

Summary
The flexible and heterogeneous nature of carbohydrate chains often renders glycoproteins refractory to traditional structure determination methods. Small Angle X-ray scattering (SAXS) can be a useful tool for obtaining structural information of these systems. All-atom modeling of glycoproteins with flexible glycan chains was applied to interpret the solution SAXS data for a set of glycoproteins. For simpler systems (single glycan, with a well defined protein structure), all-atom modeling generates models in excellent agreement with the scattering pattern, and reveals the approximate spatial occupancy of the glycan chain in solution. For more complex systems (several glycan chains, or unknown protein substructure), the approach can still provide insightful models, though the orientations of glycans become poorly determined. Ab initio shape reconstructions appear to capture the global morphology of glycoproteins, but in most cases offer little information about glycan spatial occupancy. The all-atom modeling methodology is available as a webserver at http://modbase.compbio.ucsf.edu/allosmod-foxs.
Introduction
The covalent addition of carbohydrate groups to proteins is one of the most common post-translational modifications, with over half of the human proteome containing some degree of glycosylation (Brooks, 2009). Glycosylation can act to stabilize proteins, shield the underlying protein from proteases, increase solubility, and often plays a critical role in cell-cell and host-pathogen interactions (Shental-Bechor and Levy, 2008). Despite their importance, glycoproteins as a class remain a challenging target for structural biology (Walsh, 2010). The high degree of flexibility of the glycans presented on the protein surface as well as the significant micro and macro glycan heterogeneity exhibited within a purified population of glycoprotein (Imberty and Perez, 2000), limit the efficacy of X-ray crystallography as a tool for their structural determination. Glycan modifications also limit the utility of NMR spectroscopy due to detrimental effects on rotational correlation times and significant chemical shift overlap (Slynko et al., 2009).
Small angle X-ray scattering (SAXS) has become a versatile tool for examining macromolecules in the native solution state. It can be used to study disordered systems and molecules with significant internal flexibility (Bernado, 2010; Hammel, 2012; Jacques and Trewhella, 2010; Mertens and Svergun, 2010; Pelikan et al., 2009; Putnam et al., 2007; Rambo and Tainer, 2010). Two caveats add a layer of complexity for applying SAXS to study glycoproteins, namely the different scattering contrast of the carbohydrate relative to the protein (Hammel et al., 2002) and the disproportionate layer of hydration associated with the sugars (Wittmann, 2007). Despite these limitations, the low-resolution morphologies of a number of glycoproteins determined by SAXS have been reported (Bernocco et al., 2003; Guttman et al., 2012; Lynn et al., 2005). While most of these studies are unable to directly visualize the influence or position of the glycans, in some instances the glycan positions appeared to be identifiable (Hammel et al., 2002). Rigid body refinement and molecular dynamics guided by SAXS data has been established as a method for obtaining more detailed models, but the current programs are not suited for taking glycosylation into account (Pelikan et al., 2009).
NMR studies of isolated carbohydrate chains and molecular dynamics have shown that significant internal flexibility exists within glycan chains (Woods et al., 1998). All-atom force fields have been developed for assessing the internal structures of sugars in silico (Frank and Schloissnig, 2010; Kirschner et al., 2008) and computational modeling has been useful for generating fully glycosylated structures using the non-glycosylated template (Bohne-Lang and von der Lieth, 2005; Renouf and Hounsell, 1995). However, experimental validation of these structural models has been limited.
Here, we establish the approach of assessing large ensembles of glycoprotein models against SAXS data to test the validity of the models. Concurrently, we examine the reliability of SAXS ab initio reconstruction for glycoproteins. Lastly, we demonstrate the accuracy of MW determination from the SAXS intensity for glycoproteins. This report provides a guideline and documents limitations of SAXS applied to the study of glycoproteins.
Results/Discussion
Molecular mass determination of glycoproteins
Many classic techniques for molecular weight (MW) determination of proteins and complexes yield inaccurate estimates when applied to proteins with significant glycan content (Lewis and Junghans, 2000; Wen et al., 1996). In our analysis, the MWs estimated from size exclusion chromatography and native polyacrylamide gel electrophoresis (N-PAGE) are significantly off for proteins with high glycan content, in some cases by a factor of two (Table 1). MW estimates from dynamic light scattering, which assumed a spherical model, were overall more accurate than SEC and N-PAGE, but the masses of several glycoproteins were still overestimated. This is probably the effect of flexible glycans contributing disproportionately to the observed hydrodynamic radius and thus the inferred globular MW. In contrast, accurate MW values are obtained by static light scattering (SLS) for every sample examined.
Table 1.
Molecular mass calculations (kDa)
| % mass glycan | Expecteda | SLSb | DLSc | I(0) (water) | I(0) (stds) | SAXS MoWd | Autoporodd | N- PAGE | SECe | |
|---|---|---|---|---|---|---|---|---|---|---|
| Lysozyme | 0 | 14.3 | 14.3 | 10.6 +/− 0.8 | 12.7 | 10.9 | 8.6 | 11 | ND | 3.4 |
| RNAseA | 0 | 13.7 | 15.1 | 10.3 +/− 1 | 15.9 | ND | 9.9/9.9 | 10.3/10 | ND | 10.7 |
| RNAseB | 9.1 | 15.1 | 15.3 | 13 +/− 1.4 | 16.8 | 14.8 | 10.4/12.2 | 11.4/11.6 | ND | 13.5 |
| a1AGP | 40.2 | 36.1 | 35.2 | 46.1 +/− 1.5 | 35.9 | 34.0 | 41.8/39.4 | 60.7/56.5 | 38.3 | 75.2 |
| Fetuin | 23.6 | 47.6 | 48 | 65 +/− 2.1 | 46.9 | 42.7 | 49.4/44.9 | 65/65.2 | 75.8 | 111.1 |
| 17b Fc | 6.0 | 53.6 | 62.5 | 56.6 +/− 2.1 | 57.9 | 60.2 | 51.1 | 47.1 | smear | 46.2 |
| BHA | 17.2 | 203.7 | 184.7 | 204.6 +/− 6.4 | 194.5 | 183.6 | 176.0 | 252.8 | 322 | 316.2 |
MW calculated for the most abundant glycoforms
MW estimated from static forward scattering. Uncertainty in this measurement is primarily dependent on the uncertainty in concentration determination, expected to be less than 15%.
MW estimation from dynamic light scattering assuming a spherical model
MW estimation from SAXSMoW using q range of 0–0.45 (Fischer et al., 2010). The second set of values for SAXSMow and Autoporod (Petoukhov et al., 2007) were obtained from the FPLC-SAXS data sets. Uncertainty is expected to be around 10%.
