

Abstract
Cardiovascular responses to the tyrosine kinase inhibitor imatinib were investigated in the rat. Intravenous injections of 0.3–30 mg/kg imatinib produced small decreases in pulmonary arterial pressure, larger dose-dependent decreases in systemic arterial pressure, and no change or small increases in cardiac output, suggesting that the systemic vasodilator response is more pronounced under baseline conditions. When pulmonary arterial pressure was increased with U-46619 or Nω-nitro-l-arginine methyl ester (l-NAME), intravenous injections of imatinib produced larger dose-dependent decreases in pulmonary arterial pressure. Imatinib attenuated the acute hypoxic pulmonary vasoconstrictor response. Vasodilator responses to imatinib were not inhibited by meclofenamate, glybenclamide, or rolipram, suggesting that cyclooxygenase, ATP-sensitive K+ (KATP) channels, and cAMP were not involved in mediating the response. In a 21-day prevention study, imatinib treatment (50 mg/kg ip) attenuated the increase in pulmonary arterial pressure, right ventricular hypertrophy, and small vessel remodeling induced by monocrotaline. Imatinib reduced PDGF receptor phosphorylation and PDGF-stimulated thymidine incorporation in rat pulmonary artery smooth muscle cells. These data suggest that the beneficial effect of imatinib in pulmonary hypertension may involve inhibition of PDGF tyrosine kinase receptor-mediated effects on smooth muscle cell proliferation and on vasoconstrictor tone. These results indicate that imatinib has nonselective vasodilator activity in the pulmonary and systemic vascular beds similar to the Rho kinase inhibitor fasudil and the calcium entry antagonist isradipine. The present results are consistent with the hypothesis that imatinib may inhibit a constitutively active tyrosine kinase vasoconstrictor pathway in the pulmonary and systemic vascular beds in the rat.
Keywords: imatinib, PDGF, pulmonary vascular bed, tyrosine kinase inhibitor, vasodilation
receptor tyrosine kinases are important components of extracellular signal transduction pathways that play an important role in the regulation of cellular functions such as growth, proliferation, migration, and contraction that may be involved in the vascular remodeling and vasoconstriction observed in pulmonary hypertension (16). These transmembrane receptors bind growth factor ligands such as platelet-derived growth factor (PGDF), dimerize, and initiate a sequence of intracellular events that lead to alterations in cell function (5, 16, 19). In addition to the many known receptor tyrosine kinases, a number of nonreceptor tyrosine kinases are also intracellular signaling pathways that can be activated by receptor tyrosine kinases and G protein-coupled receptors and are hypothesized to be involved in the pathogenesis of pulmonary hypertension (29). Imatinib is a tyrosine kinase inhibitor developed for the treatment of chronic myelogenous leukemia (8, 30). Imatinib has been approved for the treatment of pulmonary arterial hypertension in patients with inadequate response to established therapy (15). Imatinib was initially discovered in a program designed to identify PDGF receptor inhibitors (5, 19). PDGFs are a family of homo- and heterodimers of disulfide-bonded A-, B-, and C-polypeptide chains that exist as several possible isoforms (9, 14). PDGFs exert their effect on target cells by activating the structurally related tyrosine kinase α- and β-receptors (9, 14). PDGF has been shown to be a potent mitogen in smooth muscle cells and fibroblasts and to have potent smooth muscle contractile activity (14, 33). Imatinib inhibits a number of receptor tyrosine kinases such as c-KIT and nonreceptor tyrosine kinases such as BCR-ABL that induce leukocyte expansion in chronic myelogenous leukemia (30). This tyrosine kinase inhibitor has been reported to suppress smooth muscle contraction in isolated preparations from guinea pig bladder, rabbit and human corpora cavernosa, human prostate gland, rabbit and human myometrium, and isolated pulmonary and penile arteries from the rat (1, 7, 13, 18, 22, 23, 26, 31). Imatinib has also been reported to have erectile activity in the rat and has been reported to have selective pulmonary vasodilator activity in pulmonary hypertensive rats (1, 25). The present study was undertaken to investigate the effects of imatinib on the pulmonary and systemic vascular beds of the rat and on the development of pulmonary hypertension in response to monocrotaline treatment in a prevention study. In addition, the effects of imatinib on PDGF receptor activation (phosphorylation) and PDGF-stimulated thymidine incorporation were investigated in pulmonary artery smooth muscle cells (PASMCs) from the rat. These results show that chronic imatinib treatment attenuates the increase in pulmonary arterial pressure, reduces right ventricular hypertrophy, and attenuates the remodeling of small pulmonary arteries in monocrotaline-induced pulmonary hypertension in a prevention trial. In addition, imatinib was shown to inhibit PDGF receptor phosphorylation and PDGF-stimulated thymidine incorporation in rat PASMCs and to have potent nonselective vasodilator activity in the pulmonary and systemic vascular beds in the rat.
