 131.8 | 0.6 | 32.6 | 18.77 | 1.12 | 90 | 10 | M | 143.8 | 0.7 | 31.2 | 15.09 | −0.99 | 17 |
| c. 1528G>C/c.1528G>C | 14 | M | 183.1 | 2.5 | 64.7 | 19.30 | −0.01 | 52 | 15 | M | 157.5 | −1.6 | 48.9 | 19.71 | −0.33 | 48 |
| c.1528G>C/c.274_278del | 14 | F | 162.1 | 0.2 | 69.1 | 26.30 | 1.48 | 94 | 10 | F | 150.4 | 1.7 | 44 | 19.45 | 0.84 | 81 |
| c.1528G>C/? | 15 | M | 167.4 | −0.4 | 73.6 | 26.26 | 1.47 | 94 | 13 | M | 171 | 1.8 | 81.4 | 27.84 | 1.91 | 97 |
| c.1528G>C/c.1678C>T | 16 | M | 172.6 | −0.1 | 71 | 23.83 | 0.85 | 83 | 22 | M | 182.6 | 96.1 | 28.82 | |||
| c. 1528G>C/c.1528C>T | 16 | F | 155.4 | −1.1 | 54.6 | 22.61 | 0.61 | 73 | 17 | F | 162.7 | −0.1 | 56.2 | 21.23 | 0.08 | 54 |
| c. 1528G>C/c.1528G>C | 17 | F | 155.6 | −1.1 | 67.4 | 27.84 | 1.38 | 92 | 19 | F | 170.5 | 1.1 | 75.9 | 26.11 | 0.99 | 85 |
LCHAD, long-chain 3-hydroxy acyl-CoA dehydrogenase; BMI, body mass index; M, male; F, female.
Fig. 1.

Body composition and lipid deposition. Data are presented as means ± SD. A: there is a trend for long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD)-deficient patients (n = 9; closed bars) to have less fat-free mass and more fat mass compared with control subjects (n = 9; open bars) when expressed as %body mass. B: there was no difference in fat-free or fat mass expressed as mass/surface area between groups. C: there was a trend for LCHAD-deficient patients (n = 9; closed bars) to have more extramyocellular lipid (EMCL) but no difference in intramyocellular lipid (IMCL) compared with control subjects (n = 9; open bars). Liver lipid content was not significantly different between groups. D: a representative magnetic resonance imaging (MRI) image of the abdomen and proton spectra of the liver for one LCHAD-deficient patient and the matched control subject are presented. The lipid peak is expressed as a percent of the water peak. E: a representative MRI image of the calf and proton spectra for one LCHAD-deficient patient and the matched control subject are presented. The lipid peak is expressed as a percent of the water peak.
Table 2.
Regional fat mass and blood levels of lipids, glucose, and insulin and estimates of insulin sensitivity and secretion in patients with LCHAD deficiency and control subjects
| Patients | Controls | P Value | |
|---|---|---|---|
| Regional fat mass | |||
| Body fat of trunk, % | 28 ± 7 | 26 ± 11 | 0.6 |
| Body fat of arms and legs, % | 33 ± 7 | 28 ± 9 | 0.17 |
| Visceral adipose tissue, cm2 | 3 ± 1.4 | 4 ± 3.1 | 0.15 |
| Subcutaneous adipose tissue, cm2 | 19 ± 9 | 21 ± 15 | 0.38 |
| Lipid levels | |||
| Total cholesterol, mg/dl | 139 ± 16 | 169 ± 33 | 0.005 |
| Triglyceride, mg/dl | 79 ± 20 | 60 ± 26 | 0.15 |
| LDL cholesterol, mg/dl | 86 ± 16 | 106 ± 30 | 0.18 |
| HDL cholesterol, mg/dl | 36 ± 3 | 51 ± 12 | 0.011 |
| Glucose homeostasis | |||
| Fasting glucose, mg/dl | 96 ± 13 | 106 ± 20 | 0..21 |
| Fasting insulin, μU/ml | 17 ± 10 | 14 ± 5 | 0.22 |
| HOMA-IR | 4.85 ± 2.75 | 3.61 ± 1.41 | 0.31 |
| Insulin sensitivity index | 2.87 ± 1.92 | 2.98 ± 1.52 | 0.89 |
| Insulinogenic index | 0.77 ± 0.47 | 0.53 ± 0.14 | 0.22 |
Data are means ± SD. Body composition expressed as %body fat or cm2 on magnetic resonance imaging image and blood lipid, glucose, and insulin levels expressed as mg/dl were measured after a 10-h overnight fast. LDL, low density lipoprotein; HDL, high density lipoprotein; HOMA-IR, homeostatic model assessment of insulin resistance, calculated using the following formula: fasting glucose × fsting insulin/405. The insulin sensitivity index of Matsuda and Defronzo (28) was calculated by 10,000/√(fasting glucose × fasting insulin) × (mean glucose × mean insulin) during the oral glucose tolerance test. The insulinogenic index was calculated by the area under the curve (0-120 min) of insulin/area under the curve for glucose. P value is from a 2-sided paired t-test.