MW estimated from size exclusion chromatography
An accurate MW derived from SAXS measurements is an important check for ensuring that the data is usable for structural interpretation (Jacques and Trewhella, 2010). Several methods have been established to calculate the molecular weight from SAXS data (Fischer et al., 2010; Mylonas and Svergun, 2007). The MW determined using the SAXS intensity at zero angle, I(0), relative to either water or a reference protein (in this case RNAseA), appears to be accurate for glycoproteins, provided that any contrast offset and differences in the partial specific volume (ν̄) are accounted for (Table 1). On the other hand, MW estimates of the glycoproteins from the Porod volume with autoporod (Petoukhov et al., 2007) were less accurate than those obtained from I(0) because, as expected, Porod’s law and the volume approximation breaks down when applied to non-compact molecules with non-uniform scattering contrast (Mertens and Svergun, 2010; Porod, 1982). The more recently developed SAXSMoW calculation (Fischer et al., 2010), appears to provide more accurate MW determination than autoporod. We note that SAXSMoW calculations were far less accurate when the data was truncated below q = 0.45 Å−1; therefore, data quality at the higher angles seems critical for accuracy.
From the current SAXS I(0) and SLS MW determination, it is clear that all of the proteins examined are monomeric in solution, except for IgG Fc (dimer) and BHA (trimer). In contrast, the inaccurate masses inferred from size exclusion chromatography, native gels, and even autoporod would lead one to mistakenly conclude that several of these species are dimers. The unique case is the Fc portion of human IgG, which shows a lower value with I(0) and better accuracy with the Porod based MW estimation. Unlike most glycoproteins where the glycans are at the periphery of the protein interacting with the solvent, the Fc domain has the glycans positioned at the center of the molecule, forming a part of the dimeric interface (Deisenhofer, 1981). The restrained nature of these glycans results in a more compact molecule for which the Porod-based MW approximation is suitable.
All-atom modeling with well-defined templates
RNAseA/B
Bovine RNAseA and RNAseB serve as a useful comparison for examining the scattering effect of glycans as the two proteins differ only in that RNAseB has a single high-mannose type glycan chain at N34. Furthermore both RNAseA and RNAseB have been shown by NMR spectroscopy and X-ray crystallography to have highly similar protein structures (Williams et al., 1987). SAXS analysis with online size exclusion chromatography resolved monomeric RNAseA (Figure S1A) yielding an Rg consistent with earlier reports (Mylonas and Svergun, 2007) and a Porod volume and Dmax in good agreement with those predicted from the NMR structure (Santoro et al., 1993) (Table S1). The SAXS pattern is also consistent with the theoretical scattering pattern calculated from the NMR ensemble of RNAseA with a χ of 1.47 +/− 0.05 (Figure 1A).
Figure 1.
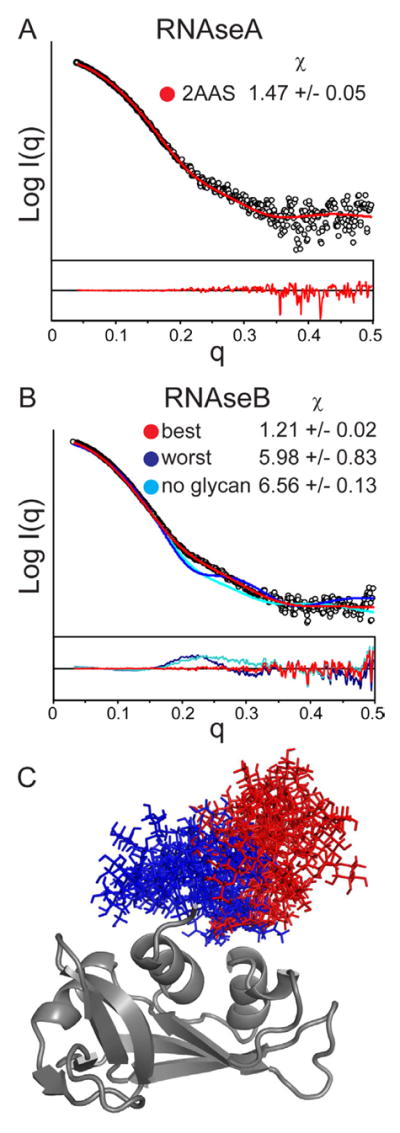
A) SAXS profile of RNAseA (black circles) and the theoretical profile of the best-fit model from the NMR ensemble (2AAS) (red). B) SAXS profile of RNAseB (black circles) with fits for the best model (lowest χ, red), poorest models (highest χ, blue), and model of RNAseB without the glycan (cyan). Residual plots are shown below. Results of the top ten averages (+/− standard deviation) of the fits are reported in the inset. FPLC-SAXS traces and Guinier plots are shown in Figures S1 and S3. C) Glycan positions are shown for the ten best (red) and poorest (blue) fitting models.
The SAXS data for RNAseB showed a slightly larger Rg, Porod volume, and Dmax, as expected due to the additional glycan chain (Table S1). The Kratky plots of RNAseA and RNAseB are similar, indicating a comparable degree of overall compactness (Putnam et al., 2007) (Figure S3). Several structural ensembles of RNAseB were generated with a range of high-mannose type glycan chain sizes from 3 to 9 mannose moieties to test the effect of glycan size on the fits to the scattering data. The single best fits and overall χ values from each ensemble show that the models with 5 mannose groups (Man5) agree most closely with the scattering data (Table S2). This is already a promising result, as it is known that the Man5 glycoform is the predominant glycoform in RNAseB, with Man6 through Man9 in much lower abundance (Guttman, 1997).
A comparison of the best and poorest fitting RNAseB structures with a Man5 glycan indicates a clear difference in glycan orientation (Figure 1B, C). In the ten best fitting models (χ of 1.21 +/− 0.02), the glycan chain protrudes outward from the protein, whereas in the models with the worst fits (χ of 5.98 +/− 0.83), the glycan chain is localized close to the protein surface. The glycan orientation revealed from the best fitting models is consistent with crystallographic studies, which concluded that the glycan is relatively disordered and faces into a solvent void in the crystal lattice (Williams et al., 1987).
Since several conformations may exist in solution, especially for flexible systems, a single structural model may not accurately fit the measured SAXS data. The minimal ensemble search (MES) has been developed to find a combination of the smallest possible number of structures that recapitulate the observed SAXS pattern (Pelikan et al., 2009). For the present analyses, MES was attempted to improve the fit, incorporating up to 5 models from each ensemble. For RNAseB, MES led to no improvement in the fit over the single best model. Taken together this indicates that while there is probably high internal flexibility within each glycosidic bond, the spatial occupancy of the full glycan chain is relatively limited. One possible explanation is the carbohydrates can achieve more favorable hydration with the glycan protruding away from the protein. Alternatively, weak interactions may constrain certain dihedrals at the base of the glycan, constraining the overall orientation. Even if some glycan dihedrals were rigid, the current methodology is of insufficient resolution for probing such details.
