

Abstract
Hepatocyte apoptosis is a hallmark of nonalcoholic steatohepatitis. We have previously observed that the saturated free fatty acids (FFAs) induce hepatocyte apoptosis in part via a death receptor 5 (DR5)-mediated signaling pathway. Cellular inhibitor of apoptosis protein 1 and 2 (cIAP-1 and cIAP-2) proteins are potent inhibitors of death receptor-mediated apoptosis. However, the role of the cIAPs in FFA-mediated hepatocyte apoptosis is unexplored. Our aim was to determine whether cIAPs are dysregulated during hepatocyte lipoapoptosis. cIAP proteins underwent rapid cellular elimination following treatment with the saturated FFAs palmitate (PA) and stearate. In contrast, PA did not decrease cIAP-1 and cIAP-2 mRNA expression in the cells. Degradation of cIAPs was dependent on their E3-ligase activity, suggesting that cIAPs undergo autoubiquitination that leads to proteasomal degradation. Huh-7 cells stably expressing shRNA targeting cIAP-1, but not cIAP-2, displayed enhanced sensitivity to PA-mediated apoptosis. Incubation with the SMAC mimetic JP1584, which induces rapid degradation of cIAPs, also enhanced PA-mediated apoptosis. Hepatocytes isolated from DR5 knockout mice exhibited reduced apoptosis following treatment with PA plus JP1584, implying that degradation of cIAPs sensitizes to DR5-mediated cell death pathways. A decrease of cIAP-1 was also observed in tissue from patients with nonalcoholic steatohepatitis compared with normal obese subjects. Collectively, these results implicate proteasomal degradation of cIAPs by FFA as a mechanism contributing to hepatocyte lipoapoptosis.
Keywords: free fatty acids, apoptosis, IAPs, SMAC mimetics, DR5
nonalcoholic fatty liver disease (NAFLD) is associated with insulin resistance, which accentuates lipolysis in adipose tissue liberating free fatty acids (FFAs) into the circulation (7). The excess of serum FFAs are taken up by the liver where they exceed the esterification and oxidation capacity of the liver, thereby impairing the capacity of the liver to neutralize these toxic mediators. Accumulation of FFAs within the hepatocyte triggers cell death by apoptosis, a process termed lipoapoptosis (7, 23, 28). The magnitude of lipoapoptosis correlates with the severity of nonalcoholic steatohepatitis (NASH), an inflammatory subset of NAFLD that may progress to cirrhosis and its sequelae (23). Saturated FFAs such as palmitate (PA), but not unsaturated FFAs, such as oleate (OA), are cytotoxic to hepatocytes (1, 22, 31). Unsaturated FFAs are more efficiently esterified and incorporated into nontoxic lipid droplets compared with saturated ones, a process minimizing the toxicity of FFAs (1). The molecular mechanisms leading to lipoapoptosis by saturated FFAs are not fully understood. Hepatocytes can undergo apoptosis either by the so-called “extrinsic” or “intrinsic” pathway (2). The extrinsic pathway is initiated by death ligands, such as Fas and TNF-related apoptosis-inducing ligand (TRAIL), and their cognate receptors (17). Once activated, death receptors recruit adaptor proteins resulting in caspase 8 activation (4). The intrinsic pathway can be activated by noxious intracellular stimuli causing endoplasmic reticulum stress or mitochondrial dysfunction (19, 21). During hepatocyte lipoapoptosis, both intrinsic and extrinsic pathways converge on mitochondria, thereby executing the mitochondrial cell death pathway (19). Although a network of proapoptotic signaling cascades have been described during hepatocyte lipoapoptosis (7, 23), death receptors appear to play a critical role via the extrinsic pathway (21, 33). Indeed, we have recently identified TRAIL receptor 2 (TRAIL-R2, also known as death receptor 5 or DR5) as one of the key mediators contributing to PA-mediated hepatocyte lipoapoptosis (8). PA triggers an increase in DR5 mRNA and protein expression and promotes its oligomerization within lipid rafts on the cell membrane, a step crucial for initiating caspase 8-dependent death-signaling events (8). Despite the likely importance of DR5 signaling in NASH, factors that can regulate DR5-dependent signaling during FFA-induced cell injury remain relatively unexplored.
Cellular inhibitor of apoptosis protein-1 (cIAP-1) and -2 (cIAP-2) are members of the antiapoptotic IAP family that regulate death receptor signaling (18, 30). cIAPs are characterized by one or more zinc-binding baculoviral IAP repeat (BIR) motifs and contain a carboxyl-terminal RING domain essential for its E3 ubiquitin ligase activity (26). Mammalian cells have a natural IAP antagonist, the mitochondrial protein SMAC (second mitochondrial activator of caspases)/DIABLO (direct IAP binding protein with low pI), which is released into the cytosol from mitochondria in response to multiple proapoptotic stimuli (10). SMAC mimetics are small pharmacological molecules that mimic the amino-terminal IAP-binding motif of mature SMAC (12, 15). These molecules are known to bind to the BIR2 and BIR3 domains of cIAPs, inducing their rapid degradation by an autoubiquitination process, which, in turn, results in their proteasomal degradation. These agents are reported to enhance death receptor-induced apoptosis (29, 30). Indeed, we have recently described that TRAIL induced apoptosis is associated with degradation of cIAP-1 by caspase 8-mediated cleavage (18). Despite their likely ability to modulate death receptor-mediated apoptosis, the regulation of cIAP-1 and cIAP-2 during lipoapoptosis is unknown.
