

Abstract
Proteases are important for multiple processes during malignant progression including tumor angiogenesis, invasion and metastasis. Recent evidence reveals that tumor-promoting proteases function as part of an extensive multidirectional network of proteolytic interactions, in contrast to the unidirectional caspase cascade. These networks involve different constituents of the tumor microenvironment, and key proteases — such as cathepsin B, urokinase-type plasminogen activator and several matrix metalloproteinases — occupy central nodes for amplifying proteolytic signals passing through the network. The proteolytic network interacts with other important signaling pathways in tumor biology, involving chemokines, cytokines, and kinases. Viewing these proteolytic interactions as a system of activating and inhibiting reactions provides insight into tumor biology and reveals relevant pharmaceutical targets. This review will examine recent advances in understanding proteases in cancer, and summarize how their network of activity is co-opted to promote tumor progression.
Keywords: protease, inhibitor, invasion, metastasis, tumor microenvironment, cascade
Introduction
Proteases, once thought of as little more than cellular garbage disposals or extracellular matrix (ECM) degraders, comprise one of the largest groups of enzymes in the human genome. There are currently 570 known human proteases [1], coupled with a smaller group of endogenous protease inhibitors that tightly regulate their activity. While proteases mediate both terminal protein degradation and ECM remodeling, recent findings have revealed their functions in tumors to be significantly more complex and varied. Several hallmarks of aggressive cancer are a direct result of proteolytic activity, including, but not limited to, tumor cell invasion into the stroma, angiogenesis and metastasis [2]. A separate group of cascading proteolytic interactions controls apoptosis, which cancer cells must escape in their progression to malignancy [3]. Finally, proteases are not only involved in protein degradation, but can also activate other proteases and/or signaling molecules through specific cleavage of pro-peptides.
There are five human protease classes categorized by their catalytic mechanism (aspartic, cysteine, metallo, serine and threonine). Likewise, endogenous inhibitors exhibit specificity in their targets – cystatins predominantly inhibit cysteine proteases, serpins are most effective against serine proteases, and tissue inhibitors of metalloproteinases (TIMPs) target metalloproteinases. There is some flexibility to these interactions, however, as certain serpins can also inhibit cathepsins, and some cystatins can inhibit metalloproteinases [4, 5]. While similarities exist among members of the same family of proteases, each protease has its own distinct pattern of expression and activity allowing for specificity of targets, interactions and effects. The progression to malignancy is often associated with deregulation of the normal mechanisms regulating proteolysis, resulting in numerous proteases having dramatically altered (and often significantly upregulated) activity in cancer. As a result, many different proteases have been implicated as potential therapeutic targets (Box 1).
Box 1.
Due to the multitude of proteases upregulated during cancer progression, several pharmaceutical companies have undertaken efforts to target different types of cancer via inhibition of proteases. Results have been mixed, with some successes but also some failures. MMPs are obvious therapeutic targets due to their upregulation in several different types of cancer; however clinical trials using broad-spectrum MMP inhibitors failed to show any significant impact on tumor development [30]. In contrast, bortezomib (Velcade, PS-341) is a proteasome inhibitor that has been approved by the FDA for the treatment of multiple myeloma and mantle cell lymphoma. Bortezomib inhibits the proteasome by blocking the activity of the 26S subunit [58]. Because of its effectiveness in treating multiple myeloma, bortezomib has additionally been tested as a single or combination therapy in solid tumors [58], and researchers are also developing improved proteasome inhibitors, some of which have been tested in clinical trials [59].
The range of proteases targeted by pharmacological inhibitors in current clinical trials is quite broad. Several companies have developed inhibitors of cathepsin K, a cysteine cathepsin that degrades bone in osteolytic metastases, with some of these inhibitors reaching phase I and II clinical trials [60]. Another commonly upregulated protease, uPA, is the target of an inhibitor being tested in a phase II clinical trial (NCT00499265) as a combination therapy with gemcitabine for patients with non-resectable pancreatic cancer. One surprising addition to the list of anti-cancer agents is the drug nelfinavir, an HIV protease inhibitor. Interestingly, nelfinavir induces apoptosis in cancer cells through a mechanism independent of the mitochondria, and is now the subject of several clinical trials [61]. This is similar to results seen with the HIV protease inhibitors indinavir and saquinavir, which were shown to inhibit angiogenesis in a murine model of Kaposi sarcoma [62]. Another phase I/II clinical trial (NCT00086723) is seeking to use the activity of tPA to generate angiostatin, an antiangiogenic peptide derived from plasminogen [63]. Surprisingly, instead of inhibiting tPA, investigators in this study are examining the ability of recombinant tPA to increase the levels of angiostatin and thereby inhibit angiogenesis. The results of this study will need to be thoroughly evaluated in light of the ability of tPA to activate tumor-promoting proteases (Figure 1). As novel proteolytic inhibitors continue to be developed, the success of bortezomib and failure of broad-spectrum MMP inhibitors will help researchers direct new therapeutic strategies and clinical trials, with the eventual goal of improved cancer treatments.
As our understanding of the scope of proteolytic activities increases, our view of a protease changes from that of an enzyme acting independently to one that considers its position in a vast network of proteolytic interactions [6, 7]. This network is influenced by activating and inhibiting reactions, and can modulate angiogenesis, tumor cell invasion, ECM composition and integrity, and signaling pathways in the tumor microenvironment. New findings highlight the multiple mechanisms by which cancer cells co-opt proteolytic networks, including interactions with the stroma (Box 2), upregulation of activating proteases, and even cellular uptake of proteases secreted by stromal cells. The focus of this review will be to discuss recent data showing how proteases interact with each other, endogenous inhibitors, and other signaling molecules in a coordinated fashion to regulate tumor progression.
Box 2.
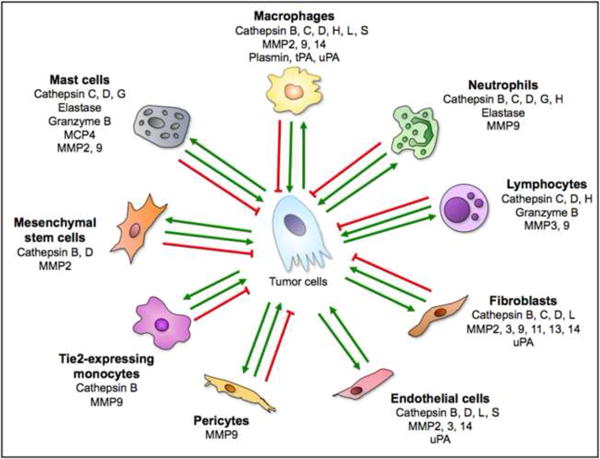
In addition to cancer cells, stromal cells are integral for the contribution of certain proteases to developing tumors, emphasizing the intricate complexity of the proteolytic network in the tumor microenvironment. Fibroblasts, endothelial cells, and infiltrating immune cells all contribute proteases, including cysteine cathepsins, various MMPs, and uPA (Figure I) [24, 29, 47, 64-68]. Infiltrating immune cells, especially tumor-associated macrophages, are some of the major contributors of stromal proteases to the tumor microenvironment. As the cellular composition of tumors varies from tumor to tumor, and within tumors, so too will the structure of the proteolytic network. Thus a full understanding of protease involvement in tumor progression will need to take into account not only proteases expressed by tumor cells, but also those expressed by stromal cells. Targeting stromal proteases has the added advantage of directing therapy toward targets provided by cell types with stable genomes, resulting in a reduced likelihood of tumors evolving resistance.
