

Abstract
Coronary collateral growth is a process involving coordination between growth factors expressed in response to ischemia and mechanical forces. Underlying this response is proliferation of vascular smooth muscle and endothelial cells, resulting in an enlargement in the caliber of arterial-arterial anastomoses, i.e., a collateral vessel, sometimes as much as an order of magnitude. An integral element of this cell proliferation is the process known as phenotypic switching in which cells of a particular phenotype, e.g., contractile vascular smooth muscle, must change their phenotype to proliferate. Phenotypic switching requires that protein synthesis occurs and different kinase signaling pathways become activated, necessitating energy to make the switch. Moreover, kinases, using ATP to phosphorylate their targets, have an energy requirement themselves. Mitochondria play a key role in the energy production that enables phenotypic switching, but under conditions where mitochondrial energy production is constrained, e.g., mitochondrial oxidative stress, this switch is impaired. In addition, we discuss the potential importance of uncoupling proteins as modulators of mitochondrial reactive oxygen species production and bioenergetics, as well as the role of AMP kinase as an energy sensor upstream of mammalian target of rapamycin, the master regulator of protein synthesis.
Keywords: angiogenesis, arteriogenesis, redox-dependent signaling, mitochondria
this article is part of a collection on Mitochondria in Cardiovascular Physiology and Disease. Other articles appearing in this collection, as well as a full archive of all collections, can be found online at http://ajpheart.physiology.org/.
Introduction
The abluminal expansion of coronary collateral vessels, to wit, the process we term collateral growth (also known as arteriogenesis), is an adaptive mechanism in the heart initiated by ischemia. The benefit of this adaptive response is striking in that patients with well-developed collaterals show reduced incidence of sudden death and smaller infarcts in the event of a coronary occlusion (45, 49, 55, 56). In their native state, coronary collateral vessels are small, high-resistance channels that have limited potential to supply flow to the myocardium downstream from an occluded, or severely stenotic, coronary artery. Indeed, in most animal models and in humans, native collateral flow is typically 70–90% less than flow to the normal zone (23, 54), which indicates their small native caliber and thus high resistance to flow. However, collateral vessels have great potential to remodel via abluminal expansion, which is reported to increase the lumen caliber between 5- and 10-fold that of native, unstimulated collaterals. The expansion results in a tremendous decrease in collateral resistance, which significantly increases blood flow to the collateral-dependent region of the myocardium. Fully developed collaterals can carry sufficient flow to prevent or minimize myocardial infarction in the event of a coronary occlusion.
In addition to stimulation of collateral growth, there is also evidence that myocardial ischemia initiates de novo angiogenesis (8, 37), which appears to parallel the presence of ischemia, i.e., as the ischemic stimulus waxes, capillary growth is evident but as the ischemic signal wanes, capillary growth stops and rarefaction occurs until capillary density has returned to basal conditions (35). In contrast, collateral growth continues as ischemia is resolved. This observation has led us to speculate that collateral growth is initiated by ischemia, but other regulatory mechanisms, e.g., shear stress, likely play a key role in the continuation of remodeling after the ischemic signals wane. Despite the fact that expansion of collateral vessels is functionally important to ameliorate or even eliminate myocardial ischemic injury, there are large interindividual differences in the number and the calibers of collateral vessels formed. These may be explained by variations in the anatomical position where the ischemic event occurred (8, 60), severity and duration of stenosis (24), and likely individual capacity to develop functional collateral vessels (i.e., as determined by redox state, expression of cytokines and growth-factors, and homing of inflammatory cells, etc.). Therefore, it is important to understand the complex genetic and molecular mechanisms leading to the development of the native collaterals as well as induced growth of these vessels. Moreover, it is equally important to determine the mechanisms underlying impaired collateral growth in models of vascular disease and endothelial dysfunction and to better design therapeutics/strategiesm enabling the induction of arteriogenesis. In this context, we believe that mitochondrial function is critical to collateral growth, and its bioenergetic component provides a common link among many factors and stimuli that affect this adaptive process in the heart. To introduce a recurring theme throughout this review, our hypothesis is that mitochondrial function provides a permissive role for collateral growth by maintaining energetic capacity and reserves, to enable phenotypic switching of cells that occurs during vascular growth. To frame this perspective, we emphasize the myriad kinase cascades that are activated during vascular growth with this important caveat: every kinase uses ATP to phosphorylate its target or substrate. Thus ATP is critical for these adaptive processes that involve kinase activation. Our goal for this review is to discuss a link among mitochondrial oxidative stress, bioenergetics and coronary collateral growth, and how energetically-based signaling, e.g., 5′-adenosine monophosphate kinase (AMPK), may be considered in therapies designed to stimulate the growth of coronary vessels in the heart, particularly in patients with metabolic syndrome.
