

Abstract
Background
Evidence in yeast indicates that gene expression is correlated with recombination activity and double-strand break (DSB) formation in some hotspots. Studies of nucleosome occupancy in yeast and mice also suggest that open chromatin influences the formation of DSBs. In Drosophila melanogaster, high-resolution recombination maps show an excess of DSBs within annotated transcripts relative to intergenic sequences. The impact of active transcription on recombination landscapes, however, remains unexplored in a multicellular organism. We then investigated the transcription profile during early meiosis in D. melanogaster females to obtain a glimpse at the relevant transcriptional dynamics during DSB formation, and test the specific hypothesis that DSBs preferentially target transcriptionally active genomic regions.
Results
Our study of transcript profiles of early- and late-meiosis using mRNA-seq revealed, 1) significant differences in gene expression, 2) new genes and exons, 3) parent-of-origin effects on transcription in early-meiosis stages, and 4) a nonrandom genomic distribution of transcribed genes. Importantly, genomic regions that are more actively transcribed during early meiosis show higher rates of recombination, and we ruled out DSB preference for genic regions that are not transcribed.
Conclusions
Our results provide evidence in a multicellular organism that transcription during the initial phases of meiosis increases the likelihood of DSB and give insight into the molecular determinants of recombination rate variation across the D. melanogaster genome. We propose that a model where variation in gene expression plays a role altering the recombination landscape across the genome could provide a molecular, heritable and plastic mechanism to observed patterns of recombination variation, from the high level of intra-specific variation to the known influence of environmental factors and stress conditions.
Keywords: Gene expression, RNA-seq, Maternal effect, Oogenesis, Germarium, Crossing-over, Recombination
Background
High-resolution transcription profiles offer insight into a wide array of biological questions including the identification of genes involved in specific molecular processes, the understanding of cellular fate and organ differentiation, the importance of genic and epigenetic factors, and the complex response to environmental conditions [1]. With the rise of sequencing technologies such as RNA-seq and supporting methodologies, researchers are now able to obtain gene expression profiles (sensu levels of transcripts), potentially identifying rare or novel transcript forms that are only present in specific cells and/or at very precise developmental times [2]. One such cell population of interest lies within the anterior portions of the Drosophila ovary, where mitotic precursor cells begin their development into functional eggs and meiotic recombination occurs.
The Drosophila ovary has served as a model for meiosis [3], embryo patterning [4], and stem cell differentiation [5,6]. Drosophila females have two ovaries comprised of 10 to 20 tube-like structures, called ovarioles, clustered together with a spatiotemporal organization of progressively developing oocytes [7]. Oogenesis in Drosophila starts within the anterior compartment of the ovariole, the germarium, where mitotic stem cells produce cystoblasts that undergo further cell division generating a large 16-cell cyst with a single cystocyte becoming the oocyte. Before exiting the germarium as a stage-1 egg chamber, the primary oocyte will have entered pachytene and undergo meiotic recombination. These anteriormost portions of the Drosophila ovariole represent a highly active community of cells, regulated with remarkable fidelity, and yet, constitute only a small fraction of the entire ovary [8]. Previous whole-genome transcriptome analyses of whole ovaries therefore offer only an amalgamated sight of its developmental and cellular complexity, limiting our understanding of the relevant gene expression activity of the germarium and early meiosis [9,10].
The process of meiotic recombination in D. melanogaster females occurs with the initiation of double-strand breaks (DSBs). At a very broad scale, crossover in Drosophila is distributed in bell-shaped fashion along chromosomes, with a maximal rate in the center of a chromosomal arm that tapers off near centromeric and telomeric regions [11]. This is also the case in many other (e.g., mice, humans, Arabidopsis, etc.) but not all (e.g., Caenorhabditis elegans and C. briggsae) eukaryotes. At finer scales, recombination maps have revealed substantial variation across chromosomes in all species analyzed, including Drosophila[12-20]. In D. melanogaster, high-resolution mapping of more than 100,000 recombination events at a scale approaching gene-level resolution showed not only extreme heterogeneity in recombination rates across chromosomes but also that these landscapes of recombination vary significantly among individuals of the same species [20]. Even within chromosomal regions traditionally assumed to have non-reduced recombination rates, crossover rates vary up to 80-fold when crossing two D. melanogaster strains, and 20-fold after combining genetic maps obtained from eight crosses of different strains [20]. Beyond the differences across genomes, between species and within species, there is an important additional layer of complexity: recombination rates are plastic and influenced by factors such as temperature, food, or maternal age [21-25].
The molecular determinants leading to DSB localization across the Drosophila genome remain obscure but a number of patterns are beginning to emerge (see [20] for details). First, unlike human and mice recombination hotspots that are strongly influenced by the presence of the PRDM9-binding DNA motif [26,27], no PRDM9 motif is detected in Drosophila[20,28-30]. Second, analyses in Drosophila reveal many different DNA motifs significantly enriched in sequences surrounding recombination events, suggesting a fundamental qualitative difference between human/mouse and Drosophila DSB localization [20,29]. Third, recombination events tend to occur within annotated transcript regions thus suggesting a possible association between transcription, chromatin accessibility, and DSBs that are repaired as recombination events [20]. This latter observation is in agreement with evidence in the yeast S. cerevisiae where some, but not all, hotspots of recombination increase activity with transcription [31]. Mapping of chromatin accessibility and nucleosome occupancy in yeast and mice [32-34] also suggests, albeit more indirectly, that the formation of DSBs could be influenced by transcription based on the known effect of transcription on chromatin remodeling and histone modifications. The impact of active transcription on meiotic DSB localization and recombination landscapes in a multicellular organism remains, however, unexplored.
Here, we employ RNA-seq to obtain and analyze the whole transcriptome of early meiotic D. melanogaster cells. We isolated germaria-stage 3 cells to substantially enrich the fraction of sample that is actively experiencing early meiosis and DSB formation and obtain a first glimpse of the potential influence of transcription on recombination localization across the Drosophila genome. Our analyses uncover genes with germarium-specific expression patterns and novel transcripts. The study of offspring from reciprocal crosses also reveals distinct parent-of-origin effects that create differences in gene expression among genetically identical individuals.
Finally, we identify a positive relationship across the genome between transcription in early meiotic cells and recombination rates. Importantly, recombination events are found to target actively transcribed genes relative to genes with no detectable transcription thus allowing us to rule out that the observed association is due to DSB preference for static gene properties at the level of DNA sequence (e.g., G + C content). These results provide insight into the molecular determinants of recombination rate variation across the D. melanogaster genome and a clear path for future studies to assess the molecular causes of recombination variation among individuals and its plastic nature.