IgG Fc
The papain-cleaved constant domain (Fc) of human IgG was also isolated and examined. This molecule is a homodimer of the Fc2 and Fc3 domains with a single N-linked glycan on each subunit. The crystal structure of IgG Fc includes the coordinates for the entire glycan chain, which form contacts at the cavity between the 2 Fc2 domains (Deisenhofer, 1981). Initial models of the Fc domains based on the full coordinates in the crystal, including the termini absent in the crystal, resulted in a χ of 1.77 +/− 0.04. Ensembles were generated to probe a diverse occupancy of the glycan chains. The resulting models with the glycans deviating from the dimeric interface showed poorer agreement with the measured SAXS data with χ of 2.17 +/− 0.21.
A wide array of fragment antigen binding (Fab) domains of IgG have been structurally characterized and have shown distinct “elbow” angles (angle between Fv and Fc1). In a recent study, these elbow motions were a requirement for Fab structures to obtain good fits to SAXS data (Schneidman-Duhovny et al., 2012). As the IgG Fc has a similar hinge between the second and third constant domains (Fc2 and Fc3), a structural ensemble was generated allowing hinge motions, while restraining glycans to the crystal coordinates. Agreement with the SAXS data was significantly improved with a χ of 1.42 +/− 0.03 (Figure 2A, Table S2). Thus, for this glycoprotein, the sampling of the N-terminal extension and elbow angle, rather than glycan positions, were critical for generating accurate models. Although the sampling of the 10 residue N-terminal extension is broad, among best fitting models it consistently protrudes away from the protein, rather than near the surface. The best models show some variance in the elbow angle, but MES did not lead to an appreciable improvement in the fit (χ of 1.35 vs. 1.38 for the single best model). This may highlight one limitation of SAXS, namely that several distinct structures can yield very similar SAXS patterns. Despite this caveat, the analysis indicates that IgG Fc is not fixed in the angle suggested by the crystal structure.
Figure 2.

SAXS profile and model fits for IgG Fc (A), a1AGP (B), BHA (C), and Fetuin (D) with residual plots shown below. The resulting χ values for each set are reported in the insets and Table S2. A–C) Theoretical profiles of the best (red), worst (blue) and model without glycans (cyan) are fit against the SAXS data. Overlays of the top 5 models of each ensemble are shown below the data sets as grey cartoons with glycans depicted as blue sticks. D) SAXS profile of Fetuin with theoretical fits for the glycosylated models using the top five structures from I-TASSER as templates. The top 10 best-fitting models are shown for each of the 5 templates. The two predicted cystatin domains (8–118 and 132–230) are shown in orange and cyan, respectively. The region spanning 237–273, predicted to be intrinsically disordered is shown in red. The protease susceptible fragment 277–318 is shown in magenta. N-linked (blue) and O-linked (purple) glycan chains are shown as sticks.
a1AGP
A step up in complexity, alpha-1-glycoprotein (a1AGP) provided another useful test of our all-atom modeling approach since it bears five complex type glycans and the non-glycosylated protein structure has been determined (Schonfeld et al., 2008). The fully glycosylated form has previously been crystallized, but diffraction was poor, presumably due to the relatively disordered glycans (McPherson et al., 1984). Since the glycans don’t seem to alter the tertiary structure (Friedman et al., 1986), models were generated to sample glycan conformations, without any flexibility permitted for the protein beyond the modified Asn residues and the N-terminal tail that was absent from the crystal structures. The resulting structural ensemble of a1AGP with glycans included several structures that were in reasonable agreement with the SAXS data (χ of 1.62 +/− 0.06) (Figure 2B, Table S2).
To test the effect of the glycan size on the resulting fits, the same experimental data was fit to an additional ensemble omitting all sialic acids (NeuAc) at the end of each glycan chain. Modeling without NeuAc generated models with significantly poorer fits than with NeuAc (2.27 +/− 0.27 vs. 1.62 +/− 0.06), with each ensemble sampling 200 models. The calculated Rg from the models without NeuAc (23.93 +/− 0.25) is also less consistent with the SAXS data than when NeuAc was included (25.56 +/− 0.20; Rg obtained from SAXS was 26.5 +/− 0.06). Therefore, similarly to the RNAseB results, including the proper glycan type/size is important for the accuracy of the resulting models.
While the modeling approach generates models consistent with the SAXS data, comparisons of the best-fit models reveal no apparent spatial convergence for any of the glycan chains. MES resulted in an improvement in the fit (χ 1.32 vs. 1.52 for the best single model), indicating that several glycan conformations more accurately describe the solution structure of this system. Unlike with RNAseB, some or all of the glycan chains in a1AGP may in fact have broad spatial sampling. Additionally, the higher number of variables (5 glycan chains and an unresolved N-terminus) will limit the likelihood of capturing a single conformation that most accurately fits the SAXS pattern, even with exhaustive ensemble sizes. However, the fact that good fits are achieved with the glycosylated models indicates that the protein structure in the non-glycosylated crystal structure is consistent with that in the fully glycosylated protein
BHA
Influenza Hemagglutinin is a trimeric viral surface protein that catalyzes host-virus membrane fusion, and contains several glycans to shield itself from the host immune response. Although the crystal structure of bromelain-released influenza hemagglutinin (BHA) has been solved in its glycosylated form, the majority of carbohydrate was not resolved, presumably due to inherent flexibility and disorder (Wilson et al., 1981). Of the 200 all-atom models generated including the full 21 glycans (per trimer), the best ten were consistent with the SAXS data with a χ of 1.77 +/− 0.03 (Figure 2C). There was no clear spatial occupancy bias for the glycans between the best and worst fitting models and MES for this system led to no significant improvement in the goodness of fit over the best individual model (χ 1.71 vs. 1.73). As glycans chains decorate most of the protein surface, changes in any single glycan may only have very minor effects on the overall fit. Therefore, for a system essentially coated in glycan chains, our all-atom modeling approach can assess whether or not the overall structure is consistent with SAXS data, but reveals little about the specific disposition of individual glycans.
Modeling with little structural information: Fetuin
Since no structural data exists for bovine Fetuin, it serves as a good system to test the limits of the all-atom modeling approach for SAXS analysis. Along with the three complex type N-linked glycans, the protein also contains 4 sites of O-linked glycosylation (Figure S2). The Kratky plot indicates that Fetuin is mostly a well-ordered globule with some disordered portions (Figure S3). Based on sequence similarity, the protein has two cystatin domains followed by a 100 residue C-terminal portion. Several algorithms (DISEMBL, DisProt, PrDOS) predict that much of the C-terminal region (residues 240–320) is intrinsically disordered (Ishida and Kinoshita, 2007; Linding et al., 2003; Sickmeier et al., 2007). HS-1, the human homolog of Fetuin, is also known to be proteolytically cleaved at the residues 280–317 (Nawratil et al., 1996). The full sequence of Fetuin was submitted to I-TASSER along with disulfide restraints for in silico structure prediction (Roy et al., 2010; Zhang, 2008). Low C scores of −4.54 and −4.95 for the top two models were achieved with a relatively low clustering density (0.0049 and 0.0033), indicating poor confidence in the models (predicted to be 0.24±0.07 (TM-score) and 18.1±2.4Å (RMSD)). Nevertheless, the coordinates from each of the five models were used as a template for generating glycosylated ensembles to compare against the SAXS data.