METHODS
The Institutional Animal Care and Use Committee of the Tulane University School of Medicine approved the experimental protocol employed in these studies, and all procedures were conducted in accordance with institutional guidelines. In hemodynamic experiments, adult male Sprague-Dawley rats (Charles River) weighing 333–415 g were anesthetized with Inactin (100 mg/kg ip; Sigma-Aldrich) and were placed in the supine position on an operating table. Supplemental doses of Inactin were administered intraperitoneally in order to maintain a uniform level of anesthesia. Body temperature was maintained with a heating lamp. The trachea was cannulated with a short segment of PE-240 tubing to maintain a patent airway. The animals spontaneously breathed room air. In experiments in which the effects of imatinib on the pulmonary vasoconstrictor response to ventilatory hypoxia were investigated, the rats breathed a 10% O2-90% N2 gas mixture from a plastic funnel placed over the opening of the endotracheal tube. A femoral artery was catheterized with PE-50 tubing for measurement of systemic arterial pressure. The left jugular and femoral veins were catheterized with PE-50 tubing for intravenous injections and infusions of agents. For pulmonary arterial pressure measurement, a specially designed 3-F single-lumen catheter with a curved tip and with radiopaque marker was passed from the right jugular vein and into the main pulmonary artery under fluoroscopic guidance (Picker-Surveyor Fluoroscope) as previously described (24). Pulmonary and systemic arterial pressures were measured with Namic Perceptor DT transducers (Boston Scientific), digitized by a Biopac MP100 data acquisition system (Biopac Systems), continuously recorded, and stored on a Dell PC. Cardiac output was measured by the thermodilution technique with a Cardiomax II computer (Columbus Instruments). A known volume (0.2 ml) of room-temperature 0.9% NaCl solution was injected into the jugular vein catheter with the tip near the right atrium, and changes in blood temperature were detected by a 1.5-F thermistor microprobe catheter (Columbus Instruments) positioned in the aortic arch from the left carotid artery. The indicator dilution data were stored on the PC.
Each experimental series was carried out in a separate group of rats. In the first set of experiments, the effects of intravenous injections of imatinib in doses of 0.3–30 mg/kg on pulmonary and systemic arterial pressures, and cardiac output, were investigated in the anesthetized intact-chest rat under baseline conditions.
In the second set of experiments, responses to intravenous injections of imatinib were investigated when pulmonary arterial pressure was increased to ∼30 mmHg by continuous intravenous infusion of the thromboxane receptor agonist U-46619. After an initial high priming rate (400 ng/min), the U-46619 infusion was adjusted (150–250 ng/min) to maintain pulmonary arterial pressure at ∼30 mmHg.
In the third set of experiments, the effect of the nitric oxide synthase (NOS) inhibitor Nω-nitro-l-arginine methyl ester (l-NAME; 50 mg/kg iv) on vasodilator responses to imatinib in the pulmonary and systemic vascular beds was investigated to determine the role of endogenous nitric oxide (NO) in modulating the response to the tyrosine kinase inhibitor.
In the fourth set of experiments, the effect of imatinib (10 mg/kg iv) on the acute hypoxic pulmonary vasoconstrictor response was investigated when pulmonary arterial pressure was increased by ventilation with a 10% O2-90% N2 gas mixture.
In the fifth set of experiments, decreases in pulmonary and systemic arterial pressures in response to intravenous injections of imatinib (10 mg/kg), the Rho kinase inhibitor fasudil (30 μg/kg), and the calcium entry antagonist isradipine (10 μg/kg) were compared when pulmonary arterial pressure was increased to a high steady level (∼30 mmHg) with intravenous infusion of U-46619.
In the sixth set of experiments, the effects of sodium meclofenamate (5 mg/kg iv), glybenclamide (5 mg/kg iv), and rolipram (1.5 mg/kg iv) on vasodilator responses to imatinib (10 mg/kg iv) were investigated to determine the role of products of cyclooxygenase, ATP-sensitive K+ (KATP) channels, and cAMP in mediating or modulating the vasodilator response to imatinib.
For mechanism studies, primary rat PASMCs were isolated as previously described (6). The cells were grown in M199 medium (Sigma-Aldrich) supplemented with 10% FBS and were used for experiments between passages 2 and 5. PDGF-AA, -AB, -BB, and -CC were obtained from PeproTech. The tyrosine kinase inhibitor imatinib was obtained from Novartis Pharma. Rabbit anti-PDGF-α and -β receptor and anti-phosphorylated PDGF-α and -β receptor antibodies were purchased from Santa Cruz Biotechnology. Insulin-transferrin-sodium selenite (ITS) supplement was purchased from Roche. Anti-rabbit secondary antibody was purchased from KPL.
The effect of imatinib on PDGF-stimulated DNA synthesis (thymidine incorporation) in rat PASMCs was investigated. Cells were plated into a 24-well plate at a density of 5 × 104 cells/well with M199 and 10% FBS overnight and then arrested in serum-free M199 supplemented with 10 mg/l ITS for 24 h. The cells were treated with 1 μM imatinib for 1.5 h, followed by 20 ng/ml PDGF-AA, -BB, or -AB. [3H]thymidine (5 μCi/ml; Amersham) was added to the cells 6 h after the growth factors were applied. The cells were harvested with lysis buffer [0.2 N NaOH, 0.1% sodium dodecyl sulfate (SDS)] 24 h after the growth factors were added. Radioactivity was counted with a scintillation counter.