Mitochondrial Function and Structure
The term, mitochondrion, was derived from combining the Greek words, mitos (thread) and chondrion (granule). Mitochondria play central roles in high energy phosphate (ATP) and heat production in eukaryotic cells (18, 36, 39). In addition to these processes, mitochondria are involved in calcium homeostasis (17), intracellular signaling (18, 20), apoptosis (15, 16), heme and steroid synthesis (42, 51), cellular proliferation, and membrane potential regulation (36), as well as oxygen sensing (7). Most of these processes are critically dependent on energy that is produced by the hydrolysis of ATP, the majority of which is produced via aerobic metabolism in the mitochondria.
Following enzymatic reactions that degrade carbohydrates, fats, and proteins into smaller molecules such as pyruvate, fatty acids, and amino acids, respectively, mitochondria further transform these energetic elements into NADH and/or FADH2, through β-oxidation and the Krebs cycle. These reducing equivalents are then used by the mitochondrial respiratory chain in a process called oxidative phosphorylation, in which electrons derived from oxidation of NADH and FADH2 are passed along electron transport chain and ultimately transferred to molecular oxygen at complex IV (53). The incremental release of energy produced by oxidation of these reducing equivalents is used to pump protons (H+) into the intermembrane space, creating an electrochemical gradient that is subsequently used by ATP synthase (complex V) for the phosphorylation of ADP into ATP. During this process, a small percentage of electrons (estimates range from 0.01 to 1–2% depending the preparation and state of respiration) “escape” from the complexes and react with oxygen to form superoxide (O2·−), a highly reactive variant of reactive oxygen species (ROS). If the rate of superoxide formation exceeds the capacity of mitochondrial antioxidant systems [e.g., superoxide dismutase-2 (SOD-2)], toxicity can occur. This leads to damage of mitochondrial DNA, proteins, membrane components and ultimately compromises key mitochondrial functions such as cellular respiration. This state of impaired molecular function due to damage inflicted by accumulation of ROS is commonly referred to as oxidative stress. Oxidative stress strongly correlates with mitochondrial dysfunction and likely contributes to the decline in mitochondrial bioenergetics associated with aging processes and many mitochondrial-related diseases (27).
Recent advances in mitochondrial imaging have revealed that mitochondria are highly dynamic and form networks in living cells. They undergo fission and fusion processes constantly. The balance between mitochondrial fission and fusion is altered in response to various stimuli, e.g., fission can be associated with apoptosis, stress, or cellular division, whereas fusion can transmit signals concerning energy and Ca2+ levels and facilitate exchange of mitochondrial DNA and proteins (32). In addition to fission and fusion, mitochondria also move along the cytoskeleton by attaching to specific motor and connector proteins. Although mitochondria share similar functions in most tissues, their morphology, dynamic behavior, and regulation vary greatly in a cell type-specific manner to achieve cell type-specific functions for particular physiological demands. For instance, formation of mitochondrial networks in neurons is regulated by local calcium concentration and directed to cellular regions of high energy demand (26), whereas mitochondria in adult cardiomyocytes and skeletal muscles are tethered to form an arrangement between myofibrils so as to provide continuous energy supply for contraction (32).
Mitochondrial Production of ROS
Cellular sources of ROS generation within the cardiovascular system include cardiomyocytes, endothelial cells, and neutrophils. ROS can be produced by several mechanisms including electrons leaked from mitochondrial complexes, NADPH oxidase, and xanthine dehydrogenase/xanthine oxidase (61). In cells and organs with high-energy demands, there is a high density of mitochondria that not only produce ATP but also ROS as a by-product of mitochondrial respiration. ROS are produced in the mitochondria because the transfer of electrons through the electron transport chain is not 100% efficient. Consequently, a small percentage of electrons escape from electron transport chain complexes and reduce O2 to produce O2·− (2). Although this is a low rate of electron leakage, considering the continual flux of electrons for energy production in metabolically active tissues, this leak translates to an enormous production of ROS by the mitochondria.
Within mitochondria, O2·− is normally neutralized by superoxide dismutase-2 (SOD-2 or Mn-SOD), which is located in the mitochondrial matrix and also by superoxide dismutase-1 (SOD-1 or Cu/Zn-SOD). SOD-1, typically considered as cytoplasmic SOD, is also located between the inner and outer mitochondrial membranes (43). Both SOD-1 and -2 dismutate O2·− to hydrogen peroxide (H2O2), which is less reactive than O2·−. Hydrogen peroxide is then converted to O2 and H2O by antioxidant enzymes, such as glutathione peroxidase-1 and catalase. However, if there is an imbalance between the mitochondrial prooxidant generation and its antioxidant defenses, mitochondrial oxidative stress may ensue (46).