2 out of the 5 ensembles produced structural models that were in loose agreement with the SAXS data (χ of 2.06 +/− 0.12 and 1.95 +/− 0.16) (Figure 2D, Table S2). Since these templates have significantly different protein structures, namely a completely different fold within the N-terminal cystatin domain, this presents a limitation of the current methodology. With so many degrees of freedom, it is possible to generate an array of distinct structures with similar consistency to the scattering data. However, all of the starting models without glycans had terrible fits to the SAXS data (χ > 15). Therefore, the incorporation of the glycans into this type of analysis is essential. With additional restraints, this approach could generate a more reliable model of this glycoprotein.
Ab initio reconstructions
The most common method of extracting structural information from SAXS data is the use of ab initio bead modeling to construct a shape envelope of the molecule consistent with the SAXS data (Petoukhov and Svergun, 2007). Typically, several bead models are calculated, aligned, and averaged to generate an average low-resolution model of the molecule. Although this approach can be useful for obtaining coarse-grain shape information, it is not well-suited for examining flexible molecules (Bernado, 2010; Hammel, 2012). Though often attached to an ordered protein substrate, glycans may contribute significant flexibility to a system. We tested the general accuracy of ab initio modeling for glycoproteins using two programs, DAMMIN and GASBOR.
Shape reconstruction was carried out for the RNAseA and RNAseB data sets to examine the overall effects of the single glycan chain. Both GASBOR and DAMMIN produced shape envelopes for RNAseA that were convergent with normalized spatial discrepancy (NSD) 0.63 +/− 0.044 and 0.72 +/− 0.01, respectively (values under 1 are considered to indicate consistency between the individual reconstructions). Docking the NMR structure into the final averaged shape reconstruction showed the expected shape and size for this protein (Figure 3A, B). The same approach for the glycosylated RNAseB also resulted in shape reconstructions with good convergence (NSD of 0.62 +/− 0.038 and 0.82 +/− 0.013). For DAMMIN, the composite, filtered shape envelope had a bulge proximal to the glycan base (Figure 3C). However, the composite GASBOR model only showed an overall larger envelope relative to RNAseA with no clear bulge (Figure 3D). Several of the individual GASBOR models showed glycan-like protrusions; however, the relative feature locations were highly variable. The effects of aligning and filtering the individual models appear to exclude the variable glycan density from the final averaged model. These results echo those reported for a study of Vitronectin, in which the carbohydrates also contributed little to the overall modeled shape (Lynn et al., 2005).
Figure 3.

Shape reconstructions of RNAseA (A, B), RNAseB (C, D), a1AGP (E, F) and Fetuin (G, H) with averaged DAMMIN (left) and GASBOR (right) models shown. Fits are shown in Figure S3. The envelopes were scaled to the measured Porod volume (listed in Table S2). For each protein the best (lowest χ) all-atom model was docked into both enantiomers of the final ab initio shape envelopes using SITUS, only the better enantiomer is shown (Wriggers and Birmanns, 2001). A–D) All-atom models RNAseA and RNAseB are shown as cartoons with glycans as sticks. E–H) The shape reconstructions calculated for the full glycoprotein (light gray) and only the protein substructure alone (dark gray) are shown for a1AGP (E, F) a1AGP and Fetuin (G, H) with N-linked glycans shown as blue sticks. The domains of Fetuin are colored as described in Figure 2, with O-linked glycans shown as purple sticks.
To test how ab initio reconstructions perform for a highly glycosylated system, we ran the same bead modeling approach on the a1AGP SAXS data. The final filtered shape envelope is clearly larger than would be expected for the protein alone, thus the glycan density seems to be reconstructed at least to some degree (Figure 3E, F). Convergence of both GASBOR and DAMMIN ab initio shape reconstructions for a1AGP is weaker than that for RNAseA and RNAseB models (NSD 0.81 +/− 0.04 and 1.50 +/− 0.082, respectively) and it appears that the glycan positions are highly ambiguous. Therefore, beyond approximate shape of the molecule, little information on the glycan arrangement is present in the bead models for this system.
Lastly, we applied this approach to the Fetuin data set, where it would potentially be the most useful in the absence of any structural data. The consistency between the models was moderate (NSDs 0.89 +/− 0.04 and 1.55 +/− 0.058 for DAMMIN and GASBOR, respectively). Both reconstruction programs revealed a similar overall elongated morphology with two lobes (Figure 3G, H). The best fitting all-atom model of Fetuin (from ensemble 4, χ 1.73) was docked into both composite envelopes. Although our all-atom model of Fetuin is speculative, the overall shape is consistent with ab initio models and therefore it may reflect the overall domain and glycan arrangement present in the solution structure.
In conclusion, even with all of the caveats limiting the ab initio approach for glycoproteins, it generates the expected size and a shape consistent with the protein substructure. For the examples studied here, the glycan positions cannot be effectively visualized. Instead, glycans appear to contribute bulk volume to the shape reconstructions or at best a bulge near the glycan attachment site. Since the scattering contrast of the carbohydrate is significantly different from that of the protein, small angle neutron scattering (SANS) with contrast matching should be an effective way to directly resolve glycans (Whitten and Trewhella, 2009). A similar technique is possible for contrast matching with SAXS; however, the high concentrations of solute required for effective contrast matching would render the solution conditions potentially non-physiological and perturbing to protein structure (Putnam et al., 2007).
Conclusions
We have demonstrated that the native molecular weight of glycoproteins can be accurately determined from SAXS, provided that the scattering contrast and partial specific volume are properly taken into account. Ab initio modeling can successfully determine the shape of glycoproteins, but provides little information about the spatial occupancy of the glycans. For systems with known coordinates for the protein component, an all-atom modeling approach is a more effective way to interpret SAXS data, potentially revealing the positioning of the glycans or regions absent from the template structures. Although nearly all glycoproteins are expected to have some degree of glycan heterogeneity, modeling with the most abundant glycoform appears sufficient for generating structures in agreement with SAXS data. We have recently used this technique to identify the disposition of the large V1/V2 loops of HIV gp120, a highly glycosylated system (Guttman et al., 2012). With the new implementation of AllosMod (Weinkam et al., 2012) and FoXS (Schneidman-Duhovny et al., 2010) servers, ensembles of glycosylated conformations can be generated and fit against the scattering data within minutes, allowing rapid sampling and assessment of a wide range of glycoprotein conformations. The webserver can be found at http://modbase.compbio.ucsf.edu/allosmod-foxs/, and has been setup to generate all types of O-linked and N-linked glycans found in mammalian systems. The approach will be valuable as SAXS data will be more readily available with the application of high throughput methodologies (Hura et al., 2009).