The effect of imatinib on PDGF receptor phosphorylation in rat PASMCs was evaluated by Western blot analysis. Cells were plated into a six-well plate at a density of 3 × 105 cells/well with M199 and 10% FBS overnight and then arrested in serum-free M199 supplemented with 10 mg/l ITS for 24 h. One micromolar imatinib or the same volume of dimethyl sulfoxide (DMSO) vehicle (Sigma) was added to the cells for 1.5 h, followed by 20 ng/ml PDGF-AA, -AB, -BB, and -CC, and the cells were incubated for an additional 1-h period. Twenty micrograms of total protein was resolved in a 8–16% gradient mini-gel (Invitrogen) and transferred to PVDF blot (Amersham). The blot was then incubated with the anti-phosphorylated PDGF-α or -β receptor antibody (1:200) for 1 h at room temperature and washed. The blot was incubated with secondary antibody (1:5,000) for another 1-h period at room temperature. The Western blot was analyzed with an ECL kit from Amersham. After the film was developed, the same blots were stripped with stripping buffer (100 mM β-mercaptoethanol, 2% SDS, 62.5 mM Tris, pH 6.8) and reprobed for anti-PDGF-α or -β receptor (1:200).
The effect of imatinib treatment in preventing (attenuating) the increase in pulmonary arterial pressure, right ventricular hypertrophy, and small vessel remodeling in monocrotaline-induced pulmonary hypertension was investigated. Male Sprague-Dawley rats weighing 250–300 g were injected with 60 mg/kg monocrotaline into the tail vein. Hemodynamic values were measured on day 21 in monocrotaline-treated rats. The monocrotaline-treated rats received daily intraperitoneal injections of imatinib (50 mg/kg) or vehicle, and treatment was continued for 21 days. On day 21, hemodynamic measurements were made, the animals were euthanized, and the heart and lungs were removed for evaluation of right ventricular hypertrophy and pulmonary vessel histology.
The effect of imatinib on medial hypertrophy in small pulmonary arteries following treatment with monocrotaline was evaluated. Elastic trichrome-stained sections from monocrotaline-treated and control animals were masked and analyzed by observers blinded to the treatments. Pulmonary artery wall thickness was measured with an Olympus microscope with digital camera interfacing with a PC utilizing Image-Pro software. Ten arteries with external diameter of 50–200 μm were selected for measurement. The wall thickness (WT) and external diameter (D) of selected pulmonary arteries were measured and “percent wall thickness” (%WT) was calculated by the formula %WT = [2WT/D] × 100. The resulting average measurements from individual animals were pooled with other animals in the same group to give the mean percentage of wall thickness as an index of medial hypertrophy of small pulmonary arteries (21, 27, 28). Right ventricular hypertrophy was assesed using the Fulton Index (RV/LV + S) as previously described (24).
Drugs.
The tyrosine kinase inhibitor imatinib mesylate (Novartis, Basel, Switzerland) was dissolved in deionized water titrated to a pH of 5. U-46619 (Cayman Chemical) was dissolved in 95% ethyl alcohol and diluted in 0.9% NaCl solution. l-NAME (Nω-nitro-l-arginine methyl ester hydrochloride), sodium meclofenamate, rolipram, glybenclamide (Sigma-Aldrich), and fasudil (LC Labs, Woburn, MA) were dissolved in 0.9% NaCl. Isradipine (Sigma-Aldrich) was dissolved in 0.9% NaCl and 1% Tween 80. Working dilutions were made daily with 0.9% NaCl and were kept on ice.
Statistics.
Data are expressed as means ± SE and were analyzed by paired and group t-tests and ANOVA with repeated measures. The criterion for statistical significance was P < 0.05.
RESULTS
Effect of imatinib on pulmonary and systemic vascular beds.
Imatinib has been approved for the treatment of pulmonary hypertension in patients with inadequate response to established therapy (10, 19). However, the relative effects of this agent on vascular resistance in the pulmonary and systemic vascular beds has not been determined. In the present study, the effects of intravenous injections of imatinib in a wide range of doses on pulmonary and systemic arterial pressures and cardiac output were investigated. Under baseline conditions intravenous injection of imatinib in doses of 0.3–30 mg/kg produced dose-related decreases in systemic arterial pressure, small decreases in pulmonary arterial pressure, and no change or small increases in cardiac output, suggesting that the major effect was on the systemic vascular bed (Fig. 1A). However, when baseline tone in the pulmonary vascular bed was increased with U-46619, intravenous injection of imatinib in the same dose range produced dose-related decreases in systemic and pulmonary arterial pressures and a small increase or no significant effect on cardiac output, indicating that imatinib had potent vasodilator effects in both the systemic and pulmonary vascular beds when tone was increased (Table 1 and Fig. 1B). The duration of the decreases in pulmonary arterial pressure in response to imatinib was longer than the duration of the decreases in systemic arterial pressure.
Fig. 1.