Uncoupling proteins (UCPs) are mitochondrial proteins that can serve to reduce production of ROS. These proteins depolarize mitochondria, which reduces the potential driving electron transfer and as such decrease ROS generation (57). The idea that metabolic uncoupling regulates mitochondrial ROS generation was initially based on evidence indicating that mitochondrial ROS production is increased in UCP-3−/− mice, although there may be other compensatory mechanism(s) to mediate this increase (63). Mitochondrial ROS levels can be effectively controlled by the rate of proton reentry into the mitochondrial matrix. This process is mediated through UCPs. UCPs are a family of inner mitochondrial membrane proteins whose main function is to allow the reentry of protons to the mitochondrial matrix, bypassing ATP synthase. Thus UCPs represent a line of antioxidant defense aimed at resolving mismatches between outward and inward proton fluxes (5, 58). Secondary to allowing proton reentry, UCPs have been shown to regulate energy metabolism as well as control body mass (3–5, 13, 41). Because of their roles in the intersection between energy efficiency and oxidative stress, UCPs have drawn our attention particularly to how they can impact oxidative metabolism positively and negatively, especially in cardiovascular function.
Mitochondria as Targets of Oxidative Stress
One of the consequences of excessive production or inadequate neutralization of ROS in the mitochondria is the oxidative modification of mitochondrial DNA (mtDNA). Mitochondrial DNA is more susceptible to oxidative damage than nuclear DNA for a number of reasons. First, mitochondria do not have complex chromatin organization consisting of histone proteins, which serve as a protective barrier against ROS. Second, mitochondria have relatively limited repair activity against DNA damage compared with the nucleus, which harbors an extensive armament of DNA repair mechanisms. Third, the O2·− formed inside the mitochondria cannot pass through the membranes, and hence, oxidative damage resulting from mitochondrial ROS may be largely confined to the mitochondria (61, 62). Fourth, the close proximity of mtDNA to the sites of ROS production in the mitochondrial matrix enables conditions conducive to oxidative modification of DNA bases (14). We do not completely ignore a possible damaging effect from cytosolic-derived ROS or the potential cross talk between the cytosolic and mitochondrial ROS, but the vulnerability of mtDNA to ROS and the high densities of mitochondria in the myocardium provide rationale, suggesting that the myocardium is more prone to oxidative damage derived from the mitochondrial electron transfer. Chronic increases in mitochondrial ROS can induce mitochondrial DNA fragmentation and rearrangement (44, 64), followed by compromised mitochondrial structure and function. By overwhelming cellular antioxidant systems, excessive mitochondrial ROS production also induces a vicious cycle of cytosolic ROS production, which in turn leads to a detrimental decline in myocardial function, progression of myocardial remodeling, and heart failure (21).
Although there is no question about the existence of mitochondrial oxidative stress as being distinct from some of the causes of hindered coronary collateral growth, the answers to understanding the precise mechanisms linking the former impairment to the latter are evasive. Previously, in a model of Zucker obese fatty (ZOF) rats with insulin resistance and impaired endothelial function, we found that resolution of mitochondrial oxidative stress restored coronary collateral growth to levels approaching those in control lean rats (47). However, our results did not delineate the precise mechanism(s) by which mitochondrial oxidative stress exerted this negative influence. We speculate that mitochondrial oxidative stress, which induces oxidative modification of mtDNA (11, 12, 50), impairs expression of the mitochondrial genome. The mitochondrial genome contains 13 genes that encode proteins that are critical for oxidative phosphorylation and energy production. The absence of these proteins leads to reduced efficiency of electron transfer, resulting in higher rates of electron leakage and consequent ROS production. The increased ROS levels lead to further mtDNA damage, thus producing a vicious cycle of mitochondrial dysfunction. One reason that alleviation of mitochondrial ROS levels may have been successful is that the administration of mitochondrial-directed free radical scavengers interfered with this vicious cycle and allowed restoration of normal mitochondrial function.
Energy Sensors and the Modulation of Cell Phenotype
5′-Adenosine monophosphate-activated protein kinase (AMPK) was first characterized as a “fuel gauge” in modulating cellular energy flux in eukaryotic cells in response to an increased ratio of AMP to ATP under stressful conditions (30, 34). AMPK is a heterodimeric protein kinase comprised of one catalytic subunit-α and two regulatory subunits-β and -γ (6, 29, 38). While AMPK is ubiquitously expressed, its activation is heavily regulated. The primary mechanism responsible for AMPK activation involves phosphorylation of threonine residues on the α-subunit by at least two known AMPK kinases, the tumor suppressor gene liver kinase B1 and calcium calmodulin-dependent kinase (67). In addition, a rise in 5′-AMP levels resulting from the adenylate kinase reaction (2 ADP → ATP + AMP) or from the hydrolysis of ATP and/or dephosphorylation of ADP will lead to allosteric activation of AMPK via AMP binding to the α-subunit (66). Once activated, AMPK will phosphorylate downstream target proteins, with its activity proportional to work and turnover of ATP (66). Activation of AMPK produces a wide range of regulatory effects on metabolism, including either inhibition or stimulation of energy expenditure in a tissue-dependent manner. In the liver, for example, AMPK activation is linked to inhibition of ATP-consuming processes, e.g., decreased protein synthesis, and promotion of energy-conserving cellular processes, e.g., increased glucose uptake, decreased expression of gluconeogenic enzymes, and stimulation of glycogen, cholesterol, fatty acid, and triacylglycerol synthesis (19). By contrast, in skeletal muscle, AMPK activation supports energy-expending processes such as increased glucose and fatty acid oxidation (thus increasing insulin sensitivity) (19), inhibition of fatty acid synthesis, and acceleration of mitochondrial biogenesis (31). AMPK has also been implicated in many additional processes such as exercise-induced vascular growth, tumorigenesis, atherosclerosis (33, 40, 68), and inflammation (28). However, in the context of coronary collateral growth, perhaps the most important role that AMPK plays is as an energy sensor.