Resting energy expenditure was similar but respiratory quotient was higher and total energy expenditure was lower among patients with LCHAD deficiency.
Resting energy expenditure was not significantly different between LCHAD-deficient patients and control subjects (Fig. 2, A and B). Respiratory quotient was significantly higher in patients with LCHAD deficiency after an overnight fast compared with control subjects (Fig. 2C). There was no significant difference in protein oxidation between the groups, but patients with LCHAD deficiency oxidized more carbohydrate and less fat at rest than controls (Fig. 2D). Only six control subjects completed the measurement of total energy expenditure measured by the DLW method. Total energy expenditure was ∼15% lower in LCHAD-deficient patients compared with control subjects (Fig. 2, E and F) regardless whether the groups were compared using a paired (n = 6 pairs) or unpaired t-test (subjects n = 9; controls n = 6; P = 0.04).
Fig. 2.

Energy expenditure and substrate oxidation. Data are presented as means ± SD. Indirect calorimetry was measured after a 10-h overnight fast. LCHAD-deficient patients (n = 9; closed bars and closed circles) have a similar resting energy expenditure as control subjects (n = 9; white bars and open squares) expressed as mean kcal/day (A) or kcal/kg of fat-free mass (B). Resting respiratory quotient was significantly higher in the LCHAD-deficient patients (n = 9; gray box plot) compared with control subjects (n = 9; white box plot). C: LCHAD-deficient patients (n = 8; closed bars) oxidized more carbohydrate and less fat than controls (n = 8; white bars). Total energy expenditure was lower in LCHAD-deficient patients (n = 9; closed bars and closed circles) compared with control subjects (n = 6; white bars and open squares) expressed as mean kcal/day (E) or as kcal/kg of fat-free mass (F).
Responses to oral glucose loads were identical in LCHAD-deficient patients and matched control subjects.
Neither fasting nor postoral plasma glucose or insulin levels (expressed as either absolute values or as a percent change from basal levels) were different between the LCHAD-deficient patients and control subjects (Fig. 3). All estimates of insulin sensitivity were likewise similar between groups (Table 2).
Fig. 3.

Plasma glucose and insulin with an oral glucose tolerance test. Data are presented as means ± SD. Participants fasted overnight (10 h), and 2 fasting blood samples were drawn 15 min apart (−15, 0 min). After the fasting blood samples were collected, participants consumed a standard glucose drink (Glucola) orally. Additional blood samples were taken at 30, 60, 90, and 120 min after the oral glucose load. LCHAD-deficient patients (n = 9; closed circles) have similar glucose concentrations (A) and percent change in plasma glucose (B) compared with control subjects (n = 9; open squares) before and after an oral glucose load. LCHAD-deficient patients (n = 9; closed circles) have similar insulin concentrations (C) and percent change in plasma insulin (D) compared with control subjects (n = 0; open squares) before and after an oral glucose load.
Plasma long-chain acylcarnitines were higher among patients with LCHAD deficiency.
LCHAD-deficient patients had similar fasting free fatty acid levels compared with controls (Fig. 4A), and, in both groups, free fatty acid levels decreased similarly after an oral glucose load. Plasma acetylcarnitine concentrations, an indirect marker of acetyl-CoA in the tissue (45), were not different between the two groups (Fig. 4B). As expected given the known enzymatic defects in the LCHAD-deficient patients, the sum of the long-chain acylcarnitines (including C12:1, C12:0, C14:2, C14:1, C14:0, C16:1, C16:0, C18:2, C18:1, and C18:0) was significantly greater, both before and after an oral glucose load, in the patients with LCHAD deficiency compared with controls (Fig. 4E). All of the long-chain acylcarnitine species were significantly greater among LCHAD-deficient patients compared with control subjects (Fig. 5). On the other hand, levels of summed short-chain acylcarnitines (including C3:0, C4:0, C5:1, C5:0, C4-OH, and C5-OH carnitines) (Fig. 4C) and medium-chain acylcarnitines (including C6:0, C8:0, C10:2, C10:1, and C10:0) (Fig. 4D) were not different between the patients and controls. Because some short-chain acylcarnitine species, such as propionylcarnitine, are derived from branched-chain amino acid (BCAA) oxidation and elevated BCAA have been associated with insulin resistance, we measured fasting plasma amino acid profiles and found there was no difference in any of the fasting plasma amino acids, including the BCAA, between patients with LCHAD deficiency and control subjects (Table 3).
Fig. 4.