Herein, our data indicate that PA induces degradation of cIAPs by a proteasome-dependent and caspase-independent process. The elimination of cIAPs appears to enhance lipoapoptosis via the DR5-triggered, extrinsic pathway. cIAP-1 was also found to be diminished in a small sample of human NASH patients. We speculate that this dysregulation of cIAPs may be a pathogenic mechanism in human NASH.
MATERIALS AND METHODS
Cells and mice.
The human hepatocellular carcinoma cell lines, Huh-7, Hep3B, HepG2 cells, were cultured in Dulbecco's modified Eagle's medium (DMEM) with high glucose (25 mM) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Stable cell lines expressing short hairpin RNA (shRNA) against cIAP-1, cIAP-2, and caspase 8 were generated and cultured as previously described by us (18). C57BL/6 wild-type mice were obtained from Jackson Laboratory (Bar Harbor, ME). TRAIL-R−/− mice were kindly provided by Dr. El-Deiry (University of Pennsylvania School of Medicine, Philadelphia, PA) (16). Mouse hepatocytes were isolated by collagenase perfusion, purified by Percoll (Sigma-Aldrich, St. Louis, MO) gradient centrifugation, and cultured on collagen-coated plates in Waymouth medium, supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum. All animal studies had prior Institutional Animal Care and Use Committee approval. Cryopreserved human primary hepatocytes obtained from Xenotech (Lenexa, KS) were rapidly thawed at 37°C in a water bath and washed and resuspended in DMEM supplemented with 10% FBS, 0.6 μg/ml insulin, 2 μg/ml hydrocortisone, 50 U/ml penicillin, and 50 μg/ml streptomycin. Cells were plated on collagen-coated cell culture plates and medium was replaced after 4 h with Williams' E medium supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 292 μg/ml l-glutamine, 10 μg/ml insulin, 5.5 mg/ml transferrin, 6.25 ng/ml selenium, and 1 μM dexamethasone. Cells were used 24 h after thawing.
Fatty acid treatment.
Palmitic acid (no. P5585), stearic acid (SA; no. S4751), and oleic acid (no. O1008) were obtained from Sigma-Aldrich. Fatty acids were dissolved in isopropanol at a concentration of 80 mM at 37°C and diluted in DMEM containing 1% bovine serum albumin at the desired final concentration. The final concentrations of FFAs used in this study (100–800 μM) are similar to the total fasting FFA plasma concentrations in human NASH (5, 24).
Plasmids.
pEGFP-N1 was purchased from Clontech (Mountain View, CA). pEBB-HA-cIAP-1H588A, in which His588 in the RING domain was mutated to alanine (Ala), was generated as described previously (18). pEBB-HA-cIAP2 (a gift from Drs. Ezra Burstein, University of Texas Southwestern, Dallas, TX and Colin Duckett, University of Michigan at Ann Arbor, MI) was subjected to site-directed mutagenesis to generate the pEBB-HA-cIAP-2H574A where His574 in the RING domain was replaced with Ala, using the QuickChange II Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) following the supplier's instructions.
Real-time PCR.
Total RNA was extracted from the cells using the TRIzol reagent (Invitrogen, Life Technologies, Grand Island, NY) and was reverse-transcribed into complementary DNA using Moloney leukemia virus reverse transcriptase (Invitrogen). Quantification of the complementary DNA template was performed with real-time PCR (LightCycler 480 II System; Roche Applied Science, Indianapolis, IN) using SYBR green (Molecular Probes, Life Technologies), as previously described by us (1). PCR primers were as follows: human cIAP-1, 5′-AGCTAGTCTGGGATCCACCTC-3′ (forward), 5′-GGGGTTAGTCCTCGATGAAG-3′ (reverse); human cIAP-2, 5′-TGGAAGCTACCTCTCAGCCTAC-3′ (forward), 5′-GGAACTTCTCATCAAGGCAGA-3′ (reverse); human DR5, 5′-GGGAAGAAGATTCTCCTGAGATGTG-3′ (forward), 5′-ACATTGTCCTCAGCCCCAGGTCG-3′ (reverse). Primers for human 18S ribosomal RNA, used as an internal control, were from Ambion (Life Technologies).
Immunoblot analysis.
Total cell lysates were obtained as previously described by us (3). Protein samples were resolved by SDS-PAGE, transferred to nitrocellulose membranes, and incubated overnight with primary antibodies at a dilution of 1:1,000. Appropriate horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biochemicals, Santa Cruz, CA) were used at a dilution of 1:3,000. Bound antibodies were visualized with chemiluminescent substrate (ECL, Amersham, Arlington Heights, IL). The primary antibodies were purchased by the following companies: goat polyclonal anti-cIAP-1 (AF8181) was from R&D Systems (Minneapolis, MN); rabbit polyclonal anti-cIAP-2 (ab 32059) was from Abcam (Cambridge, MA); rat monoclonal anti HA (11867423001) was from Roche Applied Science; rabbit polyclonal anti-poly(ADP-ribose) polymerase (PARP) antibody (9542) was from Cell Signaling Technology (Beverly, MA); goat polyclonal anti-actin (sc-1615) was from Santa Cruz Biochemicals.