As such, we believe AMPK is positioned to determine whether the energy status of a cell is sufficient to enable phenotypic switching, or in the situation of energy limitations, prevent phenotypic switching and growth. Indeed, recent work from our laboratory has shown during impaired mitochondrial bioenergetics induced by oxidative stress, AMP kinase is activated (48). Activation of this kinase has profound effects on events involved in growth, such as inhibiting the activity of mammalian target of rapamycin (mTOR), one of the master regulators of protein synthesis. We envision a scheme in which impaired mitochondrial function results in diminished production of ATP. The diminished production of ATP would increase the ratio of AMP to ATP, leading to the activation of AMP kinase. This would in turn phosphorylate mTOR, inhibiting its activity. A decrease in mTOR activity would decrease the phosphorylation of p70 S6 kinase and 4E-BP1, which would inhibit mRNA translation. Thus any biological or pathological effect that would compromise mitochondrial function may have profound consequences on the systems adaptive response to ischemia.
Coronary Collateral Growth: Roles of UCPs and AMPK Under Normal Physiological and Pathophysiological Conditions
Under pathological conditions, mitochondrial uncoupling has been implicated in tumor growth (1). Some data have shown that cancer cells favor aerobic glycolysis over mitochondrial oxidative phosphorylation to meet rapid and uncontrolled cellular proliferation, although recent data suggest that mitochondrial biogenesis is vital for tumor growth (61). This metabolic switch is known as the “Warburg effect.” UCP-2 plays a role in this metabolic switch by reducing ROS production secondary to a decreased mitochondrial membrane potential, thus protecting the tumor cells from mitochondrial oxidative stress. Although the molecular mechanisms underlying the “Warburg effect” in cancer cells are not completely understood, several lines of evidence have shown that UCP-2 seems to counteract activities of the tumor suppressor protein p53. First, overexpression of UCP-2 in colon cancer cells diminished ROS levels and interfered with phosphorylation of p53 at Ser15, Ser33, and Ser46 by stress-induced protein kinases (9). Second, cancer cells with overexpression of UCP-2 displayed an increased reliance on glycolytic metabolism (9) and siRNA knockdown of UCP-2 reversed this phenotype (52). Third, activation of p53 favors transcription of target genes involved in oxidative phosphorylation, so inhibition of p53 by UCP-2 would further promote a shift in metabolism from mitochondrial respiration toward glycolysis (65). Fourth, translocation of p53 to initiate the intrinsic apoptotic cascade was blocked by uncoupling effect of FCCP, whereas UCP-2 knockdown promotes this translocation in JB6 cells (59). Overall, these data support the notion that UCP-2 promotes cancer growth not only through inhibition of oxidative stress via reduced mitochondrial ROS production but also by inhibiting key tumor suppressor functions mediated by p53. The relevance of the following data for coronary collateral growth is based on the observation that mitochondrial dysfunction in cardiac tissue from obese rats correlates with impaired capacity for arteriogenesis. Our laboratory previously observed that the growth of coronary collaterals was impaired in ZOF rats compared with lean rats (21, 22). ZOF rats are a model of the human metabolic syndrome because these rats share many of the same afflictions including obesity, insulin resistance, hyperlipidemia, hyperinsulinemia, and hyperphagia (46). As the metabolic syndrome is tightly associated with mitochondrial dysfunction, we studied mitochondrial function in ZOF rats (47). We observed higher lipid peroxide contents and oxidative stress from mitochondrial proteins isolated from the left ventricular tissues in the obese compared with lean rats. In addition, expression of UCP-2 was higher and mitochondrial bioenergetic capacity was reduced in ZOF rats. Treatment with a mitochondrial-targeted antioxidant mitoquinone lowered lipid peroxidation levels and improved antioxidant protein expression significantly in ZOFs. More importantly, mitoquinone normalized mitochondrial respiration profiles and UCP-2 expression to levels observed in the lean rats.