Figure I shows the major cell types found in the tumor microenvironment, and lists some of the proteases (selected from the network in Figure 1) that they contribute. The green arrows represent cancer-promoting interactions; however, several of these proteases and cell types can have context-dependent tumor-suppressing effects, as indicated by the red inhibitory lines. Some of the data ascribing protease expression to particular cell types has come from studies using normal cells and tissues, and therefore the ability of all cells to express each protease listed in different tumor microenvironments is not yet firmly established. However, the potential for these cell types to contribute the listed proteases to the tumor microenvironment must be taken into consideration when examining the proteolytic network in cancer. The abundance of stromal proteases allows for increased complexity of the proteolytic network in the tumor microenvironment as proteases not usually expressed by tumor cells are now present and can cleave additional substrates. References for stromal cells expressing specific proteases are: macrophages [24, 29, 46, 57, 67-69], neutrophils [29, 67, 70-73], lymphocytes [13, 29, 70, 72, 74], fibroblasts [29, 67, 68, 75], endothelial cells [29, 64, 68, 76], pericytes [77], Tie 2-expressing monocytes [78], mesenchymal stem cells [79, 80], and mast cells [29, 72, 81].
Interactions of proteases within smaller, interconnected cascades
Individual proteases, with their broad range of targets, multitude of activation mechanisms, and proposed impact on tumor progression and metastasis, cannot be fully understood without placing them in the proper context of upstream and downstream proteases serving as activators or substrates. Proteases are synthesized as inactive (or marginally active) zymogens and require cleavage, usually by other proteases, for activation. Much like the well-studied kinase cascades that control cellular proliferation, proteases also function in vertical relation to each other, amplifying signals as targets increase at each step. As our understanding of protease biology grows, we can gain insights into tumor biology by examining their interconnectivity, as illustrated by several recognizable cascades of proteolytic interactions. Each example below features prominent proteases as central nodes that can serve as major regulatory hubs for the whole cascade. Smaller cascades centered on these nodes can then be connected to form a network of proteolytic activity (Figures 1, 2).
Figure 1.
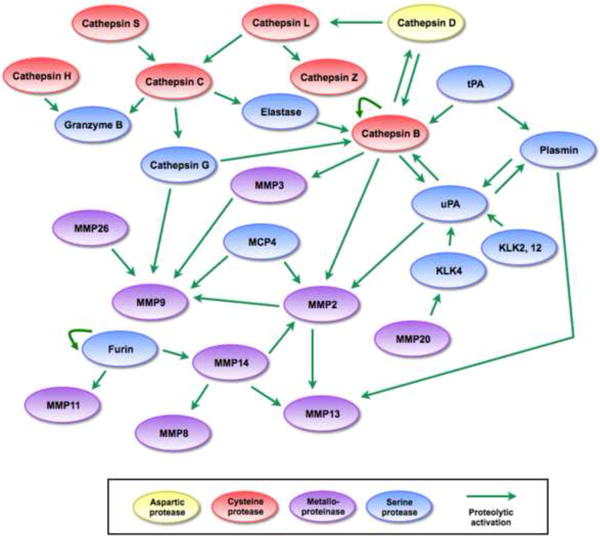
Different interactions within a proteolytic network.
Visualizing proteolytic interactions as a network of coordinated cascades reveals that the network has multiple entry points, interactions are multidirectional, and signals can be amplified in many directions. Examining protease-protease interactions specifically shows that similar to the cascade view, several proteases occupy nodes in the network and function as key regulators of proteolytic activity. Additionally, interactions involve proteases of different families and can proceed in multiple different pathways, allowing for some proteases to compensate for the absence of others. References for proteolytic interactions not described in the main text: cathepsin D activates cathepsin L [18], cathepsin L activates cathepsin Z [90], cathepsin B activates cathepsin D, MMP2, and MMP3 [18, 75], tissue-type plasminogen activator (tPA) activates cathepsin B and plasmin [91], MMP14 can activate MMP2 [92], MMP2, MMP14, and plasmin can activate MMP13 [93], MMP14 activates MMP8 [94], MMP26 can activate KLK4 [26], and furin activates MMP11 and MMP14 [31].
Figure 2.
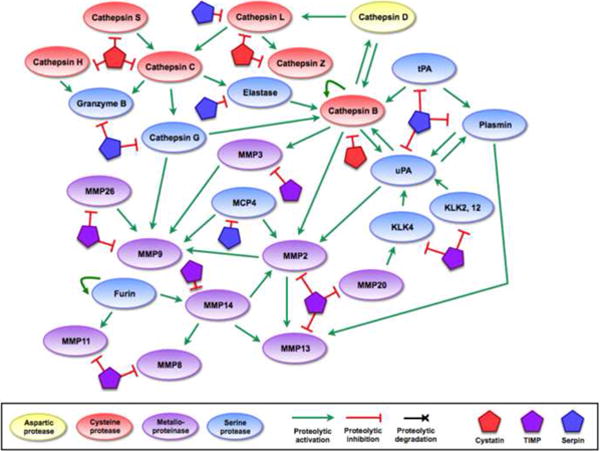
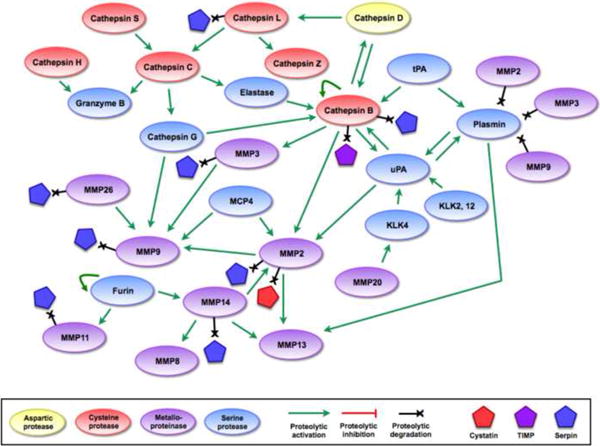
Mechanisms of proteolytic regulation within the network.