Methods
Sample preparation
Fetuin from calf serum, human α1AGP, and RNAseB from bovine pancreas were purchased from Sigma Aldrich. Lysozyme was from MP Biomedicals (Solon, OH) and bovine RNAseA was purchased from (Worthington Biochemicals). Each of these was repurified by size exclusion in PBS (20mM sodium phosphate pH 7.4, 150mM NaCl, 0.02% sodium azide, 1mM EDTA). The Fc portion of IgG 17b were prepared by treatment with papain (Thermo Scientific, Rockville MD) and isolated by size exclusion chromatography. Hemagglutinin from X31 virus particles (Charles River, Wilmington, MA), was cleaved with bromelain (Sigma Aldrich) as described in Compans et al (Compans et al., 1970). Liberated BHA was separated from remaining particles by ultracentrifugation and purified by size exclusion chromatography (Superdex 200) in HBS (10mM Hepes pH 7.4, 150mM NaCl, 0.02% azide). Proteins were concentrated using microcentricon spin filters immediately before analysis (Amicon 10 kDa cutoff, Millipore, Billerica MA). Sample purity was verified by SDS-PAGE and N-PAGE.
Dynamic light scattering
Dynamic and static light scattering measurements were performed at 20°C on a Dynapro Nanostar (Wyatt Technology, Santa Barbara, CA). 30 acquisitions of 5 seconds were collected at a range of concentrations (0.1 to 1mg/mL). Buffer viscosity was calculated with SedNterp (Laue et al., 1992). Data was fit using the Wyatt Dynamics analysis software assuming a spherical model, and a differential index of refraction (dn/dc) of 0.185.
Small-angle X-ray scattering
Small-angle X-ray scattering (SAXS) measurements were conducted on Beam Line 4-2 at the Stanford Synchrotron Radiation Laboratory (Smolsky et al., 2007). The focused 11 keV X-ray beam irradiated a thin-wall quartz capillary cell, which contained a sample aliquot, placed at 1.7 m upstream of the Rayonix MX 225HE detector (Evanston, IL). The detector pixel numbers were converted to the momentum transfer q=4π*sinθ/λ, where 2*θ is the scattering angle and λ the X-ray wavelength of 1.127 Å, using a silver behenate powder standard placed at the capillary position. A set of 16 consecutive 1 second X-ray exposures were made on each sample aliquot at 15°C with 15 μL oscillations (30μL injected) between exposures to minimize radiation damage. Protein scattering data were processed by MarParse and scaled for the transmitted beam intensity integrated for each exposure, and azimuthally averaged (Smolsky et al., 2007). Exposures were averaged with rejection criteria of 1.3 standard deviations relative to the initial exposure to omit frames exhibiting radiation damage or other artifacts. Buffer scattering data were processed in the same way and subtracted from corresponding protein scattering data.
All samples were collected over a broad concentration range (0.25 – 12mg/mL). Data sets at the highest concentration free of self association or inter-particle interference as reflected by the linearity of the Guinier regions (QRg<1.3) were used for further analysis; Lysozyme (1.5mg/mL), RNAseA (1.5mg/mL), RNAseB (1.5mg/mL), a1AGP (0.75mg/mL), Fetuin (1.5mg/mL), BHA (1.0mg/mL) and IgG Fc (1.3mg/mL). The 1-D SAXS curves were processed using GNOM to determine approximate Rg and Dmax values for the P(r) plots, the pairwise distance distribution functions (Petoukhov and Svergun, 2007; Svergun, 1991, 1992).
Molecular weights were calculated for each sample using the zero angle scattering intensity “I(0)” relative to water or RNAseA as a standard (Mylonas and Svergun, 2007). Partial specific volumes (ν̄) were calculated as the weighted average of the protein component 0.7425 (Mylonas and Svergun, 2007), and that based on the glycan from the known composition (Lewis and Junghans, 2000). Due to the higher contrast difference (Δρ) of glycans the difference in contrast between the reference protein (RNAseA) and each glycoprotein was factored using the equation:
Where I(0) is the scattering intensity at zero angle of the standard (std) and protein (prot), C is the concentration, and v is the partial specific volume. The Δρ for glycoproteins was calculated as the difference of the weighted average of protein (4.47 × 1023 e/cm3) and glycan (5.10 × 1023 e/cm3) from the buffer (3.34 × 1023 e/cm3). Molecular weights were also calculated on a relative scale using “SAXS MoW” (q up to 0.45 Å−1) (Fischer et al., 2010), as well as from autoporod (Petoukhov et al., 2007).
Size-exclusion directly online with SAXS (SEC-SAXS) was used to obtain higher quality data sets for RNAseA, RNAseB, a1AGP, and Fetuin. 100uL of each sample at 3–5mg/mL were injected onto a highres Sepharose 200 column (GE Healthcare) with a flowrate of 50uL/min in PBS. The flow was passed through the capillary cell and 1 sec exposures were collected ever 5 seconds. Rg and I(0) for each frame were batch analyzed using autoRg (Petoukhov et al., 2007), and frames with stable Rg values were merged in primus (Konarev et al., 2003). Absorbance, I(0) and Rg values for the elution profiles are shown in Figure S1. The Rg values near the peak shoulder showed no deviations, thus interparticle repulsion/association were not evident under these conditions.
All-atom modeling of glycoproteins
Ensembles of all-atom structural models were generated in MODELLER (Sali and Blundell, 1993) as an adaptation of previous work (Weinkam et al., 2012). Initial structures were generated by addition of glycan chains with ideal geometries derived from CHARMM (Guvench et al., 2008; Guvench et al., 2009), followed by a 1 Å randomization of the full atomic coordinates. The structures are first relaxed with conjugate gradient steps followed by molecular dynamics equilibration at 300K followed by several rounds of simulated annealing. The procedure is repeated to produce 200 models for each protein (unless otherwise stated). The intra-protein interactions consist of bonded (for stereochemistry) and non-bonded (for excluded volume and atomic distance) terms used in standard MODELLER. The atomic distance term is a sum of harmonic energy functions to restrain pairs of atoms relative to the template structure and this term is omitted for atoms within two residues of a glycosylation site and for loops or termini absent in the template structure. Intra-glycan interactions consist of a bonded term that is a sum of harmonic energy functions over all pairwise atomic distances within each sugar monomer. Bonds between sugar monomers move with proper dihedral rotation using distance and angle restraints. The full procedure can be accessed using our webserver at http://salilab.org/allosmod. Details of each glycoprotein template are listed in the following subsections and site-specific glycan types are illustrated in Figure S3.
Next, the theoretical profile from each structural model was calculated and compared against the raw SAXS data using FoXS (Schneidman-Duhovny et al., 2010). As there is still no “best” established method for this type of comparison, we rely on the normalized discrepancy value (χ) to evaluate the goodness of fit (Putnam et al., 2007), as well as on a visual inspection of the fit. Since χ is heavily dependent on the error associated with the measurement, the metric is useful for comparing model SAXS patterns with experimentally measured data within a given set, but less so between sets with different experimental SAXS curves. For the current data sets, we consider a χ of 1.0–1.5 a good fit, whereas χ values above 2 indicate inconsistency between the model and the SAXS data.