Assessment of cell death.
Necrosis and apoptosis were quantified by assessing the characteristic nuclear and cellular morphology after staining with the nuclear binding dye 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI, Sigma-Aldrich), using fluorescence microscopy (Zeiss LSM 510, Carl Zeiss, Jena, Germany). Apoptosis was also determined by measuring caspase 3/7 activation (DEVDase activity) by using Apo-ONE homogeneous caspase 3/7 kit (Promega, Madison, WI) following the manufacturer's instruction. In selected experiments, apoptosis was confirmed by detection of PARP cleavage by immunoblot analysis as described above.
Preparation of human samples.
The study was approved by the Mayo Institutional Review Board, and all patients gave written, informed consent for participation in medical research. Our cohort consisted of eight patients who underwent liver biopsy at the time of bariatric surgery for medically complicated obesity at Mayo Clinic, Rochester, MN. The diagnosis of NAFLD was based on liver biopsy features as assessed by an experienced hepatopathologist. Patients with body mass index >30 kg/m2 were divided into two histological groups: obese normal (normal liver biopsies, n = 3) and NASH (n = 5). Patients with NAFLD who had secondary causes of steatohepatitis (i.e., drugs, prior gastric surgery for obesity) and patients with other etiologies of chronic liver disease were excluded from this study. Liver biopsy samples of ∼10–15 mg were placed in microcentrifuge tubes containing 300 μl of protein extraction buffer (50 mm HEPES, pH 7.4, 0.05% SDS) and 20 μl of protease inhibitor mixture III (Calbiochem EMD Biosciences, La Jolla, CA). The tissue samples were then emulsified with a PRO250 Homogenizer (Pro Scientific, Oxford, CT) and centrifuged at 750 g for 6 min. The supernatant was removed and centrifuged a second time at 10,000 g for 5 min. The resulting tissue lysates were analyzed by immunoblot analysis as described above.
Reagents.
The pan-caspase inhibitor Q-VD-OPH was from Enzyme Systems Products (Solon, OH). The proteasome inhibitor MG132 was from Calbiochem EMD Biosciences The SMAC mimetic JP1584 was from Joyant Pharmaceuticals (Dallas, TX). Necrostatin-1 was purchased from Tocris Bioscience (St. Louis, MO). Human anti-TNF-α neutralizing antibody (AF-210-NA) was from R&D Systems.
Statistical analysis.
All data represent at least three independent experiments and are expressed as means ± SE. Differences between groups were compared by an unpaired two-tailed t-test, and P values <0.05 were considered statistically significant.
RESULTS
cIAP-1 and cIAP-2 undergo rapid cellular elimination following treatment with PA.
We initially examined cIAP protein levels following FFA treatment. Huh-7, Hep3B, and HepG2 cells were treated with 800 μM of the saturated FFA PA for up 16 h. In all the cell lines, both cIAP-1 and cIAP-2 underwent relatively rapid cellular elimination (Fig. 1A). Notably, cIAP-2 was almost undetectable by immunoblot analysis in HepG2 cells. The reduction in cIAP-1 and cIAP-2 protein levels was sustained over 16 h. Rapid degradation of cIAP-1 and cIAP-2 was also observed following incubation with SA, another saturated FFA, at the same concentration (Fig. 1B). In contrast, treatment with an identical concentration of the nontoxic unsaturated FFA OA did not result in sustained depletion of either cIAP-1 or cIAP-2 (Fig. 1A). PA-treatment induced also a slight reduction in the protein level of X-linked inhibitor of apoptosis protein (XIAP), the third most abundant member of the mammalian IAP family. However, XIAP decrease occurred significantly later compared with the decrease in cIAP-1 and -2, making it less likely to contribute to the apoptotic signaling (Fig. 1C). Next, we assessed whether the decrease of cIAPs was due to reduced mRNA expression. cIAP-1 and cIAP-2 mRNA expression levels were not significantly altered by PA or OA treatment in any of the cell lines examined (Fig. 1D), suggesting that FFA-mediated cellular elimination of cIAP-1 and cIAP-2 occurs via a posttranslational mechanism. We have previously reported that degradation of cIAP-1 following TRAIL treatment is mediated by caspase 8 cleavage of this protein (18). Interestingly, PA-induced cIAP-1 and cIAP-2 degradation in Huh-7 cells was not affected either by caspase 8 shRNA-targeted knockdown or by the use of a pan-caspase inhibitor (Fig. 2A). In contrast, degradation of cIAP proteins was prevented by the proteasome inhibitor MG132 (Fig. 2B). Similar results were observed in human primary hepatocytes, although cIAP-2 was preferentially degraded compared with cIAP-1 in these cells (Fig. 2C). As expected, cIAP-1 and cIAP-2 expression in human hepatocytes was also significantly lower than in cancer cells. Previous studies have also demonstrated that proteasomal degradation of cIAPs occurs through their autoubiquitination, which is mediated by the E3 ubiquitin ligase activity of their RING domains (15, 26, 30). To test whether FFA-mediated degradation of cIAPs requires their autoubiquitination, cells were transiently transfected with constructs expressing cIAP-1 and cIAP-2 mutants deficient in their E3 ligase activity due to specific replacement of critical histidine residues in their RING domains (H588A in cIAP-1 and H574A in cIAP-2). cIAP-1 and cIAP-2 RING mutants both failed to undergo degradation by FFA treatment and actually accumulated after 16 h of PA treatment (Fig. 2D), consistent with previous finding demonstrating that loss of the E3 ligase activity results in more stable cIAP-1 and cIAP-2 proteins (11). These results imply that cIAPs undergo autoubiquitination followed by proteasomal degradation upon FFA treatment.