Plasma free fatty acids and acylcarnitines with an oral glucose tolerance test. Data are presented as means ± SD. Participants fasted overnight (10 h), and a fasting blood sample (0 min) was analyzed for free fatty acids and plasma acylcarnitines. After the fasting blood sample was collected, participants consumed a standard glucose drink (Glucola) orally. Additional blood samples were taken at 30, 60, 90, and 120 min after the oral glucose load and analyzed similarly. A: LCHAD-deficient patients (n = 9; closed circles) have similar fasting free fatty acids as control subjects (n = 9; open circles). B, C, and D: acetylcarnitine, the sum of the short-chain acylcarnitines, and the sum of the medium-chain acylcarnitines were not different between LCHAD-deficient patients (n = 9; closed circles) and control subjects (n = 9; open circles) after an overnight fast or following an oral glucose load. E: the sum of the long-chain acylcarnitines was significantly higher among LCHAD-deficient patients (n = 9; closed circles) compared with control subjects (n = 9; open circles) after an overnight fast and following an oral glucose load.
Fig. 5.

Long-chain acylcarnitine species. Data are presented as means ± SD. Individual long-chain acylcarnitine species are presented from the fasting blood sample and from the sample collected 120 min following an oral glucose load. A: all of the long-chain acylcarnitine species were significantly higher among LCHAD-deficient patients (n = 9; closed bars) compared with control subjects (n = 9; white bars) following a 10-h overnight fast (Pre-OGTT) and after an oral glucose load (B; 120 min Post-OGTT). *P < 0.0001.
Table 3.
Plasma amino acid concentrations in patients with LCHAD deficiency and control subjects
| Amino Acid, μmol/l | Patients With LCHAD Deficiency | Controls | P Value |
|---|---|---|---|
| Hydroxyproline | 18 ± 9 | 20 ± 9 | 0.66 |
| Histidine | 86 ± 14. | 89 ± 8 | 0.54 |
| Asparagine | 46 ± 10 | 55 ± 9 | 0.08 |
| Taurine | 44 ± 8 | 58 ± 22 | 0.07 |
| Serine | 114 ± 16 | 120 ± 12 | 0.39 |
| Glutamine/Glutamate | 544 ± 86 | 553 ± 91 | 0.82 |
| Arginine | 91 ± 36 | 88 ± 34 | 0.84 |
| Glycine | 268 ± 71 | 262 ± 55 | 0.85 |
| Aspartic | 15 ± 7 | 11 ± 7 | 0.32 |
| Citrulline | 28 ± 6 | 28 ± 6 | 0.58 |
| Threonine | 143 ± 35 | 147 ± 22 | 0.80 |
| Alanine | 363 ± 104 | 391 ± 107 | 0.59 |
| Proline | 248 ± 93 | 217 ± 71 | 0.46 |
| Ornithine | 46 ± 8 | 57 ± 24 | 0.19 |
| Cystine | 0.39 ± 45 | 0.51 ± 1.07 | 0.71 |
| Lysine | 191 ± 43 | 179 ± 32 | 0.30 |
| Tyrosine | 61 ± 18 | 67 ± 22 | 0.61 |
| Methionine | 23 ± 4 | 23 ± 4 | 0.81 |
| Valine | 231 ± 59 | 226 ± 42 | 0.84 |
| Isoleucine | 65 ± 19 | 63 ± 17 | 0.79 |
| Leucine | 120 ± 27 | 118 ± 27 | 0.90 |
| Phenylalanine | 52 ± 6 | 55 ± 7 | 0.42 |
| Tryptophan | 55 ± 16 | 54 ± 10 | 0.81 |
Data are means ± SD expressed as μmol/l. Amino acids were measured in serum collected after a 10-h overnight fast. P value is from a 2-sided paired t-test.
Serum leptin and plasma ghrelin levels were similar but plasma high molecular weight adiponectin was lower among patients with LCHAD deficiency.
There was no difference in fasting leptin levels, either total or per kilogram of fat mass, between LCHADD-deficient patients and controls (Fig. 6, A and B). Total adiponectin was not different between groups but HMW adiponectin levels were lower in LCHAD-deficient patients compared with control subjects (Fig. 6, C and D). Plasma total and acyl-ghrelin were similar both before and after an oral glucose load between groups (Fig. 6, E and F).
Fig. 6.

Plasma leptin, adiponectin, and ghrelin concentrations. Data are presented as means ± SD. Samples collected after an overnight 10-h fast were analyzed for plasma leptin, adiponectin, and high-molecular-weight adiponectin. LCHAD-deficient patients (n = 9; closed bars) have similar fasting leptin concentrations as control subjects (n = 9; open bars) expressed as ng/ml (A) or normalized to body fat (B; ng·ml−1·kg body fat−1). Fasting total adiponectin was similar between groups (C), but high-molecular-weight adiponectin (D) levels were lower in LCHAD-deficient patients (n = 9; closed bars) compared with control subjects (n = 9; open bars). After the fasting blood sample was collected, participants consumed a standard glucose drink (Glucola) orally. Additional blood samples were taken at 30, 60, 90, and 120 min after the oral glucose load and analyzed along with the fasting sample for total and acyl-ghrelin. Total ghrelin and acyl-ghrelin concentrations were similar between LCHAD-deficient patients (n = 9; closed circles) and controls (n = 9; open squares) before and after an oral glucose load.