Results and discussion
General patterns of the Drosophila early meiotic transcriptome
We isolated mRNAs from meiotic portions of the Drosophila ovary, dissecting the germarium and stages 1–3, and compared them to later, more developed regions of the ovary, hereafter referred to as ‘Early’ and ‘Late’, respectively (see Methods). We performed ultra-deep mRNA sequencing (mRNA-seq) that obtained over 467 million (M) of 120 bp-long reads. Approximately 80% of these reads mapped correctly to the D. melanogaster genome reference sequence, with a total average coverage greater than 400× when mapped to annotated transcripts (Table 1). Each of our eight independent samples sequenced (see Methods) generated between 34.5 and 60.3 M mapped reads, with a median mapped read count of 49.8 M. There was no difference between total mapped read counts between Early and Late tissues (P = 0.59).
Table 1.
mRNA-seq statistics for each sample
| Strain | Condition* | Gross reads | Mapped reads | % mapped | Avg. depth |
|---|---|---|---|---|---|
| 208 |
Early |
44,938,695 |
34,472,998 |
76.71 |
84.3 |
| Late |
52,572,707 |
44,868,154 |
85.34 |
119.2 |
|
| 375 |
Early |
67,255,860 |
48,750,637 |
72.49 |
119.0 |
| Late |
61,344,832 |
51,546,801 |
84.03 |
121.5 |
|
| 375F×208M |
Early |
61,305,389 |
50,863,571 |
82.97 |
107.3 |
| Late |
71,871,368 |
60,296,814 |
83.9 |
122.9 |
|
| 375M×208F |
Early |
57,532,606 |
45,646,995 |
79.34 |
86.9 |
| Late |
50,982,155 |
37,072,046 |
72.72 |
93.5 |
|
| Combined | Early |
231,032,550 |
179,734,201 |
77.8 |
457.2 |
| Late | 236,771,062 | 193,783,815 | 81.84 | 397.4 |
*Early and Late indicate Drosophila Early- and Late-ovarian transcriptome, respectively.
Comparisons of Early versus Late transcript profiles show high similarity, with a strong correlation coefficient (Spearman’s R = 0.952; P < 0.001; see Figure 1). There are 7,914 genes expressed in Early regions compared to 7,557 genes in Late regions. These results suggest that roughly 50% of all genes are actively transcribed, a value similar to the typical percentage in other Drosophila tissues, based on mRNA seq [35] or array-based comparisons of germarium and testes [46% of all genes expressed; [36]]. The detection of 5% more genes being transcribed (i.e., above a FPKM threshold of 1) in Early relative to Late meiosis (P = 0.015) is accompanied by a reduced average level of transcription for active genes in early meiosis by more than 13% (average FPKM of 89.1 and 103.1 for Early and Late stages, respectively; Wilcoxon Matched Pairs Test, Z = 44.5, P < 1×10-12). Similar differences are observed when defining active genes based on FPKM greater than 0.1 (Z = 41.7, P < 1×10-12).
Figure 1.

Comparison of Log10 FPKM values for Drosophila Early- and Late- ovarian transcriptome. Orange points indicate significantly differentially expressed genes based on FDR-corrected significance level of 5% (q < 0.05). Spearman’s R = 0.952 (P < 1x10-12).
The Drosophila X chromosome is enriched in genes preferentially or uniquely expressed in females (i.e., female-biased genes) and deficient in male-biased genes [37-42]. Focusing only on the male germline, however, a recent study has shown that differences between X and autosomes are not caused by different gene content but to the lack of sex chromosome dosage compensation in Drosophila testes thus reducing transcript levels of X-linked genes [43]. In our deep-sequencing study, we see that the early ovarian transcriptome shows the expected “female” bias with actively expressed genes unequally distributed among chromosomes: 60.4% of genes on the X chromosome are transcribed compared with 55.3% in autosomes (χ2 = 20.9, P = 4.8×10-6; see Figure 2). A more extreme difference is observed when defining active genes based on FPKM > 0.1 (79.5 vs. 70.0% for X and autosomes, χ2 = 89.2, P < 1×10-12). Notably, this overrepresentation of actively transcribed genes on the X chromosome is less apparent in Late meiotic stages (e.g., 56.5% on the X compared to 53.4% in autosomes, χ2 = 7.23, P = 0.007).
Figure 2.

Transcriptional differences between autosomes and the X chromosome. (A) Mean FPKM values for autosomes and the X chromosome. Error bars represent +/− 1 standard error. (B) Percentage of total transcribed genes across each chromosome. Error bars represent 90% confidence intervals. Green: Early-ovarian transcriptome, Orange: Late-ovarian transcriptome.
Finally, we observe that expressed genes are not distributed randomly across chromosomes, but are instead physically clustered (Wald–Wolfowitz or Run’s test, P < 1×10-8 for all levels of expression analyzed). When defining actively expressed genes as FPKM > 1, clusters contain an average of 3.5 consecutive genes (5.8 genes when FPKM > 0.1). These results are in agreement with data from other Drosophila tissues and conditions, with small clusters of functionally related, highly co-expressed genes [44-46].
Differentially expressed genes in early meiotic tissues
We observed 1,191 genes with differences in FPKM between Early and Late meiotic tissues at nominal P < 0.05, with 376 genes showing a significant difference after correcting for multiple testing (q < 0.05; see Methods). The degree of differential expression ranges from +241-fold to −2060-fold in the early relative to late tissues, with a median difference of 1.66-fold among genes with significant differences. We observe a bias towards overall down-regulation of genes in Early versus Late tissues (approximately five times more genes are significantly down-regulated than up-regulated) that cannot be explained by read bias in the respective samples.
The top ten over- and under-expressed genes in the Early sample are listed in Table 2. The use of DAVID (see Methods) to classify genes into GO categories reveals that the terms ‘proteolysis’ and ‘peptidase’ are significantly enriched within our top-ten up-regulated genes in our Early sample (FDR-corrected modified Fisher exact P = 0.0001 and 0.001, respectively). Furthermore, all of the known genes (sensu annotated in the Drosophila Genome, r.5.47) within this group are serine-type endopeptidases involved in proteolysis. Why there is such a bias towards genes involved in proteolysis is difficult to explain, but a similar pattern has been noted in the apex of the testis in Drosophila[36]. We suggest that the overrepresentation of serine endopeptidases may be due to the required breakdown of many meiotic proteins following their utilization in meiosis in order to prevent erroneous aggregation of many self-assembling protein complexes that may interact with DNA. The analysis of the 312 significantly down-regulated genes suggests enrichment in the GO terms phosophoproteins, RNA splicing, nucleotide binding, phosophate metabolic processes, and ribonucleotide binding. Analysis of the top ten most extreme down-regulated genes does not indicate overrepresentation of any GO term after correcting for multiple tests.
Table 2.