(a) Endogenous protein inhibitors can regulate proteolytic activity at multiple points in the network, allowing for tight regulation (or deregulation, if inhibitor expression is downregulated) of proteolytic activity. (b) Several proteases have been shown to degrade endogenous inhibitors, giving them the ability to increase the activity of other proteases indirectly. These interactions add further complexity to the network, and demonstrate how proteolytic signals can flow in multiple directions through different mechanisms. References for proteolytic interactions not described in the main text: cathepsin B can inactivate some serpins and TIMPs [95, 96], serpinB13/hurpin and serpinB3/SCCA1 inhibit cathepsin L [97, 98], cathepsin L inactivates serpinA1 [99], and several MMPs can inactivate a variety of serpins [29].
Caspases and apoptosis
All tumors must escape from apoptosis in order to progress towards malignancy. One of the best-known proteolytic cascades involves caspases leading to apoptotic cell death. Largely separate from proteolytic networks that cancers frequently co-opt during tumor progression, this chain of interactions was one of the first characterized proteolytic cascades and serves as a prototype. Although the caspase cascade can have multiple entry points, the end result is always cell death – an outcome that does not re-activate the cascade. The finality of the caspase cascade stands in stark contrast to the proteolytic interactions that will be discussed later in this review. Even so, the caspase cascade is integral to tumor biology, and has been the subject of extensive investigation as researchers seek to understand the interplay between the caspase cascade and its regulators, and how those interactions are misregulated in cancer. Caspase regulation is incredibly complex and a detailed summary is beyond the scope of this review, as other recent literature has covered this topic [3].
At the start of the caspase cascade are the initiator caspases, such as caspases 2, 8, 9, and 10 [3, 8]. These initiator caspases can directly or indirectly activate the effector caspases 3, 6, and 7 through cleavage of their prodomains. Once activated, these effector caspases cleave a variety of cellular substrates, with the end result being apoptosis. Initiation of signaling that leads to activation of the cascade generally comes from one of two sources: the intrinsic pathway, involving cytochrome c release from the mitochondria, or the extrinsic pathway, involving activation of death domain receptors on the cell surface. The importance of the initiator caspases such as caspase 8 for induction of apoptosis [3] is demonstrated by the embryonic lethality of the caspase 8 knockout mouse [9], and by evidence showing that loss of caspase 8 leads to cell survival and promotes metastasis in neuroblastoma [10]. One potential way to bypass loss of caspase 8 is via cytotoxic T lymphocyte-mediated release of granzyme B into the cell, which can indirectly activate caspase 3 and trigger apoptosis [11]. Granzyme B itself is directly downstream of at least two other proteases, as it can be activated by cathepsin C (which is itself downstream of cathepsin L and S [12]) and cathepsin H (Figure 1), demonstrating that regulation of apoptosis occurs at many levels [13]. Positive and negative regulation of the effector caspases highlights their importance as a critical node in the caspase cascade. Inhibition of caspases is accomplished by several endogenous protease inhibitors, most notably XIAP, which can be inactivated by a number of proteases, including several cysteine cathepsins [14]. Interactions such as this allow a connection between the caspase cascade and other proteolytic networks.
Cathepsin B
In contrast to the unidirectional caspase cascade, the proteolytic network that promotes tumor progression involves a wide variety of bidirectional interactions that affect numerous tumor-promoting processes. The cysteine protease cathepsin B is one of the most prominent proteases in the example of a proteolytic network shown in Figure 1, and is upregulated in many different tumor microenvironments [15]. One protease that can convert pro-cathepsin B to active cathepsin B is cathepsin D, an aspartic cathepsin which also activates cathepsin L [16], a key cysteine cathepsin that is integral to the in vivo activation of the matrix-degrading enzyme heparanase [17]. Additionally, cathepsin B can be activated by a series of other proteases, including cathepsin G, urokinase-type plasminogen activator (uPA), tissue-type plasminogen activator (tPA), and elastase [18]. Elastase and cathepsin G can both be activated by cathepsin C [19], revealing another layer of regulation on top of the interactions of these two serine proteases with cathepsin B. Finally, cathepsin B can undergo auto-activation in some conditions, further expanding the mechanisms of cathepsin B regulation [20]. This broad spectrum of activating proteases emphasizes the central importance of cathepsin B to proteolysis, and can be used as the starting point of a small network centered on this important protease (Figure 1). The interaction between cathepsin B and uPA is reciprocal, as cathepsin B can also activate uPA [21]. This example of reciprocity underscores the view that proteolytic interactions are part of a complex network as opposed to a simpler unidirectional cascade.
Cathepsin B can cleave a wide variety of targets depending on its subcellular localization in the tumor microenvironment, as activation, secretion, or cell surface translocation of this normally lysosomal enzyme occurs in both tumor and stromal cells [22-24]. Some of the best-known cathepsin B substrates are ECM proteins (discussed later), several prominent proteases (Figure 1) and inhibitors of other proteases (Figure 2b). This ability to inactivate endogenous inhibitors provides an indirect mechanism for cathepsin B to increase the activity of other proteases (Figure 2b), and increases the complexity of the network.
Urokinase-type plasminogen activator (uPA)
Best known for its ability to convert plasminogen into plasmin, uPA has attracted attention for its wide range of targets as well as its prominent location in the proteolytic network. Activation of uPA is dependent on binding of pro-uPA to uPAR [25], and can be carried out by plasmin [18], creating a feedback loop by which plasmin and uPA can activate each other, or be activated by cathepsin B. Alternatively, kallikreins (KLK) 2, 4, and 12 can also activate pro-uPA [26] (Figure 1). Beyond plasmin and cathepsin B, some of the primary targets of uPA are MMPs – either directly or indirectly. For example, the uPAR-uPA system is involved in activation of MMP2 via a mechanism dependent on contributions from stromal fibroblasts [27]; additionally, overexpression of uPA and uPAR in the basal epidermis and hair follicles leads to increased MMP2 and MMP9 activation [28]. These findings highlight the complexity of proteolytic interactions in the tumor microenvironment, which are dependent not only on proteases provided by tumor cells, but also on proteolytic contributions from various stromal cells (Box 2).
Matrix metalloproteinases (MMPs)
In cancer biology, MMPs are some of the most extensively studied proteases due to their frequent overexpression in many different cancers and their ability to degrade a multitude of substrates [29]. However, clinical trials using broad-spectrum MMP inhibitors to treat pancreatic, brain, lung, or renal cancer were largely disappointing, possibly due to the large number of MMPs with potentially opposing functions [30]. There are 24 different MMPs in mammals, characterized by conserved pro-peptide and catalytic domains and a zinc ion in their active site [31]. Simply categorizing these genes as MMPs, however, does not do justice to the level of diversity in substrates and interactions for this family. As can be expected with such a large family of proteases, multiple mechanisms exist for the activation of MMPs, with different MMPs relying on different mechanisms. In addition to cathepsin B, one of the most commonly proposed activators for MMPs is furin, a serine protease that cleaves at the R[30] XKR or RRKR sequence contained in approximately one-third of MMPs [31].