We believe that improvement in respiration profiles and antioxidant defenses leading to partial restoration of coronary collateral growth in the obese rats (mentioned in the preceding paragraph) is directly connected to AMP kinase signaling. More specifically, in ZOF rats, mitochondria demonstrate both impaired bioenergetic capacity and a higher degree of oxidative stress (47). This compromises high-energy phosphate production, which would increase the amount of AMP, whereas that of ATP would decrease. These events would lead to activation of AMP kinase, which would inhibit protein synthesis through decreasing mTOR activity. A decrease in protein synthesis would have many adverse consequences on coronary collateral growth. Resolution of mitochondrial oxidative stress improved respiration profiles and antioxidant defenses (47), which led to partial restoration of coronary collateral growth in the obese rats in response to ischemia. Taken together, these results imply that in obese rats, mitochondrial oxidative stress and the consequential impaired bioenergetic capacity of the mitochondria impairs coronary collateral growth, whereas rectification of the oxidative stress with mitochondrial-directed free radical scavengers restores coronary collateral growth in response to repetitive ischemia (Fig. 1).
Fig. 1.
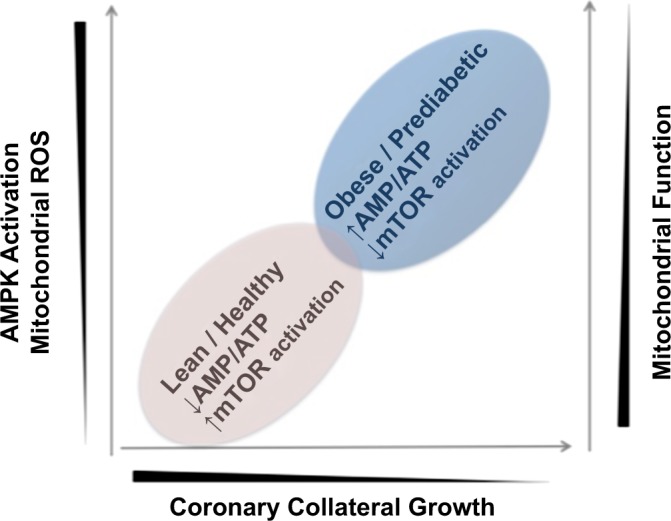
Hypothetical representation of the relationship between mitochondrial function and coronary collateral growth. When mitochondrial function is impaired, bioenergetics capacity is limited and energy production is impaired. This leads to activation of AMP kinase and inhibition of mammalian target of rapamycin (mTOR), as master regulator of protein synthesis. Also as mitochondrial function is impaired, reactive oxygen species (ROS) production by the mitochondria is increased, which can lead to fragmentation of mitochondrial DNA, further impairing mitochondrial function.
We readily admit that the review is centric from the aspect of focusing on the energetic basis of phenotypic switching in vascular growth. We would be remiss not to mention many other aspects of growth that require energy. For example, secretion of matrix, production of matrix metalloproteases, and migration all require energy. Thus a more comprehensive analysis of vascular growth and the requirement of energy is more complicated than presented in Fig. 1, but importantly, these issues only reinforce the importance of energy and bioenergetics in the process–they do not diminish it.
Future Perspectives and Conclusion
Why nature selected limited numbers of genes (a total of 37) to be encoded solely by mitochondrial DNA, which is maternally inherited, but thousands of others by the nuclear genome remains unknown. What are the underlying signaling mechanisms that fine-tune the cross talk between mitochondrial and nuclear-encoded polypeptides, allowing them to perform a highly coordinated task that meets cell type-specific energetic and signaling demands involved in the phenotypic switching from quiescence to proliferation or migration (25)? What can we learn from mitochondrial networks (i.e., tubular forms, beaded fragments, etc.), dynamics (i.e., fusion and fission), and the overall abundance of supramolecular assemblies in relation to the functional consequences for energy production? Also, do we know enough regarding mitochondrial functional complementation? It is no surprise that mutations and/or deficiencies in any of the polypeptides required for maintaining the proper functions and structures of mitochondria are one of the major causes of a variety of cardiovascular, muscular, neurological, and endocrinological diseases and aging (10).
In our opinion, mitochondria play a central role in the growth of coronary collateral vessels. Their role is largely permissive in that they supply energy to enable phenotypic switching of cells, a necessity for vascular growth. However, in the situation of mitochondrial oxidative stress, oxidative modification of mtDNA impairs transcription of the mitochondrial genome, ultimately inducing further ROS production. This not only results in limited energy production that impairs the energetic requirements for phenotypic switching of cells but also leads to a vicious cycle where mitochondrial oxidative stress begets further mitochondrial oxidative stress. We opine that mitochondrial integrity and proper mitochondrial function have a central position in the growth of coronary collaterals.
GRANTS
This work was supported by National Institute of Health Grants HL-32788, R01-83366, and RC1-HL-100828 (to W. M. Chilian) and by an American Heart Association Postdoctoral Fellowship 09POST2290021 (to Y. F. Pung).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Y.F.P. drafted manuscript; Y.F.P., W.J.S., J.P.H., L.Y., V.A.O., S.L., L.S.D., and W.M.C. edited and revised manuscript; Y.F.P., W.J.S., J.P.H., L.Y., V.A.O., S.L., L.S.D., and W.M.C. approved final version of manuscript.