We also examine the average and standard deviation among the 10 best fitting models to assess the variance in χ and establish whether or not different modeling strategies result in significantly better fits to the same experimental profile. This type of comparison depends on adequate conformational sampling, which can be gauged by monitoring the best fit given increasingly larger samples. For each data set, the sampling was doubled until the best fit was not significantly better than previous data set (average of 10 best fits minus one standard deviation). In all cases except Fetuin, 200 models were adequate by this criterion.
The ATSAS software package was used to predict Rg, Dmax, and envelope volumes for each structural model (Svergun et al., 1995). Proper glycan stereochemistry and linkage generated from MODELLER were checked with PDBcare (http://www.glycosciences.de/tools/pdb-care/) and Glycosidic linkage angles were analyzed by CARP (http://www.glycosciences.de/tools/carp) using the GlycoMaps database (Lutteke et al., 2005). Figures were made with PyMOL (DeLano, 2002) and Chimera (Pettersen et al., 2004).
RNAseB
The sequence of bovine ribonuclease (RNAse) (uniprot P61823) was modeled utilizing coordinates from the crystal structure of RNAseB (pdb: 1RBB) (Williams et al., 1987) as well as the NMR structure of RNAseA (pdb: 2AAS) (Santoro et al., 1993).
IgG Fc
The IgG Fc portion of Ab 17b was modeled using the coordinates from the crystal structure of IgG Fc (pdb: 1FC1) (Deisenhofer, 1981), together with the established disulfide connectivity for IgG. The coordinates for the complex type glycan with a single (β1–4)Gal on the (1–6)Man were included in the alignment. The hinge region (339–343) between the Fc2 and Fc3 domains was left unaligned to sample different elbow angles.
a1AGP
The sequence of a1AGP (uniprot P02763) was aligned to the sequence from the recently available unglycosylated crystal structure of a1AGP (pdb: 3KQ0, 3APU) (Schonfeld et al., 2008). Complex type tri- and tetra-antennary glycans containing (α2–6) terminal sialic acids were introduced at positions (15, 38, 54, 75, and 85(tetra)) based on site-specific glycan studies (Imre et al., 2005; Treuheit et al., 1992).
Fetuin
The mature sequence (uniprot: P12763) was submitted to I-TASSER structure prediction (Roy et al., 2010; Zhang, 2008) with additional disulfide restraints based on previous findings (Araki et al., 1989). All five I-TASSER models were used as templates for MODELLER runs, including triantennary sialylated complex glycans at Asn 81, 138, 158 and O-linked glycans at S253, T262, S264 and S323, based on previous site specific studies (Edge and Spiro, 1987; Guttman, 1997; Royle et al., 2002) (Figure S2).
Influenza Hemagglutinin (BHA)
The sequence of the bromelain released ecto domain of Influenza Hemagglutinin (BHA) (uniprot P03437) was threaded to the crystal structure (pdb 2HMG) (Wilson et al., 1981). High mannose and complex glycans were introduced at positions as shown in Figure S2. The glycosylation at each of the 7 Asn sites was based on predominant glycoforms observed from LC-MS analysis of peptic fragments (unpublished results), which agree closely with earlier findings (Mir-Shekari et al., 1997).
Ab initio shape reconstructions
The particle distance distribution function [P(r)] plot generated from GNOM (Svergun, 1992) was used for ab initio shape reconstruction using DAMMIN v5.3 and GASBOR22i (Svergun, 1999; Svergun et al., 2001). The number of dummy residues used for GASBOR calculations was increased to account for the difference in scattering of glycans as described previously (Hammel et al., 2002). The bead models were aligned using SUPCOMB13 with enantiomers considered (Kozin and Svergun, 2001), and spatially filtered using DAMFILT (Volkov and Svergun, 2003). Plots of the resulting fits, Guinier plots, and P(r) plots are presented in Figure S3.
Convergence of the models was assessed by the normalized spatial discrepancy (NSD) showing values under 1, except for the GASBOR models for a1AGP and Fetuin with NSDs of 1.50 +/− 0.082 and 1.55 +/−0.058, respectively. Further refinement resulted in no improvement in χ values and had no significant effect on the resulting averaged models. GASBOR models showed a greater variation of goodness of fit (χ2) compared to the DAMMIN models. Therefore, GASBOR models were ranked by goodness of fit (Svergun et al., 2001), and only the top quartile aligned and averaged. The resulting models were converted to a volume envelope using the program pdb2vol within the SITUS2.2 package (Wriggers and Birmanns, 2001). All-atom models were docked into both enantiomers of the volume envelope using the colores module within SITUS2.2. Ab initio reconstructions of non-glycosylated Fetuin and a1AGP were performed starting with theoretical scattering pattern of the protein substructure adding the same relative error present in the glycoprotein SAXS data.
Supplementary Material
Highlights.
MW determined from zero angle scattering is accurate for glycoproteins
All-atom modeling with SAXS data can identify structural features of glycans
SAXS ab initio reconstructions provide little structural information for glycans
Acknowledgments
We thank Tsutomu Matsui, Lester Carter and the staff at SSRL for assistance with SAXS data collection. We are grateful to Dina Schneidman-Duhovny and Michal Hammel for guidance with all-atom modeling, and Dr. Byron Hetrick and Dr. Seung Joong Kim for insightful discussions. 17b IgG was a kind gift of James Robinson and Peter Kwong. We also wish to thank Thomas Lutteke for generous assistance with glycan structure analysis. This work was supported by NIH grant F32-GM097805, R00-GM080352, R01-GM099989 (KKL), and R01-GM083960 (AS). Data collection at SSRL was supported by Grant P41-RR001209 from the National Center for Research Resources.