Top ten differentially expressed genes by fold-change
| Transcript | Biological process | Early FPKM | Late FPKM | Fold change | q value | |
|---|---|---|---|---|---|---|
| Over-regulated in Early
*
|
CG17475-RA |
Proteolysis |
13.95 |
0.06 |
240.9 |
1.02×10-8 |
| |
CG31267-RA |
Proteolysis |
8.22 |
0.06 |
140.7 |
1.81×10-6 |
| |
CG32833-RA |
Proteolysis |
2.91 |
0.04 |
65.3 |
9.43×10-3 |
| |
CG42704-RA |
Unknown |
62.64 |
0.98 |
64.2 |
2.08×10-8 |
| |
CG18417-RA |
Proteolysis |
1.16 |
0.02 |
48.7 |
7.64×10-3 |
| |
CG43074-RA |
Unknown |
11.81 |
0.24 |
48.7 |
3.06×10-5 |
| |
CG47205-RA |
Unknown |
7.12 |
0.15 |
46.3 |
1.40×10-3 |
| |
CG31266-RB |
Proteolysis |
3.44 |
0.09 |
40.1 |
1.74×10-3 |
| |
CG31681-RA |
Proteolysis |
2.74 |
0.07 |
39.1 |
4.71×10-3 |
| |
CG15254-RA |
Proteolysis |
2.42 |
0.06 |
37.9 |
1.40×10-3 |
| Under-regulated in Early |
Vml-RA |
d/v axis specification |
0.11 |
221.40 |
−2,059.9 |
<1×10-12 |
| |
λTry-RA |
Proteolysis |
0.07 |
3.27 |
−46.0 |
4.39×10-3 |
| |
CG8997-RA |
Unknown |
1.16 |
47.99 |
−41.4 |
4.91×10-10 |
| |
CG7916-RA |
Unknown |
0.78 |
31.21 |
−39.8 |
1.48×10-9 |
| |
CG12057-RA |
Unknown |
1.74 |
68.09 |
−39.1 |
6.71×10-7 |
| |
CG7953-RA |
Unknown |
0.68 |
26.10 |
−38.2 |
2.34×10-9 |
| |
CG33306-RA |
Unknown |
0.10 |
3.71 |
−37.7 |
1.62×10-4 |
| |
chrUextra:28564682-777 |
Probable rRNA |
20.61 |
709.30 |
−34.4 |
2.00×10-2 |
| |
CG11911-RA |
Proteolysis |
0.75 |
21.88 |
−29.0 |
7.91×10-8 |
| CG18585-RA | Proteolysis | 0.07 | 1.72 | −24.9 | 5.45×10-3 |
*Early indicates Drosophila Early-ovarian transcriptome.
These results indicate an enrichment of serine proteases in early versus late ovarian development and a concurrent down-regulation of the majority of genes in Early tissues. Interestingly, many of the top ten up-regulated genes were shown to be down-regulated in array experiments based comparisons between the whole ovary and whole fly [35]. This result emphasizes that whole-ovary experiments might have lacked sufficient power to detect important genes involved in subregions of the developing ovary.
New genes and isoforms
We applied the Cufflinks algorithm to our combined data sets and identified up to 6,004 transcript forms (genes, exons, or noncoding RNAs) that were absent from the D. melanogaster genome annotation (r. 5.47, September 2012). When we conservatively restricted the list to only those items that were detected in two or more samples and further removed items with FPKM < 1, the set still contained 220 entries. Notably, 47 high-confidence items were identified with lengths greater than 300 bp, minimally repetitive sequences and reads that reliably mapped to predicted splice junctions. Additionally, a visual inspection shows that a minimum of 13 of these new transcripts are independent of other annotated gene entries and have clear exon-intron structures and are thus strong candidates for new genes, while the rest are either novel splicing forms or putative ncRNAs.
To validate some of these new transcripts, we designed transcript-specific primers, extracted total RNA from ovaries and were able to reliably produce RT-PCR products from seven of ten haphazardly selected novel candidates. We thus, conservatively, estimate a contribution of ~30-35 novel items to the existing D. melanogaster genome annotation. Notably, a number of putative novel transcribed sequences mapped uniquely to the so-called chromosome U that consists of unordered, unoriented scaffolds not present in the D. melanogaster genome (euchomatic or heterochromatic) sequences. These results add to the notion that the actual number of unnotated genes and isoforms is still high in this model organism. Ultra-deep sequencing studies focusing on specific cell populations and variable conditions are therefore needed to fill this annotation gap that can have important consequences in genomic and evolutionary analyses.
Parent-of-origin effects in the early meiotic tissue
Differences in gene expression between genetically identical offspring from reciprocal crosses indicate that maternal and/or paternal effects alter expression. The molecular causes of these parent-of-origin effects include genomic imprinting (through epigenetic modification during gametogenesis), cytoplasmic effects of the egg and sperm, or mitochondrial contributions to nuclear transcription. To investigate parent-of-origin effects in the Early meiotic tissue in females, we studied two homozygous D. melanogaster parental strains (strains RAL-208 and RAL-375 from Raleigh, NC [47]) and the heterozygous offspring from reciprocal crosses. We identify genes with a parent-of-origin transcription pattern as those genes that show differential expression between offspring of reciprocal crosses and focused on the subset of these genes that change in transcript levels between offspring of reciprocal crosses in the same direction as maternal strains differ between them (i.e., parent-of origin effects with maternal-like transcript levels).
The comparison of offspring of reciprocal crosses reveals that there are more genes with parent-of origin effects with maternal-like transcript levels in the Early- than in the Late-ovarian development tissues (1041 and 554 genes, respectively; P < 1×10-12).Interestingly, there is an excess of genes with parent-of origin effects with maternal-like transcript levels that resemble transcription in the RAL-208 maternal strain than genes with transcription pattern resembling the RAL-375 maternal strain (P < 1×10-12). We expanded this study by investigating allele-specific transcript ratios of heterozygous offspring and observed an excess of the RAL-208 allele in both reciprocal crosses (P < 1×10-12) that is higher when the maternal strain is RAL-208 (Wilcoxon Matched Pairs Test, P = 0.004). These results not only reveal the presence of variable parent-of-origin effects acting on transcript abundance but also an overrepresentation of dominant effects in RAL-208 relative to RAL-375.
We also identified an enrichment of a common set of GO terms associated with genes showing parent-of origin effects with maternal-like transcript levels, many of which are involved in development and differentiation (Additional file 1: Table S1). When the Early and Late datasets are combined, we recover similar GO term hits as were obtained for Early tissues alone (Additional file 1: Table S2). We thus interpret this pattern as a clear signal of parent-of-origin effects in the transcriptome of the Drosophila ovary, with maternal-like gene expression that is mostly relevant to Early-ovary development.