Our understanding of MMP activation is further complicated by the fact that individual MMPs can be activated through multiple distinct mechanisms. An example of this is MMP9. In one pathway, plasmin activates MMP3, which can in turn activate MMP9 [32]. Alternatively, MMP9 can be activated in wound healing by a chymotrypsin-like serine protease (recently identified as cathepsin G [33]) through a process that is inhibited by serpinA3 [34]. Two other possible mechanisms for MMP activation are autoactivation [35] and activation via oxidation [36]. Interestingly, MMP2 is unable to autoactivate under the same conditions that lead to activation of MMP9, demonstrating the different properties of two relatively similar members of the MMP family. Finally, mast cell chymase protease-4 (MCP-4) can potentially also activate MMP9 (and MMP2) (Figure 1) as MCP-4 null mice have increased abundance of proMMP2 and proMMP9 [37]. Finally, other MMPs have displayed the ability to activate different MMP family members (Figure 1).
The activity of the MMP family is quite complex and can vary from one MMP to the next, as illustrated by the recent finding that deletion of Mmp2, Mmp7, or Mmp9 in a mouse model of prostate cancer had differential effects on tumor burden, metastasis, and angiogenesis [38]. Additionally, membrane-type MMPs, such as MMP14, are integrally important for matrix remodeling, allowing cancer cells to migrate across the basement membrane [39]. Over the past few years, new proteomic techniques, such as the combination of terminal amine isotopic labeling of substrates (TAILS) with iTRAQ (isobaric tag for relative and absolute quantitation), have been developed that allow for broad, unbiased, and even in vivo, analysis of proteolytic substrates with greater sensitivity and specificity than previously achieved [40]. These techniques have first been utilized in the analysis of MMP biology, resulting in a more comprehensive understanding of physiologically relevant MMP interactions. For example, a recent cell-based proteomic screen has revealed novel substrates for MMP2, including cystatin C (resulting in decreased inhibition of cathepsin L) [41], and proteases such as cathepsins B and L [42]. As these proteomic approaches are expanded to other proteases, the wealth of information generated will provide a more complete picture of the proteolytic interactions in the tumor microenvironment. Strategies such as these are currently of utmost importance as much of the available proteolytic interaction data has been determined through in vitro assays, making the complete in vivo picture of the proteolytic network difficult to assemble. These wide-ranging unbiased screens will allow for a much more detailed and accurate analysis of the dynamic nature of the proteome in the tumor microenvironment (reviewed in [43]).
Fitting nodes into cascades: who's on top?
The interactions of caspases in a cascade leading to cell death has been established for some time, and will not be discussed further here reference(see [3] for a more extensive review of caspases). Even though some proteases with pro-tumor functions can enhance the caspase cascade, the overall cascade is still independent from the tumor-promoting interactions described above.
The major tumor-promoting roles ascribed to proteases in the tumor microenvironment usually include migration, invasion, angiogenesis, modulation of signaling pathways, and metastasis. Genetic, pharmaceutical, and molecular studies have implicated many proteases in each tumor-promoting process, both individually and as larger groups, indicating that proteases either have high levels of redundancy in terms of substrates and activity, or that proteases function in a coordinated manner and that perturbation of one part of the cascade can impact the overall network. For example, several proteases, including cathepsins B, H, L, S, uPA (or its receptor), MMP2, MMP14, and MMP26 have been implicated in tumor angiogenesis and invasion [44-48], indicating the importance of exploring approaches to simultaneously target multiple proteases and underscoring the potential of blocking multiple nodes in the proteolytic network.
Combining in vivo experiments with the abundance of proteolytic interactions identified from the available in vitro data suggests that proteases function in a coordinated cascade in cancer biology [18, 49, 50]. In contrast to a global network view, depicting proteolytic interactions solely as cascades requires placing some proteases earlier in the cascade than others; a task that is challenging due to the plethora of possible interactions. One possibility is to view initiation of the cascade as interactions between proteases that are likely intracellular, such as cathepsin D activating cathepsin B (Figure 1), and then have the cascade proceed from there. One major challenge to this view is the data showing that many cysteine cathepsins, including cathepsin B, are translocated to the cell surface and even secreted in many tumors, although translocated proteases could still be activated by interactions en route to the cell surface. Additionally, several “downstream” proteases have the ability to activate cathepsin B themselves, making cathepsin B an imperfect candidate for being placed early in the chain of events. Recent data has provided evidence that a protease, in this case neutrophil elastase, is secreted by one cell type (neutrophils) and taken up intracellularly by another cell type (tumor cells), suggesting that proteases that are typically intracellular could still interact with secreted proteases [51].
Another possibility is to give prominence to proteases that have the ability to activate a wide range of other proteases, such as uPA (Figure 1). However, uPA can also be activated by proteases that are later in the cascade, including cathepsin B and plasmin. Additionally, none of these perspectives take into account proteases that can self-activate, such as furin [52] and cathepsin B (Figure 1). Self-activating proteases can provide novel entry points to the cascade and make it even more difficult to assign “upstream” proteases.
A final complication to the cascade view for proteolytic interactions is that some proteases have been found to have tumor inhibiting roles. In addition to caspases, these proteases include cysteine cathepsins, serine proteases, and MMPs (reviewed in [53]). These tumor-suppressing roles are context dependent; in different environments certain proteases can function to either inhibit or promote tumor progression. For example, loss of cathepsin L dramatically impedes pancreatic islet cell carcinogenesis [47], but enhances epidermal tumor progression [54]. This context-specificity extends to other classes of proteases, as several MMPs that can impair tumor progression, such as MMPs 3, 9, and 11 [53], also have tumor-promoting effects in other cases [29]. The ability of certain proteases to inhibit tumor progression in specific environments will need to be considered very carefully as researchers determine which proteases to target as anti-cancer therapies.
Viewing proteolytic interactions in networks provides insight into in vivo results
Given the difficulty in finding a consistent entry point to a unified cascade, an alternative is uniting the smaller cascades discussed earlier. This provides a more accurate view of proteolytic interactions in the form of a network of small, interconnected proteolytic cascades (Figure 1). This perspective allows for constant modulation of proteolytic activity by any number of proteases, with up- or downregulation of the activity of one protease potentially affecting the overall activity of the whole network [55]. The network view also allows for compensation by one protease for the loss of another, as demonstrated by the upregulation of cathepsin Z (also known as cathepsin X) in response to loss of cathepsin B in a mouse model of breast cancer [56, 57], allowing for the continuous flow of signals through the network in the absence of certain proteases.