ABBREVIATIONS
- SAXS
small-angle X-ray scattering
- RNAse
ribonuclease
- a1AGP
alpha-1-acid glycoprotein
- BHA
bromelain cleaved hemagglutinin
- Fc
fragment crystallizable region
- MES
minimal ensemble search
- Rg
radius of gyration
- Rh
radius of hydration
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Araki T, Yoshioka Y, Schmid K. The position of the disulfide bonds in human plasma alpha 2 HS-glycoprotein and the repeating double disulfide bonds in the domain structure. Biochim Biophys Acta. 1989;994:195–199. doi: 10.1016/0167-4838(89)90293-8. [DOI] [PubMed] [Google Scholar]
- Bernado P. Effect of interdomain dynamics on the structure determination of modular proteins by small-angle scattering. Eur Biophys J. 2010;39:769–780. doi: 10.1007/s00249-009-0549-3. [DOI] [PubMed] [Google Scholar]
- Bernocco S, Steiglitz BM, Svergun DI, Petoukhov MV, Ruggiero F, Ricard-Blum S, Ebel C, Geourjon C, Deleage G, Font B, et al. Low resolution structure determination shows procollagen C-proteinase enhancer to be an elongated multidomain glycoprotein. J Biol Chem. 2003;278:7199–7205. doi: 10.1074/jbc.M210857200. [DOI] [PubMed] [Google Scholar]
- Bohne-Lang A, von der Lieth CW. GlyProt: in silico glycosylation of proteins. Nucleic Acids Res. 2005;33:W214–219. doi: 10.1093/nar/gki385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks SA. Strategies for analysis of the glycosylation of proteins: current status and future perspectives. Mol Biotechnol. 2009;43:76–88. doi: 10.1007/s12033-009-9184-6. [DOI] [PubMed] [Google Scholar]
- Compans RW, Klenk HD, Caliguiri LA, Choppin PW. Influenza virus proteins. I. Analysis of polypeptides of the virion and identification of spike glycoproteins. Virology. 1970;42:880–889. doi: 10.1016/0042-6822(70)90337-5. [DOI] [PubMed] [Google Scholar]
- Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry. 1981;20:2361–2370. [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA, USA: DeLano Scientific; 2002. [Google Scholar]
- Edge AS, Spiro RG. Presence of an O-glycosidically linked hexasaccharide in fetuin. J Biol Chem. 1987;262:16135–16141. [PubMed] [Google Scholar]
- Fischer H, de Oliveira Neto M, Napolitano HB, Polikarpov I, Craievich AF. Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. Journal of Applied Crystallography. 2010;43:101–109. [Google Scholar]
- Frank M, Schloissnig S. Bioinformatics and molecular modeling in glycobiology. Cell Mol Life Sci. 2010;67:2749–2772. doi: 10.1007/s00018-010-0352-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman ML, Wermeling JR, Halsall HB. The influence of N-acetylneuraminic acid on the properties of human orosomucoid. Biochem J. 1986;236:149–153. doi: 10.1042/bj2360149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman A. Multistructure sequencing of N-linked fetuin glycans by capillary gel electrophoresis and enzyme matrix digestion. Electrophoresis. 1997;18:1136–1141. doi: 10.1002/elps.1150180719. [DOI] [PubMed] [Google Scholar]
- Guttman M, Kahn M, Garcia NK, Hu SL, Lee KK. Solution Structure, Conformational Dynamics, and CD4-Induced Activation in Full-Length, Glycosylated, Monomeric HIV gp120. J Virol. 2012;86:8750–8764. doi: 10.1128/JVI.07224-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvench O, Greene SN, Kamath G, Brady JW, Venable RM, Pastor RW, Mackerell AD., Jr Additive empirical force field for hexopyranose monosaccharides. J Comput Chem. 2008;29:2543–2564. doi: 10.1002/jcc.21004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvench O, Hatcher ER, Venable RM, Pastor RW, Mackerell AD. CHARMM Additive All-Atom Force Field for Glycosidic Linkages between Hexopyranoses. J Chem Theory Comput. 2009;5:2353–2370. doi: 10.1021/ct900242e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel M. Validation of macromolecular flexibility in solution by small-angle X-ray scattering (SAXS) Eur Biophys J. 2012;41:789–799. doi: 10.1007/s00249-012-0820-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel M, Kriechbaum M, Gries A, Kostner GM, Laggner P, Prassl R. Solution structure of human and bovine beta(2)-glycoprotein I revealed by small-angle X-ray scattering. J Mol Biol. 2002;321:85–97. doi: 10.1016/s0022-2836(02)00621-6. [DOI] [PubMed] [Google Scholar]
- Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imberty A, Perez S. Structure, conformation, and dynamics of bioactive oligosaccharides: theoretical approaches and experimental validations. Chem Rev. 2000;100:4567–4588. doi: 10.1021/cr990343j. [DOI] [PubMed] [Google Scholar]
- Imre T, Schlosser G, Pocsfalvi G, Siciliano R, Molnar-Szollosi E, Kremmer T, Malorni A, Vekey K. Glycosylation site analysis of human alpha-1-acid glycoprotein (AGP) by capillary liquid chromatography-electrospray mass spectrometry. J Mass Spectrom. 2005;40:1472–1483. doi: 10.1002/jms.938. [DOI] [PubMed] [Google Scholar]
- Ishida T, Kinoshita K. PrDOS: prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007;35:W460–464. doi: 10.1093/nar/gkm363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques DA, Trewhella J. Small-angle scattering for structural biology--expanding the frontier while avoiding the pitfalls. Protein Sci. 2010;19:642–657. doi: 10.1002/pro.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner KN, Yongye AB, Tschampel SM, Gonzalez-Outeirino J, Daniels CR, Foley BL, Woods RJ. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J Comput Chem. 2008;29:622–655. doi: 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. Journal of Applied Crystallography. 2003:1277–1282. [Google Scholar]
- Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. J App Cryst. 2001;34:33–41. [Google Scholar]
- Laue TM, Shah BD, Ridgeway TM, Pelletier SL. Analytical Ultracentrifugation in Biochemistry and Polymer Science (Royal Society of Chemistry) 1992 [Google Scholar]
- Lewis MS, Junghans RP. Ultracentrifugal analysis of molecular mass of glycoproteins of unknown or ill-defined carbohydrate composition. Methods in enzymology. 2000;321:136–149. doi: 10.1016/s0076-6879(00)21191-9. [DOI] [PubMed] [Google Scholar]
- Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB. Protein disorder prediction: implications for structural proteomics. Structure. 2003;11:1453–1459. doi: 10.1016/j.str.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Lutteke T, Frank M, von der Lieth CW. Carbohydrate Structure Suite (CSS): analysis of carbohydrate 3D structures derived from the PDB. Nucleic Acids Res. 2005;33:D242–246. doi: 10.1093/nar/gki013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn GW, Heller WT, Mayasundari A, Minor KH, Peterson CB. A model for the three-dimensional structure of human plasma vitronectin from small-angle scattering measurements. Biochemistry. 2005;44:565–574. doi: 10.1021/bi048347s. [DOI] [PubMed] [Google Scholar]
- McPherson A, Friedman ML, Halsall HB. Crystallization of alpha 1-acid glycoprotein. Biochem Biophys Res Commun. 1984;124:619–624. doi: 10.1016/0006-291x(84)91599-7. [DOI] [PubMed] [Google Scholar]
- Mertens HD, Svergun DI. Structural characterization of proteins and complexes using small-angle X-ray solution scattering. Journal of structural biology. 2010;172:128–141. doi: 10.1016/j.jsb.2010.06.012. [DOI] [PubMed] [Google Scholar]