Fig. 1.

Saturated free fatty acids (FFA), but not unsaturated FFA, induce cellular inhibitor of apoptosis 1 and 2 (cIAP-1 and cIAP-2) degradation in hepatocyte cell lines. A: Huh-7, HepG2, and Hep3B cells were treated with 800 μM of palmitate (PA) or oleate (OA) for the indicated time. Whole cell lysates were obtained from each experimental condition and subjected to immunoblot analysis for cIAP-1 and -2. Actin was used as loading control. B: Huh-7 cells were treated with 800 μM of stearate (SA) for the indicated time. Whole cell lysates were subjected to immunoblot analysis for cIAP-1 and -2. Actin was used as loading control. C: Huh-7, HepG2, and Hep3B cells were treated with 800 μM PA for the indicated time. Whole cell lysates were subjected to immunoblot analysis for X-linked inhibitor of apoptosis protein (XIAP). Actin was used as loading control. D: Huh-7, HepG2, and Hep3B cells were treated with 800 μM PA or OA for the indicated time. Total RNA was obtained at 8 h and expression of cIAP-1 and cIAP-2 was quantified by real-time PCR. Fold induction was determined after normalization to 18S. Data represent means ± SE of 3 independent experiments performed in triplicate. Veh, vehicle.
Fig. 2.
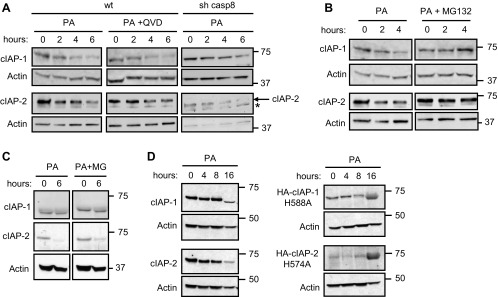
Decrease of cIAPs is caspase independent and proteasome dependent. A: wild-type (wt) Huh-7 cells were treated with PA (800 μM) in the presence (middle) or absence (left) of the pan-caspase inhibitor Q-VD-OPH (QVD; 5 μM; 30 min preincubation). Right: Huh-7 cells stably transfected with shRNA against caspase 8 (sh casp8) were treated with PA (800 μM). At the indicated time points, cell lysates were obtained and analyzed by immunoblot analysis for cIAP-1 and cIAP-2. Asterisk indicates a nonspecific band. B: Huh-7 cells were treated with 800 μM PA for the indicated time in the presence or absence of the proteasome inhibitor MG132 (MG; 10 μM). Whole cell lysates were obtained and analyzed by immunoblot analysis for cIAP-1 and cIAP-2. C: cryopreserved primary human hepatocytes were cultured for 24 h as described in materials and methods then treated with 600 μM PA for the indicated time in the presence or absence of MG132 (10 μM). Whole cell lysates were analyzed by immunoblot analysis for cIAP-1 and cIAP-2. D: Huh-7 cells were left untransfected (left) or transiently transfected with plasmids encoding HA-cIAP-1-H588A or HA-cIAP-2-H574A (right) and treated with 800 μM of PA for the indicated times; cell lysates were analyzed by immunoblot for expression of endogenous cIAP-1 and cIAP-2 (left) by using antisera against cIAP-1 and cIAP-2, or HA-cIAP-1-H588A and HA-cIAP-2-H574A (right) with an antibody against the HA tag. Actin was used as loading control.
cIAP-1 knockdown sensitizes hepatocytes to FFA-mediated apoptosis.