Transcription is associated with increased recombination rates
Ultra-high resolution mapping of recombination events in Drosophila revealed that meiotic DSBs (detected as combined non-crossover and crossover events) occur preferentially in annotated transcriptional units [20]. We thus hypothesized that gene transcription increases the probability of DSB formation in Drosophila and influence the recombination landscapes across chromosomes. Alternatively, the preference of DSB for genic units could be associated with other characteristics such nucleotide composition, reduced average nucleotide diversity relative to intergenic regions, presence of specific DNA motifs in promoter and intronic regions, etc. Although the topography of recombination landscapes in S. cerevisiae and D. melanogaster are dramatically different in terms of hotspot activity and localization, evidence based on some hotspots in yeast suggests promoter and/or transcriptional activity affects recombination activity [31]. The effects of transcription on DSB formation could be either direct via reduced nucleosome occupancy and increased chromatin accessibility, or more indirect as consequence of histone modifications.
To evaluate our hypothesis, we now focused on the expression profile during early D. melanogaster meiosis and compared the transcriptional landscape with recombination rate variation across the D. melanogaster genome. Note that we anticipate the presence of a fraction of cells other than those where DSB formation occurs in our Early-meiosis sample. We argue, however, that our sample is enriched in recombining cells and therefore, even if we may not recover the precise transcriptional profile at the time/cells where DSBs occur, genomic regions with no evidence of transcription will be particularly informative when defining coldspots of transcription during DSB formation.
To this end, we used estimates of recombination rates across the D. melanogaster genome that were experimentally obtained after genotyping 139 million informative SNPs and mapping more than 100,000 recombination events at a scale approaching gene-level resolution (see [20] for details). We then compared measures of transcription in Early meiosis with these high-resolution recombination landscapes. The analysis of adjacent 100-kb regions reveals a positive association between recombination rates and both the number of genes transcribed per interval (Spearman’s R = 0.175, P = 3.1×10-10) and the total length of the transcribed regions per interval (R = 0.122, P = 1.2×10-5; see Figure 3). To capture the possible effect of transcription levels we also obtained a measure of overall transcriptional activity within a genomic interval (OTA), defined as the Log10-transformed sum of the product FPKM × transcript length for each gene within a given genomic interval (100 kb in our study). In our study, OTA is positively associated with recombination rates across the genome (R = 0.137, P = 8.4×10-7). Multiple regression analysis shows, however, that the number (P = 0.006) and total length of transcribed sequences (P = 0.035) in a region are more relevant than OTA (P > 0.4) predicting recombination rates at the 100-kb scale.
Figure 3.
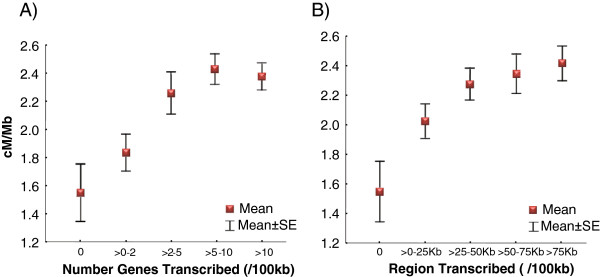
Relationship between transcription and recombination rates. (A) Mean recombination rate in cM/Mb (centimorgans per megabase) for genomic regions grouped according to the number of genes transcribed (FPKM > 0.1) within each 100-kb region. Spearman’s R = 0.168 (P = 1.5x10-9) based on non-overlaping 100-kb regions. (B) Mean recombination rate in cM/Mb for regions grouped according to the total region transcribed within each 100-kb region. R = 0.123 (P = 1.1x10-5). Error bars represent +/− 1 standard error.
Notably, the relationship between measures of transcription and recombination (Figure 3) appears to be highly contingent upon regions that lack transcription relative to regions with transcription. There is a significant difference in recombination rates between regions with no transcription and regions with one or more transcribed genes (Mann–Whitney test, P < 1×10-6). This result is consistent with the idea that our study preferentially captures the consequences of coldspots for transcription during DSB formation in our Early-meiosis sample.
The high-resolution genetic maps of D. melanogaster (see above) also allowed the localization of more than 5,000 DSBs delimited by 500 bp or less [20]. Here, we take advantage of these highly localized meiotic DSB events to investigate their distribution at the scale of single genes and intergenic regions. We observe that intergenic sequences have fewer DSBs than expected but, importantly, we detect a difference between genes transcribed and genes not transcribed (Figure 4). There is a significant excess of DSB within transcribed genes relative to random distribution (P = 5.1×10-6), while no preference/avoidance is observed for genes with no evidence of being transcribed (P > 0.4). These results show that the preference of DSB to be located within annotated genic regions in Drosophila is not merely a consequence of DNA properties of genes such higher G + C content than noncoding sequences or the presence of DNA regulatory motifs in promoter regions and introns. This result is also in agreement with the previous analysis of recombination rates and nucleotide composition showing that there is no positive association between recombination rates and G + C content (P > 0.20; [20]). Instead, the detection of recombination events targeting actively transcribed genes relative to genes with no detectable transcription strongly suggests that gene expression during early meiosis has a causal effect on DSB location and formation and, ultimately, genome-wide recombination landscapes.
Figure 4.

Relative presence of DSBs across the genome. Analyses based on the 5,610 DSB events delimited by 500 bp or less described in [20]. The relative presence is measured as the ratio of the number of DSBs observed within each category to the number expected based on a random distribution of DSBs across the genome. Conservatively, we classified genes as showing no active transcription when FPKM < 0.001 and groups of genes with low-, medium- and high-transcription represent levels of target potential associated with transcription (FPKM x transcript length); 33, 46 and 21% of active genes belong to the low-, medium- and high- transcription groups, respectively. Probabilities (shown above each bar) associated with the relative presence of DSBs were obtained based on 10,000 independent replicates of the 5,610 DSBs randomly distributed across the genome.
Finally, we investigated the effect of transcription levels on DSB presence. To this end, we divided genes with detectable transcription into three groups for low-, medium- and high-transcription levels. We observe that among genes with detectable transcription, DSBs target genes preferentially lowly transcribed genes (Figure 4), with a negative relationship between transcription levels and recombination. Again, these results may evidence the expected heterogeneity of cell populations within our Early-meiosis tissue or, alternatively, a more complex interplay between transcription, histone modification and turnover, and chromatin accessibility for DSBs.
Conclusions
We obtained and compared the transcription profiles of Early- and Late-meiosis in D. melanogaster females with mRNA-seq and ultra-deep coverage. We identified significant differences in gene expression, new genes and exons, and a pattern of parent-of-origin effects with maternal-like expression that is particularly evident in Early-meiosis stages. We also described that Early-meiosis transcription occurs more often on the X chromosome and that there is physical clustering of actively transcribed genes across chromosomes. In terms of gene categories, we report that many genes involved in proteolysis are highly expressed in early meiosis, which may be a result of the rapid degradation of meiotic proteins following their utilization in order to prevent erroneous, self-assembling aggregates [48]. Our study and results underscore the limitations of using heterogeneous cellular and tissue samples when searching for biologically relevant features specific to particular developmental times and cell sets. In our case, searching for transcriptional signals present in only meiotic oocytes benefits from not using the whole ovary—as the oocyte transitions to transcriptionally dormant following the entrance into stage 1 in D. melanogaster, vastly increasing the influence of supportive nurse cells [49].