DISCUSSION
Studying patients with LCHAD deficiency allows a unique opportunity to test relationships between mitochondrial function, accumulation of FAO intermediates, and the effects of these changes on body composition and glucose metabolism. As might be expected when the ability to oxidize fat is impaired, we found a trend for subjects with LCHAD deficiency to have higher body fat content than control subjects. Possibly contributing to this excess adiposity were lower daily energy expenditures in the LCHAD-deficient patients. We found that this deficit in 24-h energy expenditure was not due to a lower resting energy expenditure than controls, which is consistent with our finding of similar acetylcarnitine levels in the patients with LCHAD deficiency and controls. Acetyl-carnitine levels are a surrogate for tissue accumulation of acetyl-CoA and suggest that energy production at rest was normal despite having partial FAO deficiency. Instead, we suspect that the lower daily energy expenditure in the LCHAD patients is likely explained by reduced activity levels, which have been described among subjects with a FAO disorder who fear metabolic decompensation and rhabdomyolysis triggered by excessive physical activity (48). This remains speculation, though, since we did not formally measure activity in either group in this study.
Despite their FAO defect, patients with LCHAD deficiency did not accumulate excess IHL and tended to store excess lipid as EMCL rather than IMCL in the muscle compared with controls. In some animal models, decreasing FAO by pharmacological inhibition of CPT-1 with etomoxir or by reducing long-chain acyl-CoA dehydrogenase activity has been reported to lead to intracellular lipid accumulation (3, 56). Our study is the first report to demonstrate that, in humans with LCHAD deficiency, an enzyme distal to CPT-1, there may be preferentially lipid deposition in the EMCL space. The mechanism for preferential storage of lipid in the EMCL space is unknown, but a recently published study in mice treated with oxfenicine, a pharmacological inhibitor of CPT-1B, the predominant isoform of CPT-1 found in muscle, reported similar results (19). Obese mice treated with oxfenicine had lower acyl-CoA, DAG, and ceramides compared with untreated mice. The authors speculate that these reduced levels of CPT-1B were related to a compensatory decrease in fatty acid uptake at the plasma membrane because they observed lower C36, a key fatty acid transporter, in oxfenicine-treated mice. However, this mechanism would not be consistent with elevated long-chain acylcarnitines in our patients because fatty acids must enter the cell and the mitochondria before they are metabolized to these partial FAO products.
HMW adiponectin levels were lower in patients with LCHAD deficiency compared with controls. Although the regulation of adiponectin synthesis, release, and the feedback signaling that controls these processes is not fully understood, low HMW adiponectin has been associated with increased adiposity. Adiponectin signaling in muscle and liver increases FAO through activation of AMP kinase (54). Perhaps the absence of the downstream increase in FAO or accumulation of a product upstream from the FAO defect lowered adiponectin synthesis or secretion from adipose in our patients. Leptin concentrations were similar between patients with LCHAD deficiency and control subjects, so the decreased adiponectin is not related to an overall effect on adipokine synthesis and release.
A large body of evidence suggests a connection between impaired FAO and development of insulin resistance (23, 34). We show that patients with LCHAD deficiency had lower daily energy expenditures, increased percent body fat, increased long-chain acylcarnitine levels, and lower levels of HMW adiponectin, all characteristics that would be expected to be associated with impaired glucose metabolism. Yet, despite their inherent mitochondrial defect in FAO and these phenotypic characteristics, our data show that glucose and insulin responses following an oral glucose load, as well as estimated insulin sensitivity, were the same in LCHAD-deficient patients compared with normal controls. In addition, patients with LCHAD deficiency had similar suppression of free fatty acid levels after the oral glucose load as control subjects, suggesting no impairment of adipose tissue hormone-sensitive lipase responsiveness to insulin. These findings are consistent with the existing literature in which, to our knowledge, no patient with LCHAD deficiency has been reported to have developed either impaired glucose tolerance or type 2 diabetes mellitus (T2DM). In the context of glucose and lipid metabolism, the low HMW adiponectin concentration deserves additional consideration. LCHAD-deficient patients are the first patient group we are aware of who have low HMW adiponectin levels but normal estimates of insulin sensitivity, IMCL, and plasma triglyceride levels. This appears to be a unique population in which circulating HMW adiponectin levels are incongruent to, or disconnected from, both measures of glucose clearance or IMCL accumulation.