The results above suggest that depletion of cIAPs may contribute to efficient FFA-mediated apoptosis. To test this postulate, Hep3B cells were treated with PA (200 μM) with or without the SMAC mimetic JP1584 (500 nM), which causes rapid degradation of cIAPs (18). In the presence of JP1584, PA-mediated apoptosis was significantly enhanced, as demonstrated by increased apoptotic nuclear morphology and cleavage of PARP, a well-known substrate for activated caspase 3 (Fig. 3, A and B). In contrast, JP1584 did not sensitize the cells to OA-induced apoptosis (Fig. 3, A and B). To further examine whether one of the cIAPs has a potentially dominant role in protecting against lipoapoptosis, wild-type and Huh-7 cells stably expressing shRNA targeting cIAP-1 or cIAP-2 were treated with a concentration of PA (100 μM) that does not cause significant apoptosis in wild-type cells. Wild-type Huh-7 and cIAP-2 shRNA cells exhibited minimum apoptosis upon treatment with PA, whereas cIAP-1 knockdown sensitized the cells to PA-mediated apoptosis (Fig. 3, C and D). Lipoapoptosis in cIAP-1-deficient cells was blocked by QVD-OPH, a pan-caspase inhibitor, demonstrating that cell death was caspase dependent (Fig. 3D). To confirm that cIAP-1 degradation promotes PA-induced apoptosis, Huh-7 cells were transfected with cIAP-1 H588A (RING mutant) or with an empty vector (pcDNA3.1) as control, along with pEGFP-N1 expressing green fluorescent protein at a ratio of 3:1 to identify the successfully transfected cells. Cells were then treated with PA (800 μM) for 16 h and apoptosis was quantitated by fluorescence microscopy on GFP-positive cells after staining with DAPI, and by immunoblot analysis for cleaved PARP. Indeed, cells expressing the nondegradable cIAP-1 H588A displayed increased resistance to PA-induced apoptosis (Fig. 3, E and F). Finally, to further test this concept in nontransformed cells, we treated isolated primary mouse hepatocytes with PA or OA, in the presence or absence of JP1584. Consistently, JP1584 enhanced PA-mediated apoptosis in isolated mouse hepatocytes but had no effect on OA-mediated cytotoxicity (Fig. 4). Taken together, these results suggest that cellular depletion of cIAP-1 by its E3 ligase activity contributes to PA-mediated hepatocyte apoptosis.
Fig. 3.

Knockdown of cIAP-1 sensitizes hepatocyte to PA-mediated apoptosis. A: Hep3B cells were treated with vehicle or PA (200 μM) or OA (200 μM), in the presence or absence of JP1584 (500 nM) for 16 h. Apoptosis was quantitated by morphological criteria. Data are expressed as means ± SE (*P < 0.05). B: Hep3B cells treated as in A were lysed and the protein extracts were analyzed by immunoblot for poly(ADP-ribose) polymerase (PARP). C and D: Huh-7 cells stably transfected with shRNA against either cIAP-1 or cIAP-2 were treated with vehicle or PA (100 μM) for 16 h. Apoptosis was assessed morphologically after DAPI staining (C) and by measuring caspase 3/7 activation (D). RFU, rhodamine fluorescence units. Data are expressed as means ± SE (*P < 0.01). E: Huh-7 cells were transiently cotransfected with pEGFP-N1 [encoding green fluorescent protein (GFP)] and either HA-cIAP-1H588A or an empty plasmid (pcDNA3.1) for 48 h, followed by treatment with PA (800 μM) for 16 h. GFP-positive cells were scored as apoptotic on the basis of morphological criteria after staining with DAPI using a fluorescence microscope. Data are expressed as means ± SE (*P < 0.05). F: Huh-7 cells treated as in E were lysed and the protein extracts were analyzed by immunoblot for PARP.
Fig. 4.

Inhibition of inhibitors of apoptosis (IAPs) sensitizes primary mouse hepatocyte to PA-mediated apoptosis. A: primary mouse hepatocytes were isolated from wild-type mice, cultured for 4 h, then treated with vehicle, PA (200 μM), or OA (200 μM), in the presence or absence of JP1584 (JP; 500 nM), for 8 h. Apoptosis was quantitated by morphological criteria. Data are expressed as means ± SE (*P < 0.05).
Because DR5 contributes to lipoapoptosis and cIAPs modulate death receptor signaling, we sought to ascertain whether cIAPs inhibited lipoapoptosis by interfering with DR5-mediated signaling pathways. To examine this concept we first verified whether JP1584 enhances transcriptional activation of DR5. As previously reported by us, PA induced a dose-dependent increase in DR5 mRNA expression (Fig. 5A) (8). However, no changes in DR5 mRNA expression were observed in Huh-7 cells treated with 400 μM PA plus JP1584 compared with cells treated with PA alone (Fig. 5A). Next, we assessed whether the sensitizing effect of JP1584 is blocked in hepatocytes isolated from mice deficient in TRAIL-R, the only apoptosis-inducing TRAIL receptor in mice, which resembles human DR5 (32). Notably, JP1584 did not enhance PA-induced apoptosis in isolated hepatocyte from TRAIL-R−/− mice, suggesting that cIAPs exert their antiapoptotic effect downstream of DR5 (Fig. 5B). Previous studies suggested that SMAC mimetic treatment or cIAP-1 depletion induced NF-κB-mediated production of TNF-α and consequent TNF-α-induced apoptosis via an autocrine loop (30). In the same conditions, ubiquitination of the kinase RIP-1 is prevented, sensitizing the cells to RIP-1-dependent necroptosis during death receptor signaling (13, 14). To rule out the possibility that JP1584 enhances FFA toxicity by promoting necroptosis, we assessed whether necrostatin-1, an inhibitor of RIP-1 (14), can prevent the SMAC mimetic-dependent sensitization to PA-mediated cell death. However, necrostatin-1 did not confer significant protection from cell death induced by PA plus JP1584 (Fig. 5C). Similarly, addition of a neutralizing antibody against TNF-α did not attenuate cell death or caspase activation caused by PA plus JP1584 (Fig. 5, D and E), suggesting that SMAC mimetic sensitization to PA-induced cell death is not mediated by TNF-α. Collectively, these results indicate that elimination of cIAPs contributes to PA-induced apoptosis by enhancing a DR5-mediated signaling pathway.