Work in yeast has shown that chromatin accessibility and nucleosome occupancy contribute to variation in the DSB landscape, although other factors may play a more dominant role in determining the probability of DNA cleavage [33,34]. Studies of nucleosome occupancy in mice meiotic spermatocytes also suggest that open chromatin structure directs, at least in part, the formation of DSBs [32]. Indirectly, these studies suggest that recombination landscapes could be influenced by gene expression, as transcription is known to alter chromatin structure. RNA-seq has been used as a powerful method to determine transcription patterns for specific tissues, cell populations and/or conditions, but it has heretofore not been exploited as a measure to gather information underlying patterns of variation in recombination rates across whole chromosomes.
Based on our previous high-resolution genetic maps in D. melanogaster, here we investigated the specific hypothesis that DSBs preferentially target transcriptionally active genomic regions in Drosophila. To our knowledge, our results represent the first evidence in a multicellular organism that gene expression in early meiotic cells is associated with increased likelihood of DSBs. Importantly, the preference of DSB targeting annotated transcripts seems to be related to active transcription and therefore supports the model that gene expression in meiotic tissues play a role—albeit clearly not the only one—influencing the landscapes and magnitude of recombination in a particular genomic region. Indeed, although the observed association between transcription levels and recombination rates is highly significant in terms of associated probability, it is weak in terms of the variation in recombination rates that can be explained solely by transcription. As such, the proposed influence of transcription on DSB formation and recombination landscapes should be viewed as one of several determinants of DSB localization. The presence of specific DNA motifs, the vicinity to telomeres/centromeres and other high-order chromatin structures during early meiosis, are all factors likely to also play a role. Transcriptome data of specific cell types, possibly using novel transgenic methods, together with detailed genetic analyses are needed to determine the relative role of gene expression influencing DSB formation and, ultimately, recombination rates across the Drosophila genome.
Recombination rates are an evolving and variable trait with detectable differences between species as well as within species. This inter-individual and inherited variation has been documented for the total number of recombination events per meiosis or per chromosome as well as in terms of the distribution across chromosomes in a number of species, including D. melanogaster[17,20,50-60]. Further, classic Drosophila genetics studies expose clear plasticity in the distribution of recombination rates across the genome as a result of biotic and abiotic factors [21,23-25] that would also act upon inherited inter-individual variation. We propose that our model, in which variation in gene expression plays a role altering the likelihood of DSB formation and thus the landscape of recombination across chromosomes, could easily reconcile many of these observations and provide a molecular, heritable and plastic mechanism to a number of observed patterns of recombination, from the high level of intra-specific variation, to the influence of environmental factors and stress conditions. The concept that gene expression may act as a “plastic” and heritable modifier of recombination, directly or epigenetically, is particularly relevant to evolutionary models on the maintenance of recombination. Our proposed model would represent a direct and mechanistic link between stressful conditions and increased recombination (either region-specific or genome-wide), the very same circumstances where recombination may be most favorable [22,61-63].
Methods
Drosophila stocks and tissue preparation
We generated two crosses using 2 highly inbred strains (RAL-208 and −375) from the Drosophila Genetic Reference Panel (DGRP) [47] that have been previously sequenced and recombination-mapped to high resolution. These populations were collected in Raleigh (NC, USA) and subjected to 20 generations of full sib mating. Freshly eclosed virgin females were collected from both inbred lines and crosses (males RAL-208 × females RAL-375 and its reciprocal cross) and allowed to mature for 72 hours at 23.5C. Ovaries from each of the four genotypes were dissected in RNA-Later Reagent (Quiagen) using forceps and dissecting probe. Ovarioles were teased apart and early meiotic portions (Germaria to Stage 3) removed using electrolytically sharpened tungsten needles, resulting in four ‘Early’ and four ‘Late’ tissue preparations [64].
Illumina library preparation and sequencing
Total RNA was prepared from ovaries, ovaries with early meiotic regions removed, and early meiotic regions following an optimized protocol for the Quiagen RNEasy kit (Qiagen, Valencia, CA) with additional DNase treatment. mRNA was isolated using the Invitrogen Dynabead mRNA Purification kit, with two additional wash steps. mRNA was fragmented with a cation solution from New England Biolab’s NEBNext Kit, ethanol precipitated, and cDNA synthesis performed with the NEBNext Kit. End repair, dA-Tailing, and adapter ligation of custom adapters was also performed with the NEB Next kit following an optimized manufacturer’s suggested protocol. 300-350 bp adapter ligated fragments were isolated from a 2% low-melt agarose gel and PCR enriched for 13 cycles. The PCR enriched libraries were validated by running an aliquot on a standard agarose gel. Products were purified and concentration obtained with Quant-iT TM PicoGreen® dsDNA Reagent and Kits (Invitrogen, CA, USA) on a Turner BioSystems TBS-380 Fluorometer. In total we generated eight Illumina Libraries, with two independently generated libraries per genotype to obtain adequate biological and technical replicates that were also run in separate Illumina lanes. We ran two lanes with four multiplexed libraries each. Single-read 120 bp fragments were sequenced on an Illumina Hi-Seq 2000 at the University of Iowa DNA Core Facility.
Sequence alignment and expression analyses
Illumina data were separated by tag using FastX Barcode Splitter and concatenating the two lanes of data for each tag respectively. All further analyses were performed within Galaxy, an accessible bioinformatics framework capable of next-generation sequencing data analysis [65-67]. Summary statistics were gathered using FastQC. The 5′ adapter sequence was then removed from each sample and 3′ ends trimmed until reaching a quality score greater than ten using FastqTrimmer. The groomed data was then mapped to the D. melanogaster reference genome (BDGP R5/dm3) using TopHat v1.4.0 [68,69].
We then used the Cufflinks package 2.0.2 [69] to assemble transcripts, obtain their relative abundance and find differentially expressed genes. After assembling transcripts, CuffMerge was used for merging and annotation analysis and measures of expression for every transcript associated with a particular gene were obtained in FPKM (Additional file 2: Figure S1). Expression calculations for early and late ovary development were based on two sets of replicates (Early samples of RAL-375 males × RAL-208 females and its reciprocal cross, and Late samples of RAL-375 males × RAL-208 females and its reciprocal cross) with two biological replicates per genotype and condition (Early or Late). Classic-FPKM normalization was performed with pooled estimates of dispersion (negative binominal) following [70]. We then utilized the Cuffdiff 2 algorithm [70] within Cufflinks 2.0.2 to calculate differential expression at both the gene and transcript levels. In short, differential gene expression was calculated using FPKM values for every gene while incorporating expression level variances during significance testing. This was performed by first deriving a dispersion model describing variances of fragment counts across replicates, which was then used to calculate the variances on a gene’s relative expression across replicates following the method described in [70] (Additional file 3: Figure S2, Additional file 4: Figure S3 and Additional file 5: Figure S4). Genes were considered to be expressed if each sample had a minimum of ten reads mapped and were above an FPKM of 0.1 unless noted explicitly. Genes were considered to be differentially expressed if the prior expression requirements were satisfied and reached an FDR-corrected significance level of 5% (q < 0.05).