Further interactions in the network come through indirect interactions, often as a result of one protease cleaving and inactivating the inhibitor of another protease (Figure 2b), or by proteases modulating the activity and availability of a signaling factor that can affect the abundance of another protease (Figure 3). These interactions enable proteases to indirectly increase the activity of other proteases without physically interacting with them. Conversely, as evidenced by the ability of several MMPs to degrade active plasmin [29] (Figure 2b), some proteases can directly impact other proteases in certain conditions. In normal physiology, this can serve to control and prevent over-activation of proteolytic pathways; the extent to which this occurs in the tumor microenvironment is still unclear but it could potentially serve to inactivate tumor-suppressing proteases.
Figure 3.
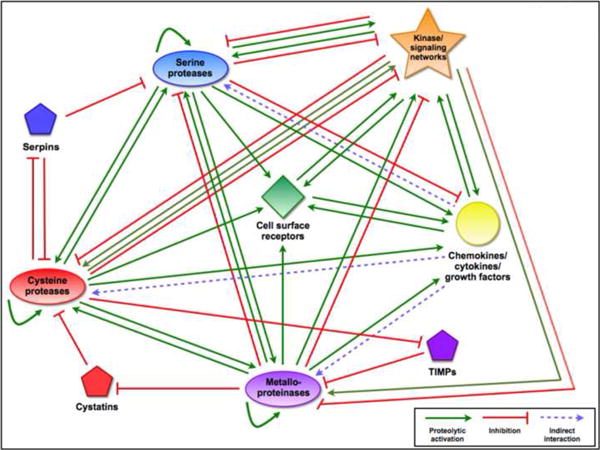
Interactions between the proteolytic network and other pathways
In addition to interactions with other proteases and endogenous inhibitors, proteases can modulate the activity of kinase signaling networks, cell surface receptors, and signaling molecules such as chemokines, cytokines, and growth factors. Depending on the protease and target protein, these can take the form of activating or degrading cleavages, allowing the proteolytic network to control the flow of signals in tumor cells as well as between tumor cells and the reactive stroma. Interactions are often bidirectional, as kinases also regulate many proteases via phosphorylation, and signaling molecules can indirectly (via signal transduction pathways) upregulate proteases in response to changes in the tumor microenvironment.
An additional level of indirect interactions in the proteolytic network is through the ability of proteases to cleave and either activate or inactivate signaling pathways and kinase cascades (Box 3) in the tumor microenvironment. This is a growing field of tumor biology, as an increasing number of growth factors, cytokines, and chemokines are found to be activated (and inactivated) via proteolytic cleavage. A final level of complexity to the network in the tumor microenvironment comes from stromal contributions to the proteolytic network (Box 2).
Box 3.
One rapidly growing area of protease research is at the interface between proteases and growth factors, cytokines, and signaling molecules. Kinases and phosphatases have long been understood to interact in regulatory networks, such as those proposed for proteases and their endogenous inhibitors. Interestingly, these two networks are intertwined at several points, as phosphorylation can regulate the activity of many proteases, and proteases can control the actions of a multitude of kinases. The interplay between kinases and proteases was the subject of a recent review [82], and only a few notable examples will be briefly highlighted here. One prominent family of proteases, the ADAMs (a disintegrin and metalloproteinase) can both regulate, and be regulated by, kinase activity. For example, ADAM10 has recently been shown to cause shedding of platelet-derived growth factor receptor (PDGFR). Additionally, PDGFR can stimulate the activity of ADAM17, although the mechanisms leading to these interactions remain to be clearly elucidated [83]. Other ADAM-kinase interactions include ADAM17-induced shedding of VEGFR2, creating soluble VEGF decoys that can impair metastasis, phosphorylation of ADAM9 by PKC and SRC, leading to ADAM9-mediated shedding of other receptors; and ADAM12-induced activation of SEC, YES1, PI3K, and PKC, which then phosphorylate ADAM12, leading to its cell-surface translocation [82]. Deubiquitylases, which remove ubiquitin from proteins, are also notably regulated via phosphorylation. Several components of the ubiquitin pathway, including ubiquitin-specific proteases (a subset of deubiquitinating enzymes) are substrates of ATM and ATR [82], providing tight control over DNA damage response pathways. A final player in the interactions between proteolytic regulation and kinases is the proteasome, although the implications of phosphorylation on proteasome activity are not fully understood. Both core and regulatory subunits of the proteasome can be phosphorylated, possibly affecting the assembly or activity of the proteasome [84]. The proteasome is also essential for the terminal degradation of many proteins in response to changes in their phosphorylation status, including several proteins implicated in tumor progression, such as cyclin-dependent kinases, MAP kinases, and AKT. Further investigation of the interactions between proteases and kinases may lead to therapeutic strategies targeting proteins in both classes of enzymes, thus allowing broader efficacy of inhibitors.
The proteases involved in interactions with cytokines are as varied as the substrates themselves, and include cathepsin G producing soluble RANK ligand, promoting osteoclast differentiation and bone resorption [85]; MMP9 activating TGF-R in bone metastasis (following cathepsin G-mediated activation of MMP9) [33]; and plasmin cleaving IL-8 and CCL14, both leading to increased chemotaxis [86], among many others (reviewed in [87]). Activation of signaling molecules by proteases can lead to feedback loops through which chemokines, cytokines, and growth factors upregulate protease expression, as demonstrated by the ability of IL-8 to upregulate MMP2 and MMP9 in bladder cancer cells [88], and the induction of cathepsin activity in tumor-associated macrophages by IL-4 [24]. Conversely, the serine protease HtrA1 can negatively regulate TGF-R signaling, by cleaving and inactivating TGF-R1 [89]. These inactivating interactions (and many others) show that proteases can potentially regulate both sides of cell signaling in the tumor microenvironment, and underscore the complexity of the proteolytic and signaling networks in cancer.
Conclusions
Our understanding of the proteolytic network in the tumor microenvironment is rapidly expanding as a result of an increased interest in protease biology and new techniques that allow for comprehensive analysis of protease activity in physiologically relevant conditions. Much like well-established kinase signaling networks, proteolytic networks consist of multiple points of entry/activation, several key nodes through which a majority of signals pass, and inhibitors/deactivators that can regulate activity of several different points in the network. The examples of protease cascades and networks illustrated in this review are not intended to be exhaustive views of all possible interactions in ‘proteolytic space’, but rather representative of recently discovered interactions that may be important in cancer. Indeed, there are still many proteases that are not currently connected to the network, but as our understanding of protease biology increases, proteases that previously were thought to act alone will likely be connected to the larger overall web. Additionally, increased understanding of how proteolytic interactions impact signaling molecules, ligand receptors, and kinase networks (Figure 3) will help direct future research into how tumor cells signal to and interact with each other.
As cancer treatment advances to more specific agents with less systemic toxicity, proteases are ideal therapeutic targets due to their frequent overexpression in many cancers, their involvement in many different stages of tumor progression, and their accessible active sites. Understanding how inhibiting one protease can affect the overall proteolytic balance of the tumor microenvironment is critical to enable researchers to design agents that target proteases with maximal impact and minimal toxicity. Inhibition of multiple proteases will likely prove more effective than targeting any individual protease, and determining how proteases are positioned relative to each other within the network will help guide effective targeting strategies. Future research should thus seek to view proteases as part of a complex network in order to achieve the goal of effective targeting of proteases in cancer.