- Mir-Shekari SY, Ashford DA, Harvey DJ, Dwek RA, Schulze IT. The glycosylation of the influenza A virus hemagglutinin by mammalian cells. A site-specific study. J Biol Chem. 1997;272:4027–4036. doi: 10.1074/jbc.272.7.4027. [DOI] [PubMed] [Google Scholar]
- Mylonas E, Svergun DI. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. Journal of Applied Crystallography. 2007;40:s245–s249. [Google Scholar]
- Nawratil P, Lenzen S, Kellermann J, Haupt H, Schinke T, Muller-Esterl W, Jahnen-Dechent W. Limited proteolysis of human alpha2-HS glycoprotein/fetuin. Evidence that a chymotryptic activity can release the connecting peptide. J Biol Chem. 1996;271:31735–31741. doi: 10.1074/jbc.271.49.31735. [DOI] [PubMed] [Google Scholar]
- Pelikan M, Hura GL, Hammel M. Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen Physiol Biophys. 2009;28:174–189. doi: 10.4149/gpb_2009_02_174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov MV, Konarev PV, Kikhney AG, Svergun DI. ATSAS 2.1 - towards automated and web-supported small-angle scattering data analysis. J Appl Cryst. 2007;40:s223–s228. [Google Scholar]
- Petoukhov MV, Svergun DI. Analysis of X-ray and neutron scattering from biomolecular solutions. Curr Opin Struc Biol. 2007;17:562–571. doi: 10.1016/j.sbi.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Porod G. General Theory. In: Kratky O, Gladder O, editors. Small-Angle X-ray Scattering. London: Academic Press; 1982. pp. 17–51. [Google Scholar]
- Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Quarterly reviews of biophysics. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA. Bridging the solution divide: comprehensive structural analyses of dynamic RNA, DNA, and protein assemblies by small-angle X-ray scattering. Curr Opin Struct Biol. 2010;20:128–137. doi: 10.1016/j.sbi.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renouf DV, Hounsell EF. Molecular modelling of glycoproteins by homology with non-glycosylated protein domains, computer simulated glycosylation and molecular dynamics. Adv Exp Med Biol. 1995;376:37–45. doi: 10.1007/978-1-4615-1885-3_4. [DOI] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royle L, Mattu TS, Hart E, Langridge JI, Merry AH, Murphy N, Harvey DJ, Dwek RA, Rudd PM. An analytical and structural database provides a strategy for sequencing O-glycans from microgram quantities of glycoproteins. Anal Biochem. 2002;304:70–90. doi: 10.1006/abio.2002.5619. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Santoro J, Gonzalez C, Bruix M, Neira JL, Nieto JL, Herranz J, Rico M. High-resolution three-dimensional structure of ribonuclease A in solution by nuclear magnetic resonance spectroscopy. J Mol Biol. 1993;229:722–734. doi: 10.1006/jmbi.1993.1075. [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M, Sali A. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010;38:W540–544. doi: 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Kim SJ, Sali A. Integrative structural modeling with small angle X-ray scattering profiles. BMC Struct Biol. 2012;12:17. doi: 10.1186/1472-6807-12-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonfeld DL, Ravelli RB, Mueller U, Skerra A. The 1.8-A crystal structure of alpha1-acid glycoprotein (Orosomucoid) solved by UV RIP reveals the broad drug-binding activity of this human plasma lipocalin. J Mol Biol. 2008;384:393–405. doi: 10.1016/j.jmb.2008.09.020. [DOI] [PubMed] [Google Scholar]
- Shental-Bechor D, Levy Y. Effect of glycosylation on protein folding: a close look at thermodynamic stabilization. Proc Natl Acad Sci U S A. 2008;105:8256–8261. doi: 10.1073/pnas.0801340105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmeier M, Hamilton JA, LeGall T, Vacic V, Cortese MS, Tantos A, Szabo B, Tompa P, Chen J, Uversky VN, et al. DisProt: the Database of Disordered Proteins. Nucleic Acids Res. 2007;35:D786–793. doi: 10.1093/nar/gkl893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slynko V, Schubert M, Numao S, Kowarik M, Aebi M, Allain FH. NMR structure determination of a segmentally labeled glycoprotein using in vitro glycosylation. J Am Chem Soc. 2009;131:1274–1281. doi: 10.1021/ja808682v. [DOI] [PubMed] [Google Scholar]
- Smolsky IL, Liu P, Niebuhr M, Ito K, Weiss TM, Tsuruta H. Biological small-angle X-ray scattering facility at the Stanford Synchrotron Radiation Laboratory. J Appl Cryst. 2007;40:s453–s458. [Google Scholar]
- Svergun D, Barberato C, Koch MHJ. CRYSOL- a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. J Appl Cryst. 1995;28:768–773. [Google Scholar]
- Svergun DI. Mathematical Methods in Small-Angle Scattering Data Analysis. J Appl Cryst. 1991;24:485–492. [Google Scholar]
- Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J App Cryst. 1992;25:495–503. [Google Scholar]
- Svergun DI. Restoring Low Resolution Structure of Biological Macromolecules from Solution Scattering Using Simulated Annealing. Biophys J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun DI, Petoukhov MV, Koch MH. Determination of domain structure of proteins from X-ray solution scattering. Biophys J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treuheit MJ, Costello CE, Halsall HB. Analysis of the five glycosylation sites of human alpha 1-acid glycoprotein. Biochem J. 1992;283 (Pt 1):105–112. doi: 10.1042/bj2830105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI. Uniqueness of ab-initio shape determination in small-angle scattering. J Appl Cryst. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh G. Biopharmaceutical benchmarks 2010. Nat Biotechnol. 2010;28:917–924. doi: 10.1038/nbt0910-917. [DOI] [PubMed] [Google Scholar]
- Weinkam P, Pons J, Sali A. Structure-based model of allostery predicts coupling between distant sites. Proc Natl Acad Sci U S A. 2012;109:4875–4880. doi: 10.1073/pnas.1116274109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Arakawa T, Philo JS. Size-exclusion chromatography with online light-scattering, absorbance, and refractive index detectors for studying proteins and their interactions. Anal Biochem. 1996;240:155–166. doi: 10.1006/abio.1996.0345. [DOI] [PubMed] [Google Scholar]
- Whitten AE, Trewhella J. Small-angle scattering and neutron contrast variation for studying bio-molecular complexes. Methods Mol Biol. 2009;544:307–323. doi: 10.1007/978-1-59745-483-4_20. [DOI] [PubMed] [Google Scholar]
- Williams RL, Greene SM, McPherson A. The crystal structure of ribonuclease B at 2.5-A resolution. J Biol Chem. 1987;262:16020–16031. doi: 10.2210/pdb1rbb/pdb. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature. 1981;289:336–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- Wittmann V. Glycopeptides and Glycoprotein; Synthesis, Structure, and Application. Vol. 267. Berlin Heidelberg New York: Springer; 2007. [Google Scholar]
- Woods RJ, Pathiaseril A, Wormald MR, Edge CJ, Dwek RA. The high degree of internal flexibility observed for an oligomannose oligosaccharide does not alter the overall topology of the molecule. European Journal of Biochemistry. 1998;258:372–386. doi: 10.1046/j.1432-1327.1998.2580372.x. [DOI] [PubMed] [Google Scholar]
- Wriggers W, Birmanns S. Using situs for flexible and rigid-body fitting of multiresolution single-molecule data. Journal of structural biology. 2001;133:193–202. doi: 10.1006/jsbi.2000.4350. [DOI] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.