REFERENCES
- 1.Baffy G, Derdak Z, Robson SC. Mitochondrial recoupling: a novel therapeutic strategy for cancer? Br J Cancer 105: 469–474, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J 134: 707–716, 1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med 37: 755–767, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Brand MD, Buckingham JA, Esteves TC, Green K, Lambert AJ, Miwa S, Murphy MP, Pakay JL, Talbot DA, Echtay KS. Mitochondrial superoxide and aging: uncoupling-protein activity and superoxide production. Biochem Soc Symp (71): 203–213, 2004 [DOI] [PubMed] [Google Scholar]
- 5.Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab 2: 85–93, 2005 [DOI] [PubMed] [Google Scholar]
- 6.Carling D, Woods A, Thornton C, Cheung PC, Smith FC, Ponticos M, Stein SC. Molecular characterization of the AMP-activated protein kinase and its role in cellular metabolism. Biochem Soc Trans 25: 1224–1228, 1997 [DOI] [PubMed] [Google Scholar]
- 7.Chandel NS, Schumacker PT. Cellular oxygen sensing by mitochondria: old questions, new insight. J Appl Physiol 88: 1880–1889, 2000 [DOI] [PubMed] [Google Scholar]
- 8.Chilian WM, Mass HJ, Williams SE, Layne SM, Smith EE, Scheel KW. Microvascular occlusions promote coronary collateral growth. Am J Physiol Heart Circ Physiol 258: H1103–H1111, 1990 [DOI] [PubMed] [Google Scholar]
- 9.Derdak Z, Mark NM, Beldi G, Robson SC, Wands JR, Baffy G. The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res 68: 2813–2819, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DiMauro S. Mitochondrial diseases. Biochim Biophys Acta 1658: 80–88, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Dobson AW, Xu Y, Kelley MR, LeDoux SP, Wilson GL. Enhanced mitochondrial DNA repair and cellular survival after oxidative stress by targeting the human 8-oxoguanine glycosylase repair enzyme to mitochondria. J Biol Chem 275: 37518–37523, 2000 [DOI] [PubMed] [Google Scholar]
- 12.Druzhyna NM, Musiyenko SI, Wilson GL, LeDoux SP. Cytokines induce nitric oxide-mediated mtDNA damage and apoptosis in oligodendrocytes. Protective role of targeting 8-oxoguanine glycosylase to mitochondria. J Biol Chem 280: 21673–21679, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Esteves TC, Brand MD. The reactions catalysed by the mitochondrial uncoupling proteins UCP2 and UCP3. Biochim Biophys Acta 1709: 35–44, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature 408: 239–247, 2000 [DOI] [PubMed] [Google Scholar]
- 15.Green DR. Apoptotic pathways: the roads to ruin. Cell 94: 695–698, 1998 [DOI] [PubMed] [Google Scholar]
- 16.Green DR, Reed JC. Mitochondria and apoptosis. Science 281: 1309–1312, 1998 [DOI] [PubMed] [Google Scholar]
- 17.Hajnoczky G, Csordas G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 40: 553–560, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays 23: 1112–1119, 2001 [DOI] [PubMed] [Google Scholar]
- 19.Hardie DG, Sakamoto K. AMPK: a key sensor of fuel and energy status in skeletal muscle. Physiology (Bethesda) 21: 48–60, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Hardie DG, Salt IP, Hawley SA, Davies SP. AMP-activated protein kinase: an ultrasensitive system for monitoring cellular energy charge. Biochem J 338: 717–722, 1999 [PMC free article] [PubMed] [Google Scholar]
- 21.Hattan N, Chilian WM, Park F, Rocic P. Restoration of coronary collateral growth in the Zucker obese rat: impact of VEGF and ecSOD. Basic Res Cardiol 102: 217–223, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Hattan N, Warltier D, Gu W, Kolz C, Chilian WM, Weihrauch D. Autologous vascular smooth muscle cell-based myocardial gene therapy to induce coronary collateral growth. Am J Physiol Heart Circ Physiol 287: H488–H493, 2004 [DOI] [PubMed] [Google Scholar]
- 23.Heil M, Schaper W. Pathophysiology of collateral development. Coron Artery Dis 15: 373–378, 2004 [DOI] [PubMed] [Google Scholar]
- 24.Helfant RH, Vokonas PS, Gorlin R. Functional importance of the human coronary collateral circulation. N Engl J Med 284: 1277–1281, 1971 [DOI] [PubMed] [Google Scholar]
- 25.Hock MB, Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol 71: 177–203, 2009 [DOI] [PubMed] [Google Scholar]