Novel gene identification
Potentially novel genes were first identified by CuffLinks as significant reads mapping to unannotated regions of the dm3 genome that fit our expression criteria. Cufflinks initially identified 6004 potentially novel items. Restricting this list to only those that were detected in two or more samples reduced the number to 1308, and then filtering for only those expressed at reliable levels above one FPKM in at least one sample reduced the set to 220 entries. From this filtered list, we manually identified 47 items with lengths greater than approximately 300 bp, were minimally repetitive, and possessed reads that reliably mapped to predicted splice junctions. We identified 13 of these items to be candidate novel genes, based on a more stringent visual inspection and identification of apparent intron-exon structures.
We performed RT-PCR on a subset of our identified potentially novel genes with probable open reading frames that were missing from both tracks in an attempt to confirm expression of the novel transcript. PCR primers were designed for regions with significant RNA-seq reads mapped that spanned more than 300 base pairs. Primers were first tested with genomic DNA as a template, and then with whole ovary cDNA as a template (primers and conditions available upon request). A gene was considered novel if the mRNA track contained it but it was unannotated, or if it was missing from both mRNA tracks and the DM3 genome annotation.
Parent-of-origin effects
To study parent-of-origin effects in early and late tissues, we first identified genes that are significantly differentially expressed between offspring of reciprocal crosses (RAL-375 males × RAL-208 females and its reciprocal cross). We then focused on those genes with parent-of-origin effect that have levels of transcription in the offspring resembling the levels of transcription observed in the crosses’ maternal strain (RAL-208 or −375). To investigate allele-specific transcription in heterozygous offspring we obtained the set of reads that uniquely map to only one of the D. melanogaster parental strains with zero mismatches but not to the other parental strain, and vice versa, using MOSAIK assembler [71]. We also removed all reads that would differentially map to one parental sequence and not to the other if one of the reference sequences contained one or more ‘N’s for this read. Additionally, we only studied genes with a minimum 100 allele-specific reads to increase accuracy in the allelic ratios.
Gene-term enrichment analyses were performed with DAVID [72], utilizing the BP_FAT subset of gene ontology (GO) terms to identify enriched biological themes. We then combined the early and late tissue GO data for each strain and repeated the analysis. We report P-values from the DAVID analysis according to the EASE score, a modified Fishers exact test [72]. FDR-corrected EASE scores are reported utilizing the Benjamini-Hochberg multiple-test correction procedure employed by DAVID.
Genomic distribution of transcribed genes
In order to test whether transcribed and untranscribed genes were distributed randomly across the genome, we performed a Run’s test for randomness. To determine the number of runs, we separated genes into two categories of transcriptional activity: genes transcribed at greater levels than 1 FPKM and those that were not. Along the length of the genome, switches from one category to another counted as a completed run. Overlapping genes were counted separately following this scheme.
Recombination vs. expression analysis
To test the hypothesis that gene expression is associated with recombination rates across the genome, we first generated landscapes of expression for each chromosome. We calculated the number of genes expressed at a threshold greater than 1 FPKM (unless noted otherwise) and the number of kilobases transcribed (counting overlapping transcript regions only once) per 100 kb adjacent intervals. We also obtained a measure of overall transcriptional activity within a genomic interval (OTA), defined as the Log10-transformed sum of the product FPKM × transcript length for each gene within a given genomic interval. High-resolution recombination landscapes for adjacent 100 kb regions across the whole genome were obtained from [20].
Availability of supporting data
The datasets supporting the results of this article are available in the NCBI SRA repository [http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?study=SRP032523].
Abbreviations
DSB: Double strand break; GO: Gene ontology; FPKM: Fragments per kilobase of transcript per million fragments mapped; FDR: False discovery rate; PCR: Polymerase chain reaction; RT-PCR: Real time polymerase chain reaction.
Competing interests
The authors declare that they have no competing interests.
Author’s contributions
AA and JC designed the study, analyzed data and wrote the manuscript. AA performed the experiments and data collection. Correspondence and requests for materials should be addressed to JC (josep-comeron@uiowa.edu). Both authors read and approved the final manuscript.
Supplementary Material
Top Enriched GO Terms among genes with parent-of origin effects with maternal-like transcript levels for Early- and Late-Ovarian samples. Table S2: Top Enriched GO Terms among genes with parent-of origin effects with maternal-like transcript levels.
FPKM distribution density across genes. Blue region: Early-ovarian transcriptome; Orange region: Late-ovarian transcriptome.
Volcano plot of genes by significance and fold change.
Early vs Late Log2 fold difference histogram following normalization.
P-value distribution following normalization and testing with CuffDiff v2.0.2 before correcting for multiple tests. This histogram displays the approximate expected distribution of significance following proper normalization—harboring signals of true positives enriched at the low-end, while enrichment in the highest bin is due to genes with low read counts.
Contributor Information
Andrew B Adrian, Email: andrew-adrian@uiowa.edu.
Josep M Comeron, Email: josep-comeron@uiowa.edu.