Fig. 5.
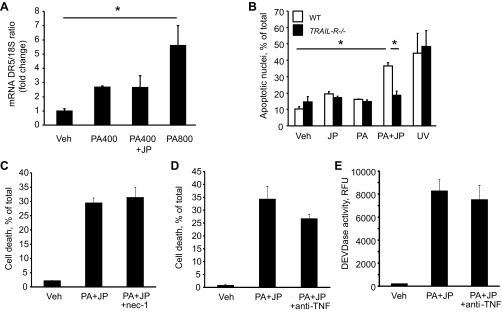
cIAP-1 prevents FFA-mediated apoptosis downstream of DR5. A: Huh-7 cells were treated with vehicle, PA (400 or 800 μM), or PA (400 μM) + JP (500 nM) for 8 h. Total RNA was obtained and DR5 mRNA expression was quantified by real-time PCR. Fold induction was determined after normalization to 18S. Data are expressed as means ± SE (*P < 0.05). B: primary mouse hepatocytes isolated from wild-type and TRAIL-R−/− mice were cultured for 4 h then treated with either vehicle, PA (200 μM), JP (500 nM), or PA + JP for 8 h, or UV light for 4 min. Apoptosis was quantified by morphological criteria. Data are expressed as means ± SE (*P < 0.05). C: Huh-7 cells were pretreated with necrostatin-1 (nec-1; 50 μM) or vehicle for 1 h, then treated with vehicle, PA (800 μM) + JP1584 (500 nM), or PA + JP1584 + nec-1 for 16 h. Cell death was assessed by morphological appearance after DAPI staining. D and E: Huh-7 cells were treated with vehicle, PA (800 μM) + JP1584 (500 nM), or PA + JP1584 + anti-TNF-α antibody (100 ng/ml) for 16 h. Cell death was assessed by morphological appearance after DAPI staining (D) and apoptosis was quantified by measuring caspase 3/7 activation. Data are expressed as means ± SE (*P < 0.05).
cIAP-1 is decreased in liver samples from NASH patients.
To verify whether cIAP-1 levels are decreased in hepatocytes exposed to high concentrations of FFA in vivo, cIAP-1 expression was assessed in human liver specimens, comparing samples from obese normal controls (n = 3) to those from patients with steatosis plus inflammation (NASH) (n = 5). As expected, cIAP-1 protein levels were significantly lower in liver tissue from patients with NASH compared with liver tissue from obese normal controls (Fig. 6). These results suggest that cIAP-1 is decreased in human NAFLD.
Fig. 6.

cIAP-1 is decreased in human nonalcoholic steatohepatitis (NASH). Tissue lysates were prepared from liver biopsies of obese normal subjects (n = 3) and NASH patients (n = 5) and analyzed by immunoblot for cIAP-1. Relative cIAP-1 protein expression was determined by densitometric analysis after normalization to actin. Data are expressed as means ± SE (*P < 0.01, NASH vs. obese normal).
DISCUSSION
The present study provides new insights into the mechanisms of FFA-induced hepatocyte injury. The principal findings of this study indicate that 1) cIAP-1 and cIAP-2 undergo proteasomal degradation in hepatocytes following treatment with PA; 2) cellular depletion of cIAPs sensitizes hepatocytes to FFA cytotoxicity; and 3) cIAP-1 is decreased in human liver specimens of NASH. These results are discussed in greater detail below.
cIAP-1 and cIAP-2 are negative regulators of apoptosis (26). Although various studies indicated that cIAPs are overexpressed in malignant cells, including hepatocellular carcinoma, they are also present in nontransformed cells (25, 30, 34). The role of cIAPs in pathological conditions other than malignancies remains largely unexplored. In the present study, we observed that cIAP-1 and cIAP-2 decreased upon treatment with saturated FFA in liver cell lines. In addition, our study revealed that depletion of cIAP-1, but not cIAP-2, contributes to lipoapoptosis in Huh-7 cells. cIAPs are regulated by transcriptional or posttranscriptional mechanisms. For example, cIAP-2 can be transcriptionally regulated by NF-κB (26). However, our results suggested that the decrease of cIAP-1 and cIAP-2 protein levels during PA treatment were not due to a decrease in steady-state mRNA levels, and, therefore, were unlikely to be due to transcriptional repression. Posttranslational degradation of cIAPs can occur in several processes. The carboxy termini of both cIAP-1 and cIAP-2 contain a highly conserved RING domain, which has a key role in posttranscriptional degradation of these proteins (26). cIAPs undergo autoubiquitination and degradation by the 26S proteasome (26). cIAP-1 can also be cleaved by caspase 8 and caspase 3 and subsequently degraded (9, 18). However, in our present observation degradation of cIAPs by FFA was not inhibited either by knockdown of caspase 8 or by a pan-caspase inhibitor, suggesting that the degradation of these proteins occurs independent of caspase activity. Instead, FFA-induced cIAP-1 and cIAP-2 degradation was proteasome dependent and required their E3 ligase activity. The precise mechanism by which FFAs induce cIAP-1 and cIAP-2 autoubiquitination remains unclear but merits further investigation. FFAs may promote assembly of protein complexes at the cell membrane either by altering membrane fluidity or via activating kinase signaling pathways (8, 17). Likely one of these mechanisms promotes cIAP-1 and cIAP-2 assembly and conformational changes necessary for their autoubiquitination.