Lipids are thought to play a key role in islet cell secretion. Pancreatic β-cell FAO is associated with decreased insulin secretion, whereas fatty acid/glucose cycling in the cytosol of the β-cell is associated with increased insulin secretion (11, 12, 50). Patients with the FAO disorder short-chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD) deficiency present with infantile hyperinsulinemic hypoglycemia (8). It was recently demonstrated that null mutations in the SCHAD gene result in loss of a protein/protein interaction between SCHAD and glutamate dehydrogenase (GDH), causing an overstimulation of GDH and increased insulin secretion from the islet cells (15, 26). The present study is the first that we are aware of to investigate insulin secretory capacity among humans with impaired long-chain FAO. We observed normal insulin secretory capacity as estimated by ISI in the LCHAD patients, suggesting the defect in long-chain FAO did not impair β-cell function. It is possible the block in long-chain FAO results in increased fatty acid/glucose cycling in the cytosol that potentiates insulin secretion in patients with LCHAD deficiency, but more detailed study of insulin secretion is needed to determine if that is true. In contrast to SCHAD deficiency, subjects with LCHAD deficiency do not exhibit hyperinsulinemia.
We can make several observations regarding accumulation of mitochondrial metabolic intermediates and glucose metabolism. Despite similar substrate availability for oxidation in target tissues, LCHAD-deficient patients had dramatically increased long-chain acylcarnitines. Increased acylcarnitines, byproducts of partial FAO, have been associated with insulin resistance in both rodent and human studies (2, 23, 30). We observed extremely elevated long-chain acylcarnitines in our LCHAD-deficient patients in the context of normal carbohydrate metabolism. With levels of long-chain acylcarnitines nearly 100-fold higher than have been reported in patients with diabetes (2, 30), our data suggest that mitochondrial dysfunction at the level of long-chain FAO that results in the accumulation of long-chain acylcarnitines is not sufficient by itself to impair glucose clearance or alter insulin secretion. Instead, our data are consistent with elevations in long-chain acylcarnitines being related to increased adiposity in diabetic populations (30) rather than resulting from insulin resistance.
Medium-chain acylcarnitine species have been reported to be elevated among subjects with T2DM and in diet-induced insulin-resistant animal models (2, 22, 23, 30, 35). We observed similar levels of medium-chain acylcarnitines and glucose tolerance in the LCHAD-deficient patients compared with controls, so we cannot draw conclusions about relationships between this FAO intermediate and glucose metabolism. Similarly, many reports have observed elevated short-chain acylcarnitines among subjects with T2DM (30, 35). Short-chain acylcarnitines can arise from the catabolism of long-chain fatty acids to shorter chain lengths as well as from various amino acids. Because subjects with LCFAO disorders do not generate short-chain acylcarnitines through oxidation of fats, we considered whether oxidation of alternative substrates that contribute to these metabolites is occurring. The primary alternative substrate is the BCAA. However, we found no difference in BCAA concentrations between the patients with LCHAD deficiency and control subjects. Because the synthesis of short-chain acylcarnitines from BCAA occurs distal to the rate-limiting step in BCAA catabolism, branched-chain α-keto acid dehydrogenase complex, it is possible that this process would not alter circulating concentrations of these abundant amino acids.
The current study has several limitations. LCHAD deficiency is very rare, and recruiting and studying subjects is both costly and difficult, resulting in small numbers of study subjects. Knowing this, we chose very sensitive methods to quantify energy expenditure and body composition, including ectopic fat accumulation. To reduce subject burden, though, we relied on parameters derived from an oral glucose tolerance test to estimate insulin sensitivity and secretion. It is possible that, with more direct measurement techniques, such as the hyperinsulinemic-euglycemic clamp or using muscle tissue derived from biopsy samples, subtle differences in insulin sensitivity could have been detected between the patients and controls. In addition, all subjects with LCHAD deficiency were treated with a fat-restricted diet and sufficient calories from carbohydrates to prevent metabolic decomposition, although the control subjects were not required to adhere to a similar diet. By studying the LCHAD subjects on a low-fat diet, our patients avoided a dietary fat challenge that would be potentially needed to sufficiently “stress” mitochondrial function and result in the accumulation of intermediate products of FAO (e.g., DAGs, BCAAs) and IMCL that would predict insulin resistance. However, a high-fat diet is contraindicated in these patients because of high risk for hypoglycemia and rhabdomyolysis. In addition, our patient group demonstrated significantly impaired fat oxidation, even in the fasting state, as evidenced by verified clinical symptomatic episodes of hypoglycemia and rhabdomyolysis, lower fat substrate oxidation, and markedly elevated long-chain acylcarnitines than controls; yet, no differences in fasting glucose or insulin levels were detected. Another confounding factor could be an adaptive response in glucose metabolism among patients dependent upon glucose for energy production, but no data on this possible difference is currently available.