- 26.Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci 118: 5411–5419, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang H, Manton KG. The role of oxidative damage in mitochondria during aging: a review. Front Biosci 9: 1100–1117, 2004 [DOI] [PubMed] [Google Scholar]
- 28.Jeong HW, Hsu KC, Lee JW, Ham M, Huh JY, Shin HJ, Kim WS, Kim JB. Berberine suppresses proinflammatory responses through AMPK activation in macrophages. Am J Physiol Endocrinol Metab 296: E955–E964, 2009 [DOI] [PubMed] [Google Scholar]
- 29.Kemp BE, Mitchelhill KI, Stapleton D, Michell BJ, Chen ZP, Witters LA. Dealing with energy demand: the AMP-activated protein kinase. Trends Biochem Sci 24: 22–25, 1999 [DOI] [PubMed] [Google Scholar]
- 30.Kemp BE, Stapleton D, Campbell DJ, Chen ZP, Murthy S, Walter M, Gupta A, Adams JJ, Katsis F, van Denderen B, Jennings IG, Iseli T, Michell BJ, Witters LA. AMP-activated protein kinase, super metabolic regulator. Biochem Soc Trans 31: 162–168, 2003 [DOI] [PubMed] [Google Scholar]
- 31.Kukidome D, Nishikawa T, Sonoda K, Imoto K, Fujisawa K, Yano M, Motoshima H, Taguchi T, Matsumura T, Araki E. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes 55: 120–127, 2006 [PubMed] [Google Scholar]
- 32.Kuznetsov AV, Hermann M, Saks V, Hengster P, Margreiter R. The cell-type specificity of mitochondrial dynamics. Int J Biochem Cell Biol 41: 1928–1939, 2009 [DOI] [PubMed] [Google Scholar]
- 33.Li X, Han Y, Pang W, Li C, Xie X, Shyy JY, Zhu Y. AMP-activated protein kinase promotes the differentiation of endothelial progenitor cells. Arterioscler Thromb Vasc Biol 28: 1789–1795, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest 116: 1776–1783, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsunaga T, Warltier DC, Moniz M, Tessmmer J, Weihrauch D, Chilian WM. Role of nitric oxide and vascular endothelial growth factor in coronary collateral growth. Circulation 102: 3098–3103, 2000 [DOI] [PubMed] [Google Scholar]
- 36.McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol 16: R551–R560, 2006 [DOI] [PubMed] [Google Scholar]
- 37.Meininger CJ, Schelling ME, Granger HJ. Adenosine and hypoxia stimulate proliferation and migration of endothelial cells. Am J Physiol Heart Circ Physiol 255: H554–H562, 1988 [DOI] [PubMed] [Google Scholar]
- 38.Mitchelhill KI, Stapleton D, Gao G, House C, Michell B, Katsis F, Witters LA, Kemp BE. Mammalian AMP-activated protein kinase shares structural and functional homology with the catalytic domain of yeast Snf1 protein kinase. J Biol Chem 269: 2361–2364, 1994 [PubMed] [Google Scholar]
- 39.Mozo J, Emre Y, Bouillaud F, Ricquier D, Criscuolo F. Thermoregulation: what role for UCPs in mammals and birds? Biosci Rep 25: 227–249, 2005 [DOI] [PubMed] [Google Scholar]
- 40.Nagata D, Mogi M, Walsh K. AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J Biol Chem 278: 31000–31006, 2003 [DOI] [PubMed] [Google Scholar]
- 41.Negre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, Salvayre R, Penicaud L, Casteilla L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J 11: 809–815, 1997 [PubMed] [Google Scholar]
- 42.Oh-hama T. Evolutionary consideration on 5-aminolevulinate synthase in nature. Orig Life Evol Biosph 27: 405–412, 1997 [DOI] [PubMed] [Google Scholar]
- 43.Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J Biol Chem 276: 38388–38393, 2001 [DOI] [PubMed] [Google Scholar]
- 44.Packer L, Fuehr K. Low oxygen concentration extends the lifespan of cultured human diploid cells. Nature 267: 423–425, 1977 [DOI] [PubMed] [Google Scholar]
- 45.Piek JJ, van Liebergen RA, Koch KT, Peters RJ, David GK. Clinical, angiographic and hemodynamic predictors of recruitable collateral flow assessed during balloon angioplasty coronary occlusion. J Am Coll Cardiol 29: 275–282, 1997 [DOI] [PubMed] [Google Scholar]