Acknowledgements
The authors thank Ana Llopart and two anonymous reviewers for critical reading of the manuscript and suggestions. The project described was supported in part by award number RC2GM092501 from the National Institute of General Medical Sciences, NIH, to J.M.C., and an NIH Bioinformatics and Computational Biology training grant T32 GM082729 to A.B.A. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A. et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6:377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- Lake CM, Hawley RS. The molecular control of meiotic chromosomal behavior: events in early meiotic prophase in Drosophila oocytes. Annu Rev Physiol. 2012;74:425–451. doi: 10.1146/annurev-physiol-020911-153342. [DOI] [PubMed] [Google Scholar]
- Roth S, Lynch JA. Symmetry breaking during Drosophila oogenesis. Cold Spring Harb Perspect Biol. 2009;1:a001891. doi: 10.1101/cshperspect.a001891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirilly D, Xie T. The Drosophila ovary: an active stem cell community. Cell Res. 2007;17:15–25. doi: 10.1038/sj.cr.7310123. [DOI] [PubMed] [Google Scholar]
- Spradling A, Fuller MT, Braun RE, Yoshida S. Germline stem cells. Cold Spring Harb Perspect Biol. 2011;3:a002642. doi: 10.1101/cshperspect.a002642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling AC. In: The Development of Drosophila melanogaster. Volume 1. Bate MaM-A A, editor. Cold Spring Harbor, N.Y: Cold Spring Harbor Press; 1993. Developmental genetics of oogenesis; pp. 1–70. [Google Scholar]
- Morris LX, Spradling AC. Long-term live imaging provides new insight into stem cell regulation and germline-soma coordination in the Drosophila ovary. Development. 2011;138:2207–2215. doi: 10.1242/dev.065508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, Nuttall R, Edwards P, Minor J, Naiman D, Lu J, Doctolero M, Vainer M, Chan C, Malley J. et al. A survey of ovary-, testis-, and soma-biased gene expression in Drosophila melanogaster adults. Genome Biol. 2004;5:R40. doi: 10.1186/gb-2004-5-6-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Q, Chepelev I, Wei G, Tarayrah L, Cui K, Zhao K, Chen X. Dynamic regulation of alternative splicing and chromatin structure in Drosophila gonads revealed by RNA-seq. Cell Res. 2010;20:763–783. doi: 10.1038/cr.2010.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley DL, Zimm GG. The genome of Drosophila melanogaster San Diego. CA: Academic Press; 1992. [Google Scholar]
- Dumont BL, Payseur BA. Genetic analysis of genome-scale recombination rate evolution in house mice. PLoS Genet. 2011;7:e1002116. doi: 10.1371/journal.pgen.1002116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fledel-Alon A, Leffler EM, Guan Y, Stephens M, Coop G, Przeworski M. Variation in human recombination rates and its genetic determinants. PLoS One. 2011;6:e20321. doi: 10.1371/journal.pone.0020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coop G, Wen X, Ober C, Pritchard JK, Przeworski M. High-resolution mapping of crossovers reveals extensive variation in fine-scale recombination patterns among humans. Science. 2008;319:1395–1398. doi: 10.1126/science.1151851. [DOI] [PubMed] [Google Scholar]
- Singer T, Fan Y, Chang HS, Zhu T, Hazen SP, Briggs SP. A high-resolution map of Arabidopsis recombinant inbred lines by whole-genome exon array hybridization. PLoS Genet. 2006;2:e144. doi: 10.1371/journal.pgen.0020144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulathinal RJ, Bennett SM, Fitzpatrick CL, Noor MA. Fine-scale mapping of recombination rate in Drosophila refines its correlation to diversity and divergence. Proc Natl Acad Sci USA. 2008;105:10051–10056. doi: 10.1073/pnas.0801848105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman MV, Kruglyak L. Recombinational landscape and population genomics of Caenorhabditis elegans. PLoS Genet. 2009;5:e1000419. doi: 10.1371/journal.pgen.1000419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter AD, Choi JY. Natural selection shapes nucleotide polymorphism across the genome of the nematode Caenorhabditis briggsae. Genome Res. 2010;20:1103–1111. doi: 10.1101/gr.104331.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaugh SE, Heil CS, Manzano-Winkler B, Loewe L, Goldstein S, Himmel TL, Noor MA. Recombination modulates how selection affects linked sites in Drosophila. PLoS Biol. 2012;10:e1001422. doi: 10.1371/journal.pbio.1001422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron JM, Ratnappan R, Bailin S. The many landscapes of recombination in Drosophila melanogaster. PLoS Genet. 2012;8:e1002905. doi: 10.1371/journal.pgen.1002905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel JV. A relation between larval nutrition and the frequency of crossing over in the third chromosome of Drosophila melanogaster. Genetics. 1941;26:506–516. doi: 10.1093/genetics/26.5.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons PA. Evolutionary rates: effects of stress upon recombination. Biol J Linn Soc. 1988;35:49–68. doi: 10.1111/j.1095-8312.1988.tb00458.x. [DOI] [Google Scholar]
- Priest NK, Galloway LF, Roach DA. Mating frequency and inclusive fitness in Drosophila melanogaster. Am Nat. 2008;171:10–21. doi: 10.1086/523944. [DOI] [PubMed] [Google Scholar]
- Redfield H. Delayed mating and the relationship of recombination to maternal age in Drosophila melanogaster. Genetics. 1966;53:593–607. doi: 10.1093/genetics/53.3.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern C. An effect of temperature and age on crossing-over in the first chromosome of Drosophila melanogaster. Proc Natl Acad Sci USA. 1926;12:530–532. doi: 10.1073/pnas.12.8.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, de Massy B. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science. 2010;327:836–840. doi: 10.1126/science.1183439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- Heil CS, Noor MA. Zinc finger binding motifs do not explain recombination rate variation within or between species of Drosophila. PLoS One. 2012;7:e45055. doi: 10.1371/journal.pone.0045055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh ND, Stone EA, Aquadro CF, Clark AG. Fine-scale heterogeneity in crossover rate in the Garnet-Scalloped region of the Drosophila melanogaster X Chromosome. Genetics. 2013;194:375–387. doi: 10.1534/genetics.112.146746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DE, Takeo S, Nandanan K, Paulson A, Gogol MM, Noll AC, Perera AG, Walton KN, Gilliland WD, Li H. et al. A whole-chromosome analysis of meiotic recombination in Drosophila melanogaster. G3 (Bethesda) 2012;2:249–260. doi: 10.1534/g3.111.001396. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petes TD. Meiotic recombination hot spots and cold spots. Nat Rev Genet. 2001;2:360–369. doi: 10.1038/35072078. [DOI] [PubMed] [Google Scholar]
- Getun IV, Wu ZK, Khalil AM, Bois PR. Nucleosome occupancy landscape and dynamics at mouse recombination hotspots. EMBO Rep. 2010;11:555–560. doi: 10.1038/embor.2010.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Sasaki M, Kniewel R, Murakami H, Blitzblau HG, Tischfield SE, Zhu X, Neale MJ, Jasin M, Socci ND. et al. A hierarchical combination of factors shapes the genome-wide topography of yeast meiotic recombination initiation. Cell. 2011;144:719–731. doi: 10.1016/j.cell.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick DT, Wang YH, Dominska M, Griffith JD, Petes TD. Control of meiotic recombination and gene expression in yeast by a simple repetitive DNA sequence that excludes nucleosomes. Mol Cell Biol. 1999;19:7661–7671. doi: 10.1128/mcb.19.11.7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintapalli VR, Wang J, Dow JA. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet. 2007;39:715–720. doi: 10.1038/ng2049. [DOI] [PubMed] [Google Scholar]