Figure I. Proteolytic contributions from cells in the tumor microenvironment.
Acknowledgments
We would like to thank members of the Joyce lab for discussion on this topic, and Dr. Lisa Sevenich for critical review of the manuscript. Research in the Joyce laboratory is supported by the National Cancer Institute, the Breast Cancer Research Foundation, and the Geoffrey Beene Foundation. SM was supported by a Ruth Kirschstein National Research Service Award from the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Quesada V, et al. The Degradome database: mammalian proteases and diseases of proteolysis. Nucleic Acids Res. 2009;37(Database issue):D239–43. doi: 10.1093/nar/gkn570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9(4):239–52. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27(48):6194–206. doi: 10.1038/onc.2008.297. [DOI] [PubMed] [Google Scholar]
- 4.Turk B, Turk D, Salvesen GS. Regulating cysteine protease activity: essential role of protease inhibitors as guardians and regulators. Curr Pharm Des. 2002;8(18):1623–37. doi: 10.2174/1381612023394124. [DOI] [PubMed] [Google Scholar]
- 5.Hedrich J, et al. Fetuin-A and Cystatin C Are Endogenous Inhibitors of Human Meprin Metalloproteases. Biochemistry. 2010 doi: 10.1021/bi1004238. [DOI] [PubMed] [Google Scholar]
- 6.Lopez-Otin C, Overall CM. Protease degradomics: a new challenge for proteomics. Nat Rev Mol Cell Biol. 2002;3(7):509–19. doi: 10.1038/nrm858. [DOI] [PubMed] [Google Scholar]
- 7.Overall CM, Dean RA. Degradomics: systems biology of the protease web. Pleiotropic roles of MMPs in cancer. Cancer Metastasis Rev. 2006;25(1):69–75. doi: 10.1007/s10555-006-7890-0. [DOI] [PubMed] [Google Scholar]
- 8.Siegel RM. Caspases at the crossroads of immune-cell life and death. Nat Rev Immunol. 2006;6(4):308–17. doi: 10.1038/nri1809. [DOI] [PubMed] [Google Scholar]
- 9.Varfolomeev EE, et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9(2):267–76. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- 10.Stupack DG, et al. Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature. 2006;439(7072):95–9. doi: 10.1038/nature04323. [DOI] [PubMed] [Google Scholar]
- 11.Lord SJ, et al. Granzyme B: a natural born killer. Immunol Rev. 2003;193:31–8. doi: 10.1034/j.1600-065x.2003.00044.x. [DOI] [PubMed] [Google Scholar]
- 12.Dahl SW, et al. Human recombinant pro-dipeptidyl peptidase I (cathepsin C) can be activated by cathepsins L and S but not by autocatalytic processing. Biochemistry. 2001;40(6):1671–8. doi: 10.1021/bi001693z. [DOI] [PubMed] [Google Scholar]
- 13.D'Angelo ME, et al. Cathepsin H is an additional convertase of pro-granzyme B. J Biol Chem. 2010;285(27):20514–9. doi: 10.1074/jbc.M109.094573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Droga-Mazovec G, et al. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J Biol Chem. 2008;283(27):19140–50. doi: 10.1074/jbc.M802513200. [DOI] [PubMed] [Google Scholar]
- 15.Sloane BF, et al. Cathepsin B and tumor proteolysis: contribution of the tumor microenvironment. Semin Cancer Biol. 2005;15(2):149–57. doi: 10.1016/j.semcancer.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 16.van der Stappen JW, et al. Activation of cathepsin B, secreted by a colorectal cancer cell line requires low pH and is mediated by cathepsin D. Int J Cancer. 1996;67(4):547–54. doi: 10.1002/(SICI)1097-0215(19960807)67:4<547::AID-IJC14>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 17.Abboud-Jarrous G, et al. Cathepsin L Is Responsible for Processing and Activation of Proheparanase through Multiple Cleavages of a Linker Segment. J Biol Chem. 2008;283(26):18167–76. doi: 10.1074/jbc.M801327200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skrzydlewska E, et al. Proteolytic-antiproteolytic balance and its regulation in carcinogenesis. World J Gastroenterol. 2005;11(9):1251–66. doi: 10.3748/wjg.v11.i9.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Methot N, et al. Inhibition of the activation of multiple serine proteases with a cathepsin C inhibitor requires sustained exposure to prevent pro-enzyme processing. J Biol Chem. 2007;282(29):20836–46. doi: 10.1074/jbc.M702615200. [DOI] [PubMed] [Google Scholar]
- 20.Caglic D, et al. Glycosaminoglycans facilitate procathepsin B activation through disruption of propeptide-mature enzyme interactions. J Biol Chem. 2007;282(45):33076–85. doi: 10.1074/jbc.M705761200. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi H, et al. Cathepsin B efficiently activates the soluble and the tumor cell receptor-bound form of the proenzyme urokinase-type plasminogen activator (Pro-uPA) J Biol Chem. 1991;266(8):5147–52. [PubMed] [Google Scholar]
- 22.Mohamed MM, et al. Interleukin-6 increases expression and secretion of cathepsin B by breast tumor-associated monocytes. Cell Physiol Biochem. 2010;25(2–3):315–24. doi: 10.1159/000276564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cavallo-Medved D, et al. Mutant K-ras regulates cathepsin B localization on the surface of human colorectal carcinoma cells. Neoplasia. 2003;5(6):507–19. doi: 10.1016/s1476-5586(03)80035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gocheva V, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010;24(3):241–55. doi: 10.1101/gad.1874010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Behrendt N, Ronne E, Dano K. The structure and function of the urokinase receptor, a membrane protein governing plasminogen activation on the cell surface. Biol Chem Hoppe Seyler. 1995;376(5):269–79. [PubMed] [Google Scholar]
- 26.Sotiropoulou G, Pampalakis G, Diamandis EP. Functional roles of human kallikrein-related peptidases. J Biol Chem. 2009;284(48):32989–94. doi: 10.1074/jbc.R109.027946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He Y, et al. Interaction between cancer cells and stromal fibroblasts is required for activation of the uPAR-uPA-MMP-2 cascade in pancreatic cancer metastasis. Clin Cancer Res. 2007;13(11):3115–24. doi: 10.1158/1078-0432.CCR-06-2088. [DOI] [PubMed] [Google Scholar]
- 28.Zhou HM, et al. Urokinase-type plasminogen activator and its receptor synergize to promote pathogenic proteolysis. Embo J. 2000;19(17):4817–26. doi: 10.1093/emboj/19.17.4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Overall CM, Kleifeld O. Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006;6(3):227–39. doi: 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- 31.Ra HJ, Parks WC. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 2007;26(8):587–96. doi: 10.1016/j.matbio.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramos-DeSimone N, et al. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J Biol Chem. 1999;274(19):13066–76. doi: 10.1074/jbc.274.19.13066. [DOI] [PubMed] [Google Scholar]