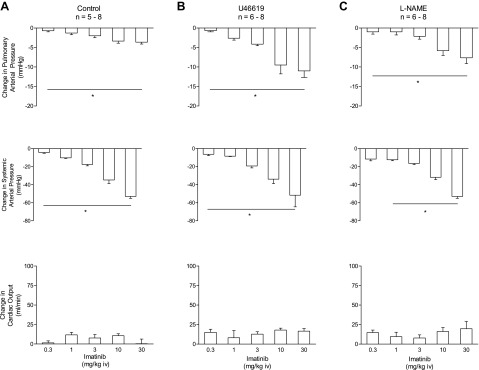
Changes in pulmonary and systemic arterial pressures and cardiac output in response to intravenous injections of the tyrosine kinase inhibitor imatinib in doses of 0.3–30 mg/kg under baseline tone conditions [baseline pulmonary arterial pressure (PAP) 19 ± 1 mmHg, mean arterial pressure (MAP) 114 ± 3 mmHg, and cardiac output (CO) 103 ± 10 ml/min; A], when PAP is increased with U-46619 infusion (baseline PAP 20 ± 1 mmHg, MAP 98 ± 5 mmHg, and CO 118 ± 7 ml/min; B), and after nitric oxide synthase (NOS) inhibition with Nω-nitro-l-arginine methyl ester (l-NAME, 50 mg/kg iv) (PAP 32 ± 3 mmHg, MAP 149 ± 5 mmHg, and CO 53 ± 5 ml/min; C). n, No. of experiments. *P < 0.05. The decrease in PAP in response to intravenous injection of imatinib (3 mg/kg) was significantly less in l-NAME-treated animals compared with U-46619-infused animals (P = 0.02, unpaired t-test).
Table 1.
Effect of U-46619 on systemic and pulmonary arterial pressure and on cardiac output
| Systemic Arterial Pressure, mmHg | Pulmonary Arterial Pressure, mmHg | Cardiac Output, ml/min | |
|---|---|---|---|
| Control | 98 ± 5 | 19 ± 1 | 118 ± 7 |
| U-46619 | 104 ± 3 | 30 ± 2* | 96 ± 5* |
Values are means ± SE for n = 12 experiments.
P < 0.05 compared with control.
Pulmonary and systemic vasodilator responses to imatinib were investigated when NOS was inhibited with l-NAME, and these data are summarized in Fig. 1C. Treatment with l-NAME (50 mg/kg iv) increased pulmonary and systemic arterial pressure and decreased cardiac output (Table 2). Intravenous injection of imatinib in doses of 0.3–30 mg/kg produced dose-related decreases in both pulmonary and systemic arterial pressures and small increases in cardiac output in the l-NAME-treated animal (Fig. 1C).
Table 2.
Effect of l-NAME on systemic and pulmonary arterial pressure and on cardiac output
| Systemic Arterial Pressure, mmHg | Pulmonary Arterial Pressure, mmHg | Cardiac Output, ml/min | |
|---|---|---|---|
| Control | 106 ± 5 | 20 ± 1 | 109 ± 10 |
| l-NAME (50 mg/kg) | 149 ± 5* | 32 ± 3* | 53 ± 5* |
Values are means ± SE for n = 8 experiments. l-NAME, Nω-nitro-l-arginine methyl ester.
P < 0.05 compared with control.
Vasodilator responses to intravenous injections of imatinib were compared in l-NAME-treated and U-46619-infused animals, and unpaired t-tests indicate that there was no significant difference between responses, with the exception that the decrease in pulmonary arterial pressure in response to the intravenous 3 mg/kg dose of imatinib was significantly greater in the U-46619-infused group, suggesting that NOS and NO may play a small role in mediating the pulmonary vasodilator response (Fig. 1, B and C).
The response to imatinib was also investigated when pulmonary arterial pressure was increased on an acute basis by ventilatory hypoxia, and these data are summarized in Fig. 2A. Ventilation with a 10% O2-90% N2 gas mixture increased pulmonary arterial pressure, and intravenous injection of 10 mg/kg imatinib attenuated the hypoxic pulmonary vasoconstrictor response (Fig. 2A). These data indicate that imatinib will induce vasodilation when pulmonary vascular resistance is increased by acute ventilatory hypoxia without significantly affecting cardiac output.
Fig. 2.

A: effect of ventilation with a 10% O2-90% N2 gas mixture on pulmonary arterial pressure and the effect of intravenous injection of 10 mg/kg imatinib on the hypoxia-induced increase in pulmonary arterial pressure. B: effects of cyclooxygenase inhibition with sodium meclofenamate (5 mg/kg iv), phosphodiesterase type 4 inhibition with rolipram (1.5 mg/kg iv), and inhibition of ATP-sensitive K+ channels with glybenclamide (5 mg/kg iv). n, No. of experiments. *P < 0.05.
The effects of sodium meclofenamate, glybenclamide, and rolipram on vasodilator responses to imatinib were investigated, and these data are summarized in Fig. 2B. The decreases in pulmonary and systemic arterial pressure in response to intravenous injection of 10 mg/kg imatinib were not altered by treatment with sodium meclofenamate (5 mg/kg iv), glybenclamide (5 mg/kg iv), or rolipram (1.5 mg/kg iv). These results indicate that responses to imatinib are not dependent on cyclooxygenase products, the opening of KATP channels, or increases in cAMP levels.
Cardiovascular responses to imatinib were compared with responses to the Rho kinase inhibitor fasudil and the calcium entry blocking agent isradipine, and these data are summarized in Fig. 3. Under elevated tone conditions in U-46619-infused animals intravenous injections of 10 mg/kg imatinib, 300 μg/kg fasudil, or 10 μg/kg isradipine produced significant decreases in pulmonary arterial and systemic arterial pressures and small changes in cardiac output (Fig. 3). Intravenous injection of the three agents produced rapid-onset decreases (5–15 s) in pulmonary and systemic arterial pressures. The duration of the decreases in pulmonary and systemic arterial pressures was greatest with isradipine (data not shown). The percent decreases in pulmonary and systemic arterial pressure in response to the three agents were similar when tone was increased with U-46619.