In conclusion, we found that, despite markedly lower FAO, decreased total energy expenditure, and lower HMW adiponectin levels compared with controls, LCHAD-deficient patients have normal glucose tolerance and estimated insulin sensitivity and secretion. Interestingly, patients with LCHAD deficiency did not store more lipid in the IMCL but preferentially stored more lipid in the EMCL compared with controls. Long-chain acylcarnitines were significantly elevated among LCHAD-deficient patients but medium-chain acylcarnitines and BCAAs were not different between groups. Increased levels of long-chain acylcarnitines did not alter glucose tolerance among our subjects, which suggests that the specific acylcarnitine species resulting from incomplete LCFAO are biomarkers of incomplete FAO and potentially of mitochondrial dysfunction but do not independently induce insulin resistance. Inherited defects of long-chain FAO with an obligatory requirement for glucose to produce ATP did not lead to impaired glucose metabolism, suggesting a substantial decrease in the FAO pathway by itself is not sufficient to induce insulin resistance or impaired glucose tolerance in humans.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants K01-DK-071869 (M. B. Gillingham) and R01-DK-071161 (J. Q. Purnell), the American Diabetes Association Grant 01-06-JF-19 (M. B. Gillingham), and Oregon Clinical & Translational Research Institute Grant UL1TR000128 from the National Center for Advancing Translational Sciences.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: M.B.G., C.O.H., and J.Q.P. conception and design of research; M.B.G., C.O.H., D.A.S., and D.M. performed experiments; M.B.G., C.O.H., and J.Q.P. analyzed data; M.B.G., C.O.H., D.A.S., D.M., and J.Q.P. interpreted results of experiments; M.B.G. prepared figures; M.B.G. drafted manuscript; M.B.G., C.O.H., D.A.S., D.M., and J.Q.P. edited and revised manuscript; M.B.G., C.O.H., D.A.S., D.M., and J.Q.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Michael O. Thorner, University of Virginia, Charlottesville, VA, for assistance in completing the total and acyl ghrelin level measurements. We thank Dr. Dawn Peters for statistical guidance and Dr. Rachel Dresbeck, Oregon Health & Science University, Portland, OR, for critical review of this manuscript.
REFERENCES
- 1. Abu-Elheiga L, Oh W, Kordari P, Wakil SJ. Acetyl-CoA carboxylase 2 mutant mice are protected against obesity and diabetes induced by high-fat/high-carbohydrate diets. Proc Natl Acad Sci USA 100: 10207–10212, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adams SH, Hoppel CL, Lok KH, Zhao L, Wong SW, Minkler PE, Hwang DH, Newman JW, Garvey WT. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J Nutr 139: 1073–1081, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alam N, Saggerson ED. Malonyl-CoA and the regulation of fatty acid oxidation in soleus muscle. Biochem J 334: 233–241, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arnold GL, Van Hove J, Freedenberg D, Strauss A, Longo N, Burton B, Garganta C, Ficicioglu C, Cederbaum S, Harding C, Boles RG, Matern D, Chakraborty P, Feigenbaum A. A Delphi clinical practice protocol for the management of very long chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab 96: 85–90, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Befroy DE, Petersen KF, Dufour S, Mason GF, de Graaf RA, Rothman DL, Shulman GI. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes 56: 1376–1381, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bruce CR, Hoy AJ, Turner N, Watt MJ, Allen TL, Carpenter K, Cooney GJ, Febbraio MA, Kraegen EW. Overexpression of carnitine palmitoyltransferase-1 in skeletal muscle is sufficient to enhance fatty acid oxidation and improve high-fat diet-induced insulin resistance. Diabetes 58: 550–558, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bruce CR, Mertz VA, Heigenhauser GJ, Dyck DJ. The stimulatory effect of globular adiponectin on insulin-stimulated glucose uptake and fatty acid oxidation is impaired in skeletal muscle from obese subjects. Diabetes 54: 3154–3160, 2005 [DOI] [PubMed] [Google Scholar]
- 8. Clayton PT, Eaton S, Aynsley-Green A, Edginton M, Hussain K, Krywawych S, Datta V, Malingre HE, Berger R, van den Berg IE. Hyperinsulinism in short-chain l-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of beta-oxidation in insulin secretion. J Clin Invest 108: 457–465, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 50: 1714–1719, 2001 [DOI] [PubMed] [Google Scholar]
- 10. Davidson A. Blocking. In: Statistical Models. Cambridge, UK: Cambridge Univ Pres2, 2003, chapt. 9A, sect. 1 [Google Scholar]
- 11. Dobbins RL, Chester MW, Daniels MB, McGarry JD, Stein DT. Circulating fatty acids are essential for efficient glucose-stimulated insulin secretion after prolonged fasting in humans. Diabetes 47: 1613–1618, 1998 [DOI] [PubMed] [Google Scholar]