cIAPs are capable of regulating proteins through either degradative K48-linked or nondegradative K63-linked polyubiquitination (20). cIAP-1 and cIAP-2 are known to ubiquitinate RIP-1, resulting in activation of prosurvival pathways through canonical activation of NF-κB (6). However, when cIAP-1 and cIAP-2 are degraded by SMAC mimetics, TNF-α is generated through noncanonical activation of NF-κB, switching the signal to a proapoptotic autocrine loop (30). This mechanism of cytotoxicity was excluded in lipoapoptosis by demonstrating that TNF-α neutralizing antibodies do not protect against PA-mediated cell killing. The proapoptotic effect of cIAP-1 elimination in lipoapoptosis was not apparent in hepatocytes isolated from TRAIL-R−/− mice, implying that cIAPs restrain DR5-mediated cytotoxicity during lipoapoptosis. Likely, the depletion of cIAPs by toxic FFA augment DR5 cytotoxic signaling by enhancing formation of a caspase 8-containing protein complex, promoting activation of this proapoptotic protease (6). This interpretation of our data is supported by the observation that FFA cytotoxicity is caspase 8 dependent (8). Although the absence of cIAPs and therefore RIP-1 ubiquitination could also potentially promote RIP-1-dependent necroptosis, the RIP-1 inhibitor necrostatin-1 did not protect from cell death in our study (14). Therefore, necroptosis does not appear to occur during FFA-mediated cell death.
Although both cIAP-1 and cIAP-2 decreased upon PA treatment, our study suggests that depletion of cIAP-1, but not cIAP-2, contributes to FFA-induced apoptosis. In agreement with our findings, Qi and others (25) recently described the importance of cIAP-1 in protecting against FFA-induced cell death in pancreatic cells. Of note, it has been previously shown that elimination of cIAP-2 upon SMAC mimetic treatment is dependent on cIAP-1 E3 ligase activity (12). Thus degradation of cIAP-2 may be downstream of cIAP-1 activation, rendering cIAP-1 the dominant inhibitor in this model. We also note that in isolated, previously cryopreserved human hepatocytes, cIAP-2 was preferentially degraded compared with cIAP-1, and thus the precise cIAP regulating lipoapoptosis remains ambiguous. Since expression of IAP proteins seems to vary among the cell types, the relative roles of cIAP-1 and cIAP-2 are likely cell type and stimulus specific (27, 34).
Collectively, our results provide new insights regarding the role of cIAPs in FFA-mediated apoptosis. Our results suggest that elimination of cIAPs by their autoubiquitination and proteasomal degradation contributes to lipoapoptosis, likely enhancing DR5-mediated hepatocyte injury.
GRANTS
This work was supported by NIH Grant OK41876 to G. J. Gores, JSPS KAKENHI Grant Number 24790709 (to Y. Akazawa), and the Mayo Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Y.A., M.E.G., and K.N. conception and design of research; Y.A., M.E.G., S.C.C., S.F.B., N.W.W., and K.K. performed experiments; Y.A., M.E.G., and G.J.G. analyzed data; Y.A., M.E.G., and G.J.G. interpreted results of experiments; Y.A. and M.E.G. prepared figures; Y.A. and M.E.G. drafted manuscript; Y.A., M.E.G., and G.J.G. edited and revised manuscript; Y.A., M.E.G., K.N., and G.J.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We appreciate Courtney Hoover for excellent secretarial assistance.