Fig. 3.

Comparison of decreases in pulmonary and systemic arterial pressures and cardiac output in response to injection of imatinib (10 mg/kg iv), the Rho kinase inhibitor fasudil (300 μg/kg iv), and the calcium entry antagonist isradipine (10 μg/kg iv) when pulmonary arterial pressure is increased to ∼30 mmHg with U-46619 infusion. n, No. of experiments. *P < 0.05 compared with baseline values.
Effect of imatinib on development of monocrotaline-induced pulmonary hypertension.
The effect of imatinib treatment on the development of monocrotaline-induced pulmonary hypertension, right ventricular hypertrophy, and small pulmonary vessel remodeling was investigated in a prevention study, and these data are shown in Fig. 4. Imatinib (50 mg/kg ip for 21 days) significantly reduced the increases in pulmonary arterial pressure, right ventricular hypertrophy, and small pulmonary artery medial hypertrophy that occurred in response to monocrotaline treatment (60 mg/kg iv).
Fig. 4.

Effect of chronic imatinib (IB) treatment (50 mg/kg ip) for 21 days on the attenuation of increases in pulmonary arterial pressure, right ventricular hypertrophy, and medial wall thickness of small pulmonary arteries in monocrotaline (MCT)-induced pulmonary hypertension in a prevention trial in the rat. n, No. of experiments. RV, right ventricle; LV, left ventricle; S, septum. *P < 0.05.
In evaluating the effect of imatinib on vascular remodeling, wall thickness was measured in small pulmonary arteries with an external diameter of ∼50–200 μm and the percent change in wall thickness was compared in the different treatment groups. There was a significant increase in percent wall thickness in small pulmonary arteries from animals with monocrotaline-induced pulmonary hypertension (Fig. 4). There was a significant decrease in the percent wall thickness in small pulmonary arteries from animals treated with monocrotaline and imatinib compared with values in animals treated with monocrotaline alone (Fig. 4).
Effect of imatinib on PDGF receptor phosphorylation and PDGF-stimulated thymidine incorporation in rat pulmonary artery smooth muscle cells.
Experiments were carried out to investigate the effect of imatinib on PDGF-stimulated arterial smooth muscle cell DNA synthesis as measured by thymidine incorporation and PDGF receptor phosphorylation in rat PASMCs, and these data are summarized in Fig. 5. There was a significant increase in thymidine incorporation (DNA synthesis) in PASMCs treated with PDGF-AA, -BB, and -AB (Fig. 5C). Phosphorylation of PDGF-α and -β receptors was assessed by Western blot analysis, and exposure to PDGF-AB and -BB induced strong phosphorylation of both PDGF receptors (Fig. 5, A and B). Exposure to PDGF-CC and -AA induced weaker phosphorylation of PDGF-α and -β receptors (Fig. 5, A and B). The phosphorylation of PDGF receptors in rat PASMCs was inhibited by treatment with 1 μM imatinib (Fig. 5, A and B). Treatment of PASMCs with 1 μM imatinib significantly decreased thymidine incorporation in response to PDGF-AA, -BB, and -AB (Fig. 5C). These data indicate that imatinib treatment can decrease PASMC proliferation (DNA synthesis) when exposed to PDGF-AA, - BB, and -AB as measured by thymidine incorporation. This inhibitory effect on thymidine incorporation correlated with the inhibition of PDGF receptor phosphorylation induced by imatinib.
Fig. 5.
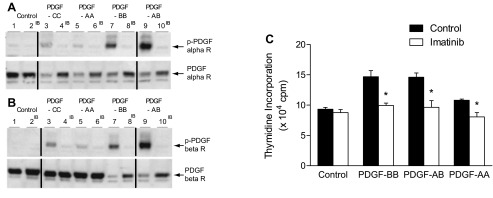
Western blots showing inhibition of PDGF-α receptor (A) and PDGF-β receptor (B) phosphorylation by imatinib. Even-numbered lanes received imatinib (IB). Lanes 1 and 2, control; lanes 3 and 4, PDGF-CC; lanes 5 and 6, PDGF-AA, lanes 7 and 8, PDGF-BB; lanes 9 and 10, PDGF-AB. Black lines separate different Western blot membranes using samples from the same culture of vascular smooth muscle cells. C: comparison of effect of tyrosine kinase inhibition with imatinib on increases in thymidine incorporation (DNA synthesis) in rat PASMCs exposed to PDGF-BB, -AB, and -AA. n = 3–5 experiments. *P < 0.05.
DISCUSSION
Imatinib has been approved for the treatment of pulmonary arterial hypertension in patients with inadequate response to established therapy with endothelin receptor antagonists, PDE5 inhibitors, and prostacyclin or analogs (15). Although it has been reported that imatinib has smooth muscle relaxant and vasodilator properties, the relative effects of this tyrosine kinase inhibitor on the pulmonary and systemic vascular beds have not been compared in animals without pulmonary hypertension (1, 29). The results of the present study indicate that imatinib has vasodilator activity in both the pulmonary and systemic vascular beds and that the systemic vasodilator effects are more pronounced under baseline conditions. However, when vasoconstrictor tone in the pulmonary vascular bed was increased with U-46619 or l-NAME, imatinib had potent vasodilator activity in both the pulmonary and systemic vascular beds, and in this regard imatinib was similar to the Rho kinase inhibitor fasudil and the calcium entry antagonist isradipine. The present data indicate that imatinib, fasudil, and isradipine decrease pulmonary arterial pressure and right ventricular afterload; however, all three agents also produce a decrease in systemic arterial pressure.