- 12. Dobbins RL, Chester MW, Stevenson BE, Daniels MB, Stein DT, McGarry JD. A fatty acid- dependent step is critically important for both glucose- and non-glucose-stimulated insulin secretion. J Clin Invest 101: 2370–2376, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gillingham M, Van Calcar S, Ney D, Wolff J, Harding C. Dietary management of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD). A case report and survey. J Inherit Metab Dis 22: 123–131, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gillingham MB, Connor WE, Matern D, Rinaldo P, Burlingame T, Meeuws K, Harding CO. Optimal dietary therapy of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Mol Genet Metab 79: 114–123, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heslegrave AJ, Hussain K. Novel insights into fatty acid oxidation, amino acid metabolism, and insulin secretion from studying patients with loss of function mutations in 3-hydroxyacyl-CoA dehydrogenase. J Clin Endocrinol Metab 98: 496–501, 2013 [DOI] [PubMed] [Google Scholar]
- 16. Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440: 944–948, 2006 [DOI] [PubMed] [Google Scholar]
- 17. Iglesias MA, Ye JM, Frangioudakis G, Saha AK, Tomas E, Ruderman NB, Cooney GJ, Kraegen EW. AICAR administration causes an apparent enhancement of muscle and liver insulin action in insulin-resistant high-fat-fed rats. Diabetes 51: 2886–2894, 2002 [DOI] [PubMed] [Google Scholar]
- 18. Jequier E, Acheson K, Schutz Y. Assessment of energy expenditure and fuel utilization in man. Annu Rev Nutr 7: 187–208, 1987 [DOI] [PubMed] [Google Scholar]
- 19. Keung W, Ussher JR, Jaswal JS, Raubenheimer M, Lam VH, Wagg CS, Lopaschuk GD. Inhibition of carnitine palmitoyltransferase-1 activity alleviates insulin resistance in diet-induced obese mice. Diabetes 62: 711–720, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kirkwood B, Sterne J. Paired measurements In: Essential Medical Statistics. Oxford, UK: Blackwell, 2003, chapt. 7, sect. 6 [Google Scholar]
- 21. Klopfenstein BJ, Kim MS, Krisky CM, Szumowski J, Rooney WD, Purnell JQ. Comparison of 3 T MRI and CT for the measurement of visceral and subcutaneous adipose tissue in humans. Br J Radiol 85: e826–e830, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koves TR, Li P, An J, Akimoto T, Slentz D, Ilkayeva O, Dohm GL, Yan Z, Newgard CB, Muoio DM. Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J Biol Chem 280: 33588–33598, 2005 [DOI] [PubMed] [Google Scholar]
- 23. Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 7: 45–56, 2008 [DOI] [PubMed] [Google Scholar]
- 24. Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, Roden M, Shulman GI. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia 42: 113–116, 1999 [DOI] [PubMed] [Google Scholar]
- 25. Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, Zhang D, Woo DK, Shadel GS, Ladiges W, Rabinovitch PS, Santos JH, Petersen KF, Samuel VT, Shulman GI. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab 12: 668–674, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li C, Chen P, Palladino A, Narayan S, Russell LK, Sayed S, Xiong G, Chen J, Stokes D, Butt YM, Jones PM, Collins HW, Cohen NA, Cohen AS, Nissim I, Smith TJ, Strauss AW, Matschinsky FM, Bennett MJ, Stanley CA. Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency involves activation of glutamate dehydrogenase. J Biol Chem 285: 31806–31818, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu J, Prudom CE, Nass R, Pezzoli SS, Oliveri MC, Johnson ML, Veldhuis P, Gordon DA, Howard AD, Witcher DR, Geysen HM, Gaylinn BD, Thorner MO. Novel ghrelin assays provide evidence for independent regulation of ghrelin acylation and secretion in healthy young men. J Clin Endocrinol Metab 93: 1980–1987, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 22: 1462–1470, 1999 [DOI] [PubMed] [Google Scholar]
- 29. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28: 412–419, 1985 [DOI] [PubMed] [Google Scholar]
- 30. Mihalik SJ, Goodpaster BH, Kelley DE, Chace DH, Vockley J, Toledo FG, DeLany JP. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity (Silver Spring) 18: 1695–1700, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miyazaki Y, Matsuda M, DeFronzo RA. Dose-response effect of pioglitazone on insulin sensitivity and insulin secretion in type 2 diabetes. Diabetes Care 25: 517–523, 2002 [DOI] [PubMed] [Google Scholar]
- 32. Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, Neschen S, White MF, Bilz S, Sono S, Pypaert M, Shulman GI. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest 115: 3587–3593, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Muoio DM. Intramuscular triacylglycerol and insulin resistance: guilty as charged or wrongly accused? Biochim Biophys Acta 1801: 281–288, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Muoio DM, Newgard CB. Fatty acid oxidation and insulin action: when less is more. Diabetes 57: 1455–1456, 2008 [DOI] [PubMed] [Google Scholar]