- 33.Wilson TJ, Nannuru KC, Singh RK. Cathepsin G-mediated activation of pro-matrix metalloproteinase 9 at the tumor-bone interface promotes transforming growth factor-beta signaling and bone destruction. Mol Cancer Res. 2009;7(8):1224–33. doi: 10.1158/1541-7786.MCR-09-0028. [DOI] [PubMed] [Google Scholar]
- 34.Han YP, Yan C, Garner WL. Proteolytic activation of matrix metalloproteinase-9 in skin wound healing is inhibited by alpha-1-antichymotrypsin. J Invest Dermatol. 2008;128(9):2334–42. doi: 10.1038/jid.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makowski GS, Ramsby ML. Autoactivation profiles of calcium-dependent matrix metalloproteinase-2 and -9 in inflammatory synovial fluid: effect of pyrophosphate and bisphosphonates. Clin Chim Acta. 2005;358(1–2):182–91. doi: 10.1016/j.cccn.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 36.Gu Z, et al. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297(5584):1186–90. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- 37.Tchougounova E, et al. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J Biol Chem. 2005;280(10):9291–6. doi: 10.1074/jbc.M410396200. [DOI] [PubMed] [Google Scholar]
- 38.Littlepage LE, et al. Matrix metalloproteinases contribute distinct roles in neuroendocrine prostate carcinogenesis, metastasis, and angiogenesis progression. Cancer Res. 2010;70(6):2224–34. doi: 10.1158/0008-5472.CAN-09-3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rowe RG, Weiss SJ. Breaching the basement membrane: who, when and how? Trends Cell Biol. 2008;18(11):560–74. doi: 10.1016/j.tcb.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 40.Auf dem Keller U, et al. A statistics based platform for quantitative N-terminome analysis and identification of protease cleavage products. Mol Cell Proteomics. 2010 doi: 10.1074/mcp.M000032-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dean RA, et al. Identification of candidate angiogenic inhibitors processed by matrix metalloproteinase 2 (MMP-2) in cell-based proteomic screens: disruption of vascular endothelial growth factor (VEGF)/heparin affin regulatory peptide (pleiotrophin) and VEGF/Connective tissue growth factor angiogenic inhibitory complexes by MMP-2 proteolysis. Mol Cell Biol. 2007;27(24):8454–65. doi: 10.1128/MCB.00821-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kleifeld O, et al. Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nat Biotechnol. 2010;28(3):281–8. doi: 10.1038/nbt.1611. [DOI] [PubMed] [Google Scholar]
- 43.Doucet A, et al. Quantitative degradomics analysis of proteolytic post-translational modifications of the cancer proteome. Mol Cell Proteomics. 2008 doi: 10.1074/mcp.R800012-MCP200. [DOI] [PubMed] [Google Scholar]
- 44.Deng Y, et al. Expression of Matrix Metalloproteinase-26 promotes human glioma U251 cell invasion in vitro and in vivo. Oncol Rep. 2010;23(1):69–78. [PubMed] [Google Scholar]
- 45.Devy L, et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009;69(4):1517–26. doi: 10.1158/0008-5472.CAN-08-3255. [DOI] [PubMed] [Google Scholar]
- 46.Gocheva V, et al. Deletion of cathepsin H perturbs angiogenic switching, vascularization and growth of tumors in a mouse model of pancreatic islet cell cancer. Biol Chem. 2010;391(8):937–45. doi: 10.1515/BC.2010.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gocheva V, et al. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006;20(5):543–56. doi: 10.1101/gad.1407406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woenne EC, et al. MMP inhibition blocks fibroblast-dependent skin cancer invasion, reduces vascularization and alters VEGF-A and PDGF-BB expression. Anticancer Res. 2010;30(3):703–11. [PubMed] [Google Scholar]
- 49.Ovaere P, et al. The emerging roles of serine protease cascades in the epidermis. Trends Biochem Sci. 2009;34(9):453–63. doi: 10.1016/j.tibs.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 50.Affara NI, Andreu P, Coussens LM. Delineating protease functions during cancer development. Methods Mol Biol. 2009;539:1–32. doi: 10.1007/978-1-60327-003-8_1. [DOI] [PubMed] [Google Scholar]
- 51.Houghton AM, et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med. 2010;16(2):219–23. doi: 10.1038/nm.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anderson ED, et al. Activation of the furin endoprotease is a multiple-step process: requirements for acidification and internal propeptide cleavage. EMBO J. 1997;16(7):1508–18. doi: 10.1093/emboj/16.7.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lopez-Otin C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 2007;7(10):800–8. doi: 10.1038/nrc2228. [DOI] [PubMed] [Google Scholar]
- 54.Dennemarker J, et al. Deficiency for the cysteine protease cathepsin L promotes tumor progression in mouse epidermis. Oncogene. 2010;29(11):1611–21. doi: 10.1038/onc.2009.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morrison CJ, et al. Matrix metalloproteinase proteomics: substrates, targets, and therapy. Curr Opin Cell Biol. 2009;21(5):645–53. doi: 10.1016/j.ceb.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 56.Sevenich L, et al. Synergistic antitumor effects of combined cathepsin B and cathepsin Z deficiencies on breast cancer progression and metastasis in mice. Proc Natl Acad Sci U S A. 2010;107(6):2497–502. doi: 10.1073/pnas.0907240107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vasiljeva O, et al. Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res. 2006;66(10):5242–50. doi: 10.1158/0008-5472.CAN-05-4463. [DOI] [PubMed] [Google Scholar]
- 58.Caravita T, et al. Bortezomib: efficacy comparisons in solid tumors and hematologic malignancies. Nat Clin Pract Oncol. 2006;3(7):374–87. doi: 10.1038/ncponc0555. [DOI] [PubMed] [Google Scholar]
- 59.Dick LR, Fleming PE. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today. 2010 doi: 10.1016/j.drudis.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 60.Palermo C, Joyce JA. Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharmacol Sci. 2008;29(1):22–8. doi: 10.1016/j.tips.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 61.Bruning A, et al. New prospects for nelfinavir in non-HIV-related diseases. Curr Mol Pharmacol. 2010;3(2):91–7. doi: 10.2174/1874467211003020091. [DOI] [PubMed] [Google Scholar]