Pulmonary hypertension is a disorder characterized by excess vasoconstriction and vascular remodeling in the pulmonary vascular bed (20). Imatinib is an antiproliferative agent developed to inhibit the BCR-ABL tyrosine kinase in patients with chronic myelogenous leukemia (5). Imatinib was discovered in a program to screen inhibitors of PDGF receptor activation, and this agent inhibits a number of receptor and nonreceptor tyrosine kinases (12, 19). Imatinib has been shown to reverse monocrotaline- and chronic hypoxia-induced pulmonary hypertension and has been approved as “add-on” therapy for patients with pulmonary hypertension who do not have adequate responses to standard therapy (10, 11, 15, 29).
In the present study imatinib treatment attenuated monocrotaline-induced pulmonary hypertension, right ventricular hypertrophy, and medial hypertrophy of small pulmonary arteries in a 21-day prevention study. The ability of imatinib to reduce medial hypertrophy in small arteries correlated with the inhibitory effect of the drug on PDGF-stimulated DNA synthesis in rat PASMCs as assessed by reduced thymidine incorporation. Imatinib inhibited PDGF receptor phosphorylation induced by PDGF-AA, -AB, -BB, and -CC. The data showing decreased PDGF-stimulated thymidine incorporation and decreased PDGF receptor activation (phosphorylation) provide information in regard to the mechanism of the beneficial effect on vascular smooth muscle cell proliferation and vascular remodeling in the pulmonary vascular bed. However, the mechanism by which imatinib induces vasodilation is uncertain. Imatinib inhibits PDGF receptor signaling, and PDGF has been shown to have potent contractile activity in aortic strips and potent vasoconstrictor activity in the rabbit ear artery (3, 17). Imatinib also inhibits other receptor tyrosine kinases such as c-KIT and has been shown to relax isolated smooth muscle preparations from the bladder, uterus, corpora cavernosa, and small penile arteries and to have potent erectile activity in the rat (4, 6, 7, 13, 18, 22, 23, 25, 26, 31, 32). The results of the present study and of previous studies in which three different tyrosine kinase inhibitors have been shown to have potent vasorelaxant activity in isolated pulmonary arteries and studies with agents such as genistein that inhibit tyrosine kinases are consistent with the hypothesis that tyrosine kinase signaling is involved in the regulation of vascular and genitourinary smooth muscle tone (1, 29, 31, 32). The observation that imatinib decreases pulmonary and systemic arterial pressure is consistent with the hypothesis that an imatinib-sensitive tyrosine kinase is playing a role in maintaining pulmonary and systemic arterial pressures at baseline levels and when this putative vasoconstrictor tyrosine kinase pathway is inhibited by imatinib, vasodilation occurs. The results of pharmacological studies suggest that cyclooxygenase products, KATP channels, cAMP, and NO are not involved in mediating vasodilator responses to imatinib in the intact-chest rat. It has been reported that inhibition of Rho kinase is involved in mediating vasorelaxant responses to imatinib in isolated pulmonary arteries contracted with U-46619, and the present results comparing responses to imatinib and the Rho kinase inhibitor fasudil are consistent with this reported mechanism (1, 2, 29). However, the role of receptor and nonreceptor tyrosine kinases in regulating vascular tone and the exact mechanism by which imatinib induces vasodilation are unknown. In addition, it is uncertain whether a decrease in contractile protein calcium sensitivity or smooth muscle intracellular calcium levels are involved in mediating vasodilator responses to imatinib in the pulmonary and systemic vascular beds. However, the ability of imatinib to inhibit vascular smooth muscle proliferation and to decrease pulmonary vascular resistance are beneficial effects in the treatment of pulmonary hypertension.
Imatinib is an antiproliferative agent developed to inhibit BCR-ABL nonreceptor tyrosine kinase in patients with chronic myelogenous leukemia (5, 8, 19, 30). Moreover, the inhibitory effects of imatinib on PDGF, c-KIT, and Src signaling suggest that these effects of imatinib may be beneficial in the treatment of pulmonary hypertension (11).
In the present study the effect of imatinib in a prevention trial was assessed, and treatment with imatinib at an intraperitoneal dose of 50 mg/kg for 21 days significantly reduced the increases in pulmonary arterial pressure, right ventricular hypertrophy, and small vessel medial hypertrophy in rats with monocrotaline-induced pulmonary hypertension.
The effect of imatinib on vascular smooth muscle cell proliferation was investigated, and exposure of rat PASMCs to imatinib attenuated increases in thymidine incorporation in responses to PDGF-AA, -BB, and -AB stimulation. The weak phosphorylation response to PDGF-AA in these cells may be due to the low level of PGDF-AA receptor in rat PASMCs. The inhibitory effect of imatinib on thymidine incorporation was correlated with results showing inhibition of PDGF receptor activation (phosphorylation). These data suggest that imatinib inhibits tyrosine kinase receptor activation and attenuates smooth muscle cell proliferation in rat PASMCs. It is therefore possible that inhibition of PDGF receptor signaling is involved in mediating the beneficial effects of imatinib in the treatment of monocrotaline-induced pulmonary hypertension, although other mechanisms cannot be excluded.