REFERENCES
- 1.Akazawa Y, Cazanave S, Mott JL, Elmi N, Bronk SF, Kohno S, Charlton MR, Gores GJ. Palmitoleate attenuates palmitate-induced Bim and PUMA up-regulation and hepatocyte lipoapoptosis. J Hepatol 52: 586–593, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akazawa Y, Gores GJ. Death receptor-mediated liver injury. Semin Liver Dis 27: 327–338, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Akazawa Y, Mott JL, Bronk SF, Werneburg NW, Kahraman A, Guicciardi ME, Meng XW, Kohno S, Shah VH, Kaufmann SH, McNiven MA, Gores GJ. Death receptor 5 internalization is required for lysosomal permeabilization by TRAIL in malignant liver cell lines. Gastroenterology 136: 2365–2376e1–7, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 281: 1305–1308, 1998 [DOI] [PubMed] [Google Scholar]
- 5.Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, Balas B, Gastaldelli A, Tio F, Pulcini J, Berria R, Ma JZ, Dwivedi S, Havranek R, Fincke C, DeFronzo R, Bannayan GA, Schenker S, Cusi K. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 355: 2297–2307, 2006 [DOI] [PubMed] [Google Scholar]
- 6.Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 30: 689–700, 2008 [DOI] [PubMed] [Google Scholar]
- 7.Cazanave SC, Gores GJ. Mechanisms and clinical implications of hepatocyte lipoapoptosis. Clin Lipidol 5: 71–85, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cazanave SC, Mott JL, Bronk SF, Werneburg NW, Fingas CD, Meng XW, Finnberg N, El-Deiry WS, Kaufmann SH, Gores GJ. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J Biol Chem 286: 39336–39348, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clem RJ, Sheu TT, Richter BW, He WW, Thornberry NA, Duckett CS, Hardwick JM. c-IAP1 is cleaved by caspases to produce a proapoptotic C-terminal fragment. J Biol Chem 276: 7602–7608, 2001 [DOI] [PubMed] [Google Scholar]
- 10.Creagh EM, Murphy BM, Duriez PJ, Duckett CS, Martin SJ. Smac/Diablo antagonizes ubiquitin ligase activity of inhibitor of apoptosis proteins. J Biol Chem 279: 26906–26914, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Csomos RA, Wright CW, Galban S, Oetjen KA, Duckett CS. Two distinct signalling cascades target the NF-kappaB regulatory factor c-IAP1 for degradation. Biochem J 420: 83–91, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darding M, Feltham R, Tenev T, Bianchi K, Benetatos C, Silke J, Meier P. Molecular determinants of Smac mimetic induced degradation of cIAP1 and cIAP2. Cell Death Differ 18: 1376–1386, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darding M, Meier P. IAPs: guardians of RIPK1. Cell Death Differ 19: 58–66, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1: 112–119, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Dueber EC, Schoeffler AJ, Lingel A, Elliott JM, Fedorova AV, Giannetti AM, Zobel K, Maurer B, Varfolomeev E, Wu P, Wallweber HJ, Hymowitz SG, Deshayes K, Vucic D, Fairbrother WJ. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science 334: 376–380, 2011 [DOI] [PubMed] [Google Scholar]
- 16.Finnberg N, Gruber JJ, Fei P, Rudolph D, Bric A, Kim SH, Burns TF, Ajuha H, Page R, Wu GS, Chen Y, McKenna WG, Bernhard E, Lowe S, Mak T, El-Deiry WS. DR5 knockout mice are compromised in radiation-induced apoptosis. Mol Cell Biol 25: 2000–2013, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guicciardi ME, Gores GJ. Life and death by death receptors. FASEB J 23: 1625–1637, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guicciardi ME, Mott JL, Bronk SF, Kurita S, Fingas CD, Gores GJ. Cellular inhibitor of apoptosis 1 (cIAP-1) degradation by caspase 8 during TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. Exp Cell Res 317: 107–116, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ibrahim SH, Kohli R, Gores GJ. Mechanisms of lipotoxicity in NAFLD and clinical implications. J Pediatr Gastroenterol Nutr 53: 131–140, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mace PD, Riedl SJ. Molecular cell death platforms and assemblies. Curr Opin Cell Biol 22: 828–836, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malhi H, Barreyro FJ, Isomoto H, Bronk SF, Gores GJ. Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut 56: 1124–1131, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem 281: 12093–12101, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Malhi H, Gores GJ. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver Dis 28: 360–369, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nehra V, Angulo P, Buchman AL, Lindor KD. Nutritional and metabolic considerations in the etiology of nonalcoholic steatohepatitis. Dig Dis Sci 46: 2347–2352, 2001 [DOI] [PubMed] [Google Scholar]
- 25.Qi Y, Xia P. Cellular inhibitor of apoptosis protein-1 (cIAP1) plays a critical role in beta-cell survival under endoplasmic reticulum stress: promoting ubiquitination and degradation of C/EBP homologous protein (CHOP). J Biol Chem 287: 32236–32245, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol 3: 401–410, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Shrikhande SV, Kleeff J, Kayed H, Keleg S, Reiser C, Giese T, Buchler MW, Esposito I, Friess H. Silencing of X-linked inhibitor of apoptosis (XIAP) decreases gemcitabine resistance of pancreatic cancer cells. Anticancer Res 26: 3265–3273, 2006 [PubMed] [Google Scholar]
- 28.Unger RH. Lipotoxic diseases. Annu Rev Med 53: 319–336, 2002 [DOI] [PubMed] [Google Scholar]
- 29.Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, Fairbrother WJ, Vucic D. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem 283: 24295–24299, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, Benetatos CA, Chunduru SK, Condon SM, McKinlay M, Brink R, Leverkus M, Tergaonkar V, Schneider P, Callus BA, Koentgen F, Vaux DL, Silke J. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 131: 682–693, 2007 [DOI] [PubMed] [Google Scholar]
- 31.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab 291: E275–E281, 2006 [DOI] [PubMed] [Google Scholar]
- 32.Wu GS, Burns TF, Zhan Y, Alnemri ES, El-Deiry WS. Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res 59: 2770–2775, 1999 [PubMed] [Google Scholar]
- 33.Wueest S, Rapold RA, Schoenle EJ, Konrad D. Fas activation in adipocytes impairs insulin-stimulated glucose uptake by reducing Akt. FEBS Lett 584: 4187–4192, 2010 [DOI] [PubMed] [Google Scholar]
- 34.Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, Mu D, Lucito R, Powers S, Lowe SW. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 125: 1253–1267, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]