- 46.Pung YF, Chilian WM. Corruption of coronary collateral growth in metabolic syndrome: Role of oxidative stress. World J Cardiol 2: 421–427, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pung YF, Rocic P, Murphy MP, Smith RA, Hafemeister J, Ohanyan V, Guarini G, Yin L, Chilian WM. Resolution of mitochondrial oxidative stress rescues coronary collateral growth in zucker obese fatty rats. Arterioscler Thromb Vasc Biol 32: 325–334, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pung YF, Sam WJ, Stevanov K, Enrick M, Chen CL, Kolz C, Thakker P, Hardwick JP, Chen YR, Dyck JR, Yin L, Chilian WM. Mitochondrial oxidative stress corrupts coronary collateral growth by activating AMPK-α signaling. Arterioscler Thromb Vasc Biol 33: 1911–1919, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rentrop KP, Feit F, Sherman W, Thornton JC. Serial angiographic assessment of coronary artery obstruction and collateral flow in acute myocardial infarction. Report from the second Mount Sinai-New York University Reperfusion Trial. Circulation 80: 1166–1175, 1989 [DOI] [PubMed] [Google Scholar]
- 50.Ricci C, Pastukh V, Leonard J, Turrens J, Wilson G, Schaffer D, Schaffer SW. Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis. Am J Physiol Cell Physiol 294: C413–C422, 2008 [DOI] [PubMed] [Google Scholar]
- 51.Rossier MF. T channels and steroid biosynthesis: in search of a link with mitochondria. Cell Calcium 40: 155–164, 2006 [DOI] [PubMed] [Google Scholar]
- 52.Samudio I, Fiegl M, McQueen T, Clise-Dwyer K, Andreeff M. The warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation. Cancer Res 68: 5198–5205, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saraste M. Oxidative phosphorylation at the fin de siecle. Science 283: 1488–1493, 1999 [DOI] [PubMed] [Google Scholar]
- 54.Schaper W. The Collateral Circulation of the Heart. New York: Elsevier, 1971 [Google Scholar]
- 55.Schaper W, De Brabander M, Lewi P. DNA synthesis and mitoses in coronary collateral vessels of the dog. Circ Res 28: 671–679, 1971 [DOI] [PubMed] [Google Scholar]
- 56.Schaper W, Ito WD. Molecular mechanisms of coronary collateral vessel growth. Circ Res 79: 911–919, 1996 [DOI] [PubMed] [Google Scholar]
- 57.Skulachev VP. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q Rev Biophys 29: 169–202, 1996 [DOI] [PubMed] [Google Scholar]
- 58.Skulachev VP. Uncoupling: new approaches to an old problem of bioenergetics. Biochim Biophys Acta 1363: 100–124, 1998 [DOI] [PubMed] [Google Scholar]
- 59.Su WP, Lo YC, Yan JJ, Liao IC, Tsai PJ, Wang HC, Yeh HH, Lin CC, Chen HH, Lai WW, Su WC. Mitochondrial uncoupling protein 2 regulates the effects of paclitaxel on Stat3 activation and cellular survival in lung cancer cells. Carcinogenesis 33: 2065–2075, 2012 [DOI] [PubMed] [Google Scholar]
- 60.Toyota E, Warltier DC, Brock T, Ritman E, Kolz C, O'Malley P, Rocic P, Focardi M, Chilian WM. Vascular endothelial growth factor is required for coronary collateral growth in the rat. Circulation 112: 2108–2113, 2005 [DOI] [PubMed] [Google Scholar]
- 61.Tsutsui H. Oxidative stress in heart failure: the role of mitochondria. Intern Med 40: 1177–1182, 2001 [DOI] [PubMed] [Google Scholar]
- 62.Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and mitochondrial DNA damage in heart failure. Circ J 72: A31–A37, 2008 [DOI] [PubMed] [Google Scholar]
- 63.Vidal-Puig AJ, Grujic D, Zhang CY, Hagen T, Boss O, Ido Y, Szczepanik A, Wade J, Mootha V, Cortright R, Muoio DM, Lowell BB. Energy metabolism in uncoupling protein 3 gene knockout mice. J Biol Chem 275: 16258–16266, 2000 [DOI] [PubMed] [Google Scholar]
- 64.von Zglinicki T, Saretzki G, Docke W, Lotze C. Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: a model for senescence? Exp Cell Res 220: 186–193, 1995 [DOI] [PubMed] [Google Scholar]
- 65.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer 9: 691–700, 2009 [DOI] [PubMed] [Google Scholar]
- 66.Winder WW, Taylor EB, Thomson DM. Role of AMP-activated protein kinase in the molecular adaptation to endurance exercise. Med Sci Sports Exerc 38: 1945–1949, 2006 [DOI] [PubMed] [Google Scholar]
- 67.Woods A, Cheung PC, Smith FC, Davison MD, Scott J, Beri RK, Carling D. Characterization of AMP-activated protein kinase beta and gamma subunits. Assembly of the heterotrimeric complex in vitro. J Biol Chem 271: 10282–10290, 1996 [DOI] [PubMed] [Google Scholar]
- 68.Zang M, Xu S, Maitland-Toolan KA, Zuccollo A, Hou X, Jiang B, Wierzbicki M, Verbeuren TJ, Cohen RA. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes 55: 2180–2191, 2006 [DOI] [PubMed] [Google Scholar]