In summary, the results of the present study indicate that imatinib has substantial vasodilator activity in the pulmonary and systemic vascular beds and in this regard is similar to the Rho kinase inhibitor fasudil and the calcium entry antagonist isradipine. Imatinib attenuated the acute hypoxic pulmonary vasoconstrictor response, and chronic treatment with the tyrosine kinase inhibitor attenuated monocrotaline-induced pulmonary hypertension, right ventricular hypertrophy, and small pulmonary vessel remodeling in a prevention study. Imatinib inhibited thymidine incorporation and PDGF receptor phosphorylation induced by PDGF isoforms in rat PASMCs. These results may be interpreted to suggest that receptor tyrosine kinase signaling may have a role in regulating smooth muscle cell proliferation and vasoconstrictor tone in the lung and the peripheral vascular bed, although a role for nonreceptor tyrosine kinases and other mechanisms cannot be excluded.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-62000 and HL-77421 and the Wetmore Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: E.A.P., J.A.L., and P.J.K. conception and design of research; E.A.P., S.T., G.F.L., and S.B. performed experiments; E.A.P., S.T., G.F.L., and P.J.K. analyzed data; E.A.P., S.T., G.F.L., S.B., J.A.L., and P.J.K. interpreted results of experiments; E.A.P., S.T., and G.F.L. prepared figures; E.A.P., G.F.L., J.A.L., and P.J.K. drafted manuscript; E.A.P., G.F.L., J.A.L., and P.J.K. edited and revised manuscript; E.A.P., S.T., G.F.L., S.B., J.A.L., and P.J.K. approved final version of manuscript.
REFERENCES
- 1.Abe K, Toba M, Alzoubi A, Koubsky K, Ito M, Ota H, Gairhe S, Gerthoffer WT, Fagan KA, McMurtry IF, Oka M. Tyrosine kinase inhibitors are potent acute pulmonary vasodilators in rats. Am J Respir Cell Mol Biol 45: 804–808, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badejo AM, Jr, Dhaliwal JS, Casey DB, Gallen TB, Greco AJ, Kadowitz PJ. Analysis of pulmonary vasodilator responses to the Rho-kinase inhibitor fasudil in the anesthetized rat. Am J Physiol Lung Cell Mol Physiol 295: L828–L836, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Berk BC, Brock TA, Webb RC, Taubman MB, Atkinson WJ, Gimbrone MA, Jr, Alexander RW. Epidermal growth factor, a vascular smooth muscle mitogen, induces rat aortic contraction. J Clin Invest 75: 1083–1086, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biers SM, Reynard JM, Doore T, Brading AF. The functional effects of a c-kit tyrosine inhibitor on guinea-pig and human detrusor. BJU Int 97: 612–616, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Carroll M, Ohno-Jones S, Tamura S, Buchdunger E, Zimmermann J, Lydon NB, Gilliland DG, Druker BJ. CGP 57148, a tyrosine kinase inhibitor, inhibits the growth of cells expressing BCR-ABL, TEL-ABL, and TEL-PDGFR fusion proteins. Blood 90: 4947–4952, 1997 [PubMed] [Google Scholar]
- 6.Chattergoon NN, D'Souza FM, Deng W, Chen H, Hyman AL, Kadowitz PJ, Jeter JR., Jr Antiproliferative effects of calcitonin gene-related peptide in aortic and pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 288: L202–L211, 2005 [DOI] [PubMed] [Google Scholar]
- 7.Cretoiu SM, Simionescu AA, Caravia L, Curici A, Cretoiu D, Popescu LM. Complex effects of imatinib on spontaneous and oxytocin-induced contractions in human non-pregnant myometrium. Acta Physiol Hung 98: 329–338, 2011 [DOI] [PubMed] [Google Scholar]
- 8.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 2: 561–566, 1996 [DOI] [PubMed] [Google Scholar]
- 9.Eriksson A, Nister M, Leveen P, Westermark B, Heldin CH, Claesson-Welsh L. Induction of platelet-derived growth factor alpha- and beta-receptor mRNA and protein by platelet-derived growth factor BB. J Biol Chem 266: 21138–21144, 1991 [PubMed] [Google Scholar]
- 10.Ghofrani HA, Morrell NW, Hoeper MM, Olschewski H, Peacock AJ, Barst RJ, Shapiro S, Golpon H, Toshner M, Grimminger F, Pascoe S. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am J Respir Crit Care Med 182: 1171–1177, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghofrani HA, Seeger W, Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med 353: 1412–1413, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Gur S, Kadowitz PJ, Hellstrom WJ. A protein tyrosine kinase inhibitor, imatinib mesylate (Gleevec), improves erectile and vascular function secondary to a reduction of hyperglycemia in diabetic rats. J Sex Med 7: 3341–3350, 2010 [DOI] [PubMed] [Google Scholar]
- 13.Gur S, Sikka SC, Abdel-Mageed AB, Abd Elmageed ZY, Rezk B, Pankey E, Kadowitz PJ, Hellstrom WJ. Imatinib mesylate (Gleevec) induces human corpus cavernosum relaxation by inhibiting receptor tyrosine kinases (RTKs): identification of new RTK targets. Urology 82: 745.e11–745.e16, 2013 [DOI] [PubMed] [Google Scholar]