- 35. Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, Rochon J, Gallup D, Ilkayeva O, Wenner BR, Yancy WS, Jr, Eisenson H, Musante G, Surwit RS, Millington DS, Butler MD, Svetkey LP. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 9: 311–326, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 350: 664–671, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, Cline GW, DePaoli AM, Taylor SI, Gorden P, Shulman GI. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest 109: 1345–1350, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Petersen KF, Shulman GI. Pathogenesis of skeletal muscle insulin resistance in type 2 diabetes mellitus. Am J Cardiol 90: 11G–18G, 2002 [DOI] [PubMed] [Google Scholar]
- 39. Potter BK, Little J, Chakraborty P, Kronick JB, Evans J, Frei J, Sutherland SC, Wilson K, Wilson BJ. Variability in the clinical management of fatty acid oxidation disorders: results of a survey of Canadian metabolic physicians. J Inherit Metab Dis 35: 115–123, 2012 [DOI] [PubMed] [Google Scholar]
- 40. Rinaldo P, Matern D, Bennett MJ. Fatty acid oxidation disorders. Annu Rev Physiol 64: 477–502, 2002 [DOI] [PubMed] [Google Scholar]
- 41. Saudubray JM, Martin D, de Lonlay P, Touati G, Poggi-Travert F, Bonnet D, Jouvet P, Boutron M, Slama A, Vianey-Saban C, Bonnefont JP, Rabier D, Kamoun P, Brivet M. Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis 22: 488–502, 1999 [DOI] [PubMed] [Google Scholar]
- 42. Schmidt-Sommerfeld E, Penn D, Duran M, Rinaldo P, Bennett MJ, Santer R, Stanley CA. Detection and quantitation of acylcarnitines in plasma and blood spots from patients with inborn errors of fatty acid oxidation. Prog Clin Biol Res 375: 355–362, 1992 [PubMed] [Google Scholar]
- 43. Schoeller DA. Recent advances from application of doubly labeled water to measurement of human energy expenditure. J Nutr 129: 1765–1768, 1999 [DOI] [PubMed] [Google Scholar]
- 44. Schooneman MG, Vaz FM, Houten SM, Soeters MR. Acylcarnitines: reflecting or inflicting insulin resistance? Diabetes 62: 1–8, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schroeder MA, Atherton HJ, Dodd MS, Lee P, Cochlin LE, Radda GK, Clarke K, Tyler DJ. The cycling of acetyl-coenzyme A through acetylcarnitine buffers cardiac substrate supply: a hyperpolarized 13C magnetic resonance study. Circ Cardiovasc Imaging 5: 201–209, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smith EH, Matern D. Acylcarnitine analysis by tandem mass spectrometry. Curr Protoc Hum Genet Chapter 17: 11–20, 2010 [DOI] [PubMed] [Google Scholar]
- 47. Soeters MR, Sauerwein HP, Duran M, Wanders RJ, Ackermans MT, Fliers E, Houten SM, Serlie MJ. Muscle acylcarnitines during short-term fasting in lean healthy men. Clin Sci (Lond) 116: 585–592, 2009 [DOI] [PubMed] [Google Scholar]
- 48. Spiekerkoetter U, Bastin J, Gillingham M, Morris A, Wijburg F, Wilcken B. Current issues regarding treatment of mitochondrial fatty acid oxidation disorders. J Inherit Metab Dis 33: 555–561, 2010 [DOI] [PubMed] [Google Scholar]
- 49. Spiekerkoetter U, Lindner M, Santer R, Grotzke M, Baumgartner MR, Boehles H, Das A, Haase C, Hennermann JB, Karall D, de Klerk H, Knerr I, Koch HG, Plecko B, Roschinger W, Schwab KO, Scheible D, Wijburg FA, Zschocke J, Mayatepek E, Wendel U. Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis 32: 498–505, 2009 [DOI] [PubMed] [Google Scholar]
- 50. Stein DT, Esser V, Stevenson BE, Lane KE, Whiteside JH, Daniels MB, Chen S, McGarry JD. Essentiality of circulating fatty acids for glucose-stimulated insulin secretion in the fasted rat. J Clin Invest 97: 2728–2735, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Szczepaniak LS, Babcock EE, Schick F, Dobbins RL, Garg A, Burns DK, McGarry JD, Stein DT. Measurement of intracellular triglyceride stores by H spectroscopy: validation in vivo. Am J Physiol Endocrinol Metab 276: E977–E989, 1999 [DOI] [PubMed] [Google Scholar]
- 52. Van Hove JL, Kahler SG, Feezor MD, Ramakrishna JP, Hart P, Treem WR, Shen JJ, Matern D, Millington DS. Acylcarnitines in plasma and blood spots of patients with long-chain 3- hydroxyacyl-coenzyme A dehydrogenase defiency. J Inherit Metab Dis 23: 571–582, 2000 [DOI] [PubMed] [Google Scholar]
- 53. Violante S, Ijlst L, van Lenthe H, de Almeida IT, Wanders RJ, Ventura FV. Carnitine palmitoyltransferase 2: New insights on the substrate specificity and implications for acylcarnitine profiling. Biochim Biophys Acta 1802: 728–732, 2010 [DOI] [PubMed] [Google Scholar]
- 54. Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 8: 1288–1295, 2002 [DOI] [PubMed] [Google Scholar]
- 55. Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 7: 941–946, 2001 [DOI] [PubMed] [Google Scholar]
- 56. Zhang D, Liu ZX, Choi CS, Tian L, Kibbey R, Dong J, Cline GW, Wood PA, Shulman GI. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc Natl Acad Sci USA 104: 17075–17080, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]