- Cash AC, Andrews J. Fine scale analysis of gene expression in Drosophila melanogaster gonads reveals Programmed cell death 4 promotes the differentiation of female germline stem cells. BMC Dev Biol. 2012;12:4. doi: 10.1186/1471-213X-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assis R, Zhou Q, Bachtrog D. Sex-biased transcriptome evolution in Drosophila. Genome Biol Evol. 2012;4:1189–1200. doi: 10.1093/gbe/evs093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, Nuttall R, Naiman D, Bouffard G, Malley J, Andrews J, Eastman S, Oliver B. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science. 2003;299:697–700. doi: 10.1126/science.1079190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgill D, Zhang Y, Parisi M, Oliver B. Demasculinization of X chromosomes in the Drosophila genus. Nature. 2007;450:238–241. doi: 10.1038/nature06330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranz JM, Castillo-Davis CI, Meiklejohn CD, Hartl DL. Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science. 2003;300:1742–1745. doi: 10.1126/science.1085881. [DOI] [PubMed] [Google Scholar]
- Llopart A. The rapid evolution of X-linked male-biased gene expression and the large-X effect in Drosophila yakuba, D. santomea, and their hybrids. Mol Biol Evol. 2012;29:3873–3886. doi: 10.1093/molbev/mss190. [DOI] [PubMed] [Google Scholar]
- Meisel RP, Malone JH, Clark AG. Disentangling the relationship between sex-biased gene expression and X-linkage. Genome Res. 2012;22:1255–1265. doi: 10.1101/gr.132100.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Presgraves DC. Little evidence for demasculinization of the Drosophila X chromosome among genes expressed in the male germline. Genome Biol Evol. 2012;4:1007–1016. doi: 10.1093/gbe/evs077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman PT, Rubin GM. Evidence for large domains of similarly expressed genes in the Drosophila genome. J Biol. 2002;1:5. doi: 10.1186/1475-4924-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutanaev AM, Kalmykova AI, Shevelyov YY, Nurminsky DI. Large clusters of co-expressed genes in the Drosophila genome. Nature. 2002;420:666–669. doi: 10.1038/nature01216. [DOI] [PubMed] [Google Scholar]
- Weber CC, Hurst LD. Support for multiple classes of local expression clusters in Drosophila melanogaster, but no evidence for gene order conservation. Genome Biol. 2011;12:R23. doi: 10.1186/gb-2011-12-3-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay TF, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu D, Casillas S, Han Y, Magwire MM, Cridland JM. et al. The Drosophila melanogaster Genetic Reference Panel. Nature. 2012;482:173–178. doi: 10.1038/nature10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringrose L, Paro R. Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development. 2007;134:223–232. doi: 10.1242/dev.02723. [DOI] [PubMed] [Google Scholar]
- Liu JL, Buszczak M, Gall JG. Nuclear bodies in the Drosophila germinal vesicle. Chromosome Res. 2006;14:465–475. doi: 10.1007/s10577-006-1062-5. [DOI] [PubMed] [Google Scholar]
- Yandeau-Nelson MD, Nikolau BJ, Schnable PS. Effects of trans-acting genetic modifiers on meiotic recombination across the a1-sh2 interval of maize. Genetics. 2006;174:101–112. doi: 10.1534/genetics.105.049270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler KE, Cherry JP, Lynn A, Hunt PA, Hassold TJ. Genetic control of mammalian meiotic recombination. I. Variation in exchange frequencies among males from inbred mouse strains. Genetics. 2002;162:297–306. doi: 10.1093/genetics/162.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey C, Baudat F, de Massy B. Genome-wide control of the distribution of meiotic recombination. PLoS Biol. 2009;7:e35. doi: 10.1371/journal.pbio.1000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Moran E, Armstrong SJ, Santos JL, Franklin FCH, Jones GH. Variation in chiasma frequency among eight accessions of Arabidopsis thaliana. Genetics. 2002;162:1415–1422. doi: 10.1093/genetics/162.3.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont BL, White MA, Steffy B, Wiltshire T, Payseur BA. Extensive recombination rate variation in the house mouse species complex inferred from genetic linkage maps. Genome Res. 2011;21:114–125. doi: 10.1101/gr.111252.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esch E, Szymaniak JM, Yates H, Pawlowski WP, Buckler ES. Using crossover breakpoints in recombinant inbred lines to identify quantitative trait loci controlling the global recombination frequency. Genetics. 2007;177:1851–1858. doi: 10.1534/genetics.107.080622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell MG. Genetic change of recombination value in Drosophila melanogaster. II. Simulated natural selection. Genetics. 1972;70:433–443. doi: 10.1093/genetics/70.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks LD, Marks RW. The organization of genetic variation for recombination in Drosophila melanogaster. Genetics. 1986;114:525–547. doi: 10.1093/genetics/114.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdullah NF, Charlesworth B. Selection for reduced crossing over in Drosophila melanogaster. Genetics. 1974;76:447–451. doi: 10.1093/genetics/76.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CG, Goodman MM, Stuber CW. Comparative recombination distances among Zea mays L. inbreds, wide crosses and interspecific hybrids. Genetics. 1995;141:1573–1581. doi: 10.1093/genetics/141.4.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann R, Jeffreys AJ. Polymorphism in the activity of human crossover hotspots independent of local DNA sequence variation. Hum Mol Genet. 2006;15:1401–1411. doi: 10.1093/hmg/ddl063. [DOI] [PubMed] [Google Scholar]
- Hadany L, Beker T. On the evolutionary advantage of fitness-associated recombination. Genetics. 2003;165:2167–2179. doi: 10.1093/genetics/165.4.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadany L, Beker T. Fitness-associated recombination on rugged adaptive landscapes. J Evol Biol. 2003;16:862–870. doi: 10.1046/j.1420-9101.2003.00586.x. [DOI] [PubMed] [Google Scholar]
- Agrawal AF, Hadany L, Otto SP. The evolution of plastic recombination. Genetics. 2005;171:803–812. doi: 10.1534/genetics.105.041301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalistratov GF, Bashkirov AA. Use of a serial portable apparatus for electrolytic sharpening of surgical instruments in the preparation of metal microelectrodes. Biull Eksp Biol Med. 1964;58:122–123. [PubMed] [Google Scholar]
- Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. 2010;Chapter 19(Unit 19 10):11–21. doi: 10.1002/0471142727.mb1910s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J. et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27:2325–2329. doi: 10.1093/bioinformatics/btr355. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosaik-Aligner. http://code.google.com/p/mosaik-aligner/
- da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Top Enriched GO Terms among genes with parent-of origin effects with maternal-like transcript levels for Early- and Late-Ovarian samples. Table S2: Top Enriched GO Terms among genes with parent-of origin effects with maternal-like transcript levels.
FPKM distribution density across genes. Blue region: Early-ovarian transcriptome; Orange region: Late-ovarian transcriptome.
Volcano plot of genes by significance and fold change.
Early vs Late Log2 fold difference histogram following normalization.
P-value distribution following normalization and testing with CuffDiff v2.0.2 before correcting for multiple tests. This histogram displays the approximate expected distribution of significance following proper normalization—harboring signals of true positives enriched at the low-end, while enrichment in the highest bin is due to genes with low read counts.