- 14.Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev 79: 1283–1316, 1999 [DOI] [PubMed] [Google Scholar]
- 15.Hoeper MM, Barst RJ, Bourge RC, Feldman J, Frost AE, Galie N, Gomez-Sanchez MA, Grimminger F, Grunig E, Hassoun PM, Morrell NW, Peacock AJ, Satoh T, Simonneau G, Tapson VF, Torres F, Lawrence D, Quinn DA, Ghofrani HA. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation 127: 1128–1138, 2013 [DOI] [PubMed] [Google Scholar]
- 16.Hubbard SR, Miller WT. Receptor tyrosine kinases: mechanisms of activation and signaling. Curr Opin Cell Biol 19: 117–123, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes AD. Increase in tone and intracellular Ca2+ in rabbit isolated ear artery by platelet-derived growth factor. Br J Pharmacol 114: 138–142, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hutchings G, Deprest J, Nilius B, Roskams T, De Ridder D. The effect of imatinib mesylate on the contractility of isolated rabbit myometrial strips. Gynecol Obstet Invest 62: 79–83, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Manley PW, Stiefl N, Cowan-Jacob SW, Kaufman S, Mestan J, Wartmann M, Wiesmann M, Woodman R, Gallagher N. Structural resemblances and comparisons of the relative pharmacological properties of imatinib and nilotinib. Bioorg Med Chem 18: 6977–6986, 2010 [DOI] [PubMed] [Google Scholar]
- 20.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, Mathier MA, McGoon MD, Park MH, Rosenson RS, Rubin LJ, Tapson VF, Varga J. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. J Am Coll Cardiol 53: 1573–1619, 2009 [DOI] [PubMed] [Google Scholar]
- 21.Meyrick B, Reid L. Hypoxia-induced structural changes in the media and adventitia of the rat hilar pulmonary artery and their regression. Am J Pathol 100: 151–178, 1980 [PMC free article] [PubMed] [Google Scholar]
- 22.Min Y, He P, Wang Q, Jin X, Song B, Li L. The effects of the c-kit blocker Glivec on the contractile response of urinary bladder. J Surg Res 171: e193–e199, 2011 [DOI] [PubMed] [Google Scholar]
- 23.Ozgur-Akdemir A, Demirturk K, Karabakan M, Volkan-Oztekin C, Abdulkadir NA, Cetinkaya M, Gur S, Hellstrom WJ. Imatinib mesylate (Gleevec) as protein-tyrosine kinase inhibitor elicits smooth muscle relaxation in isolated human prostatic tissue. Urology 78: 968.e961–968.e966, 2011 [DOI] [PubMed] [Google Scholar]
- 24.Pankey EA, Bhartiya M, Badejo AM, Jr, Haider U, Stasch JP, Murthy SN, Nossaman BD, Kadowitz PJ. Pulmonary and systemic vasodilator responses to the soluble guanylyl cyclase activator, BAY 60-2770, are not dependent on endogenous nitric oxide or reduced heme. Am J Physiol Heart Circ Physiol 300: H792–H802, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pankey EA, Lasker GF, Gur S, Hellstrom WJ, Kadowitz PJ. Analysis of erectile responses to imatinib in the rat. Urology 82: 253.e217–253.e224, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Popescu LM, Vidulescu C, Curici A, Caravia L, Simionescu AA, Ciontea SM, Simion S. Imatinib inhibits spontaneous rhythmic contractions of human uterus and intestine. Eur J Pharmacol 546: 177–181, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Rabinovitch M, Gamble W, Nadas AS, Miettinen OS, Reid L. Rat pulmonary circulation after chronic hypoxia: hemodynamic and structural features. Am J Physiol Heart Circ Physiol 236: H818–H827, 1979 [DOI] [PubMed] [Google Scholar]
- 28.Rabinovitch M, Konstam MA, Gamble WJ, Papanicolaou N, Aronovitz MJ, Treves S, Reid L. Changes in pulmonary blood flow affect vascular response to chronic hypoxia in rats. Circ Res 52: 432–441, 1983 [DOI] [PubMed] [Google Scholar]
- 29.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest 115: 2811–2821, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schiffer CA. BCR-ABL tyrosine kinase inhibitors for chronic myelogenous leukemia. N Engl J Med 357: 258–265, 2007 [DOI] [PubMed] [Google Scholar]
- 31.Villalba N, Kun A, Stankevicius E, Simonsen U. Role for tyrosine kinases in contraction of rat penile small arteries. J Sex Med 7: 2086–2095, 2010 [DOI] [PubMed] [Google Scholar]
- 32.Wang LD, Qiu XQ, Tian ZF, Zhang YF, Li HF. Inhibitory effects of genistein and resveratrol on guinea pig gallbladder contractility in vitro. World J Gastroenterol 14: 4955–4960, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamawaki H, Sato K, Hori M, Ozaki H, Nakamura S, Nakayama H, Doi K, Karaki H. Impairment of EDR by a long-term PDGF treatment in organ-cultured rabbit mesenteric artery. Am J Physiol Heart Circ Physiol 277: H318–H323, 1999 [DOI] [PubMed] [Google Scholar]