- 62.Sgadari C, et al. HIV protease inhibitors are potent anti-angiogenic molecules and promote regression of Kaposi sarcoma. Nat Med. 2002;8(3):225–32. doi: 10.1038/nm0302-225. [DOI] [PubMed] [Google Scholar]
- 63.Wahl ML, et al. Angiostatin's molecular mechanism: aspects of specificity and regulation elucidated. J Cell Biochem. 2005;96(2):242–61. doi: 10.1002/jcb.20480. [DOI] [PubMed] [Google Scholar]
- 64.Joyce JA, et al. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell. 2004;5(5):443–53. doi: 10.1016/s1535-6108(04)00111-4. [DOI] [PubMed] [Google Scholar]
- 65.Coussens LM, et al. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell. 2000;103(3):481–90. doi: 10.1016/s0092-8674(00)00139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest. 2004;114(5):623–33. doi: 10.1172/JCI22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer. 2006;6(10):764–75. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- 68.Hofmeister V, Schrama D, Becker JC. Anti-cancer therapies targeting the tumor stroma. Cancer Immunol Immunother. 2008;57(1):1–17. doi: 10.1007/s00262-007-0365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.O'Connell PA, et al. S100A10 regulates plasminogen-dependent macrophage invasion. Blood. 2010;116(7):1136–46. doi: 10.1182/blood-2010-01-264754. [DOI] [PubMed] [Google Scholar]
- 70.Hamilton G, et al. Cystatin F is a cathepsin C-directed protease inhibitor regulated by proteolysis. Embo J. 2008;27(3):499–508. doi: 10.1038/sj.emboj.7601979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Conus S, et al. Caspase-8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J Exp Med. 2008;205(3):685–98. doi: 10.1084/jem.20072152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heutinck KM, et al. Serine proteases of the human immune system in health and disease. Mol Immunol. 2010;47(11–12):1943–55. doi: 10.1016/j.molimm.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 73.Kominami E, et al. Distribution of cathepsins B and H in rat tissues and peripheral blood cells. J Biochem. 1985;98(1):87–93. doi: 10.1093/oxfordjournals.jbchem.a135277. [DOI] [PubMed] [Google Scholar]
- 74.Bidere N, et al. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem. 2003;278(33):31401–11. doi: 10.1074/jbc.M301911200. [DOI] [PubMed] [Google Scholar]
- 75.Liaudet-Coopman E, et al. Cathepsin D: newly discovered functions of a long-standing aspartic protease in cancer and apoptosis. Cancer Lett. 2006;237(2):167–79. doi: 10.1016/j.canlet.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 76.Haendeler J, et al. Cathepsin D and H2O2 stimulate degradation of thioredoxin-1: implication for endothelial cell apoptosis. J Biol Chem. 2005;280(52):42945–51. doi: 10.1074/jbc.M506985200. [DOI] [PubMed] [Google Scholar]
- 77.van Hinsbergh VW, Engelse MA, Quax PH. Pericellular proteases in angiogenesis and vasculogenesis. Arterioscler Thromb Vasc Biol. 2006;26(4):716–28. doi: 10.1161/01.ATV.0000209518.58252.17. [DOI] [PubMed] [Google Scholar]
- 78.Coffelt SB, et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. 2010;70(13):5270–80. doi: 10.1158/0008-5472.CAN-10-0012. [DOI] [PubMed] [Google Scholar]
- 79.Li G, et al. Comparative proteomic analysis of mesenchymal stem cells derived from human bone marrow, umbilical cord, and placenta: implication in the migration. Proteomics. 2009;9(1):20–30. doi: 10.1002/pmic.200701195. [DOI] [PubMed] [Google Scholar]
- 80.Silva WA, Jr, et al. The profile of gene expression of human marrow mesenchymal stem cells. Stem Cells. 2003;21(6):661–9. doi: 10.1634/stemcells.21-6-661. [DOI] [PubMed] [Google Scholar]
- 81.Lilla JN, Werb Z. Mast cells contribute to the stromal microenvironment in mammary gland branching morphogenesis. Dev Biol. 2010;337(1):124–33. doi: 10.1016/j.ydbio.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lopez-Otin C, Hunter T. The regulatory crosstalk between kinases and proteases in cancer. Nat Rev Cancer. 2010;10(4):278–92. doi: 10.1038/nrc2823. [DOI] [PubMed] [Google Scholar]
- 83.Mendelson K, et al. Stimulation of platelet-derived growth factor receptor beta (PDGFRbeta) activates ADAM17 and promotes metalloproteinase-dependent cross-talk between the PDGFRbeta and epidermal growth factor receptor (EGFR) signaling pathways. J Biol Chem. 2010;285(32):25024–32. doi: 10.1074/jbc.M110.102566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang F, et al. Metabolic control of proteasome function. Physiology (Bethesda) 2007;22:373–9. doi: 10.1152/physiol.00026.2007. [DOI] [PubMed] [Google Scholar]
- 85.Wilson TJ, et al. Cathepsin G enhances mammary tumor-induced osteolysis by generating soluble receptor activator of nuclear factor-kappaB ligand. Cancer Res. 2008;68(14):5803–11. doi: 10.1158/0008-5472.CAN-07-5889. [DOI] [PubMed] [Google Scholar]
- 86.Van Damme J, Struyf S, Opdenakker G. Chemokine-protease interactions in cancer. Semin Cancer Biol. 2004;14(3):201–8. doi: 10.1016/j.semcancer.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 87.Mortier A, Van Damme J, Proost P. Regulation of chemokine activity by posttranslational modification. Pharmacol Ther. 2008;120(2):197–217. doi: 10.1016/j.pharmthera.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 88.Mian BM, et al. Fully human anti-interleukin 8 antibody inhibits tumor growth in orthotopic bladder cancer xenografts via down-regulation of matrix metalloproteases and nuclear factor-kappaB. Clin Cancer Res. 2003;9(8):3167–75. [PubMed] [Google Scholar]
- 89.Launay S, et al. HtrA1-dependent proteolysis of TGF-beta controls both neuronal maturation and developmental survival. Cell Death Differ. 2008;15(9):1408–16. doi: 10.1038/cdd.2008.82. [DOI] [PubMed] [Google Scholar]
- 90.Sivaraman J, et al. Crystal structure of human procathepsin X: a cysteine protease with the proregion covalently linked to the active site cysteine. J Mol Biol. 2000;295(4):939–51. doi: 10.1006/jmbi.1999.3410. [DOI] [PubMed] [Google Scholar]
- 91.Dalet-Fumeron V, Boudjennah L, Pagano M. Competition between plasminogen and procathepsin B as a probe to demonstrate the in vitro activation of procathepsin B by the tissue plasminogen activator. Arch Biochem Biophys. 1996;335(2):351–7. doi: 10.1006/abbi.1996.0516. [DOI] [PubMed] [Google Scholar]
- 92.Wang Z, Juttermann R, Soloway PD. TIMP-2 is required for efficient activation of proMMP-2 in vivo. J Biol Chem. 2000;275(34):26411–5. doi: 10.1074/jbc.M001270200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Knauper V, et al. Cellular mechanisms for human procollagenase-3 (MMP-13) activation. Evidence that MT1-MMP (MMP-14) and gelatinase a (MMP-2) are able to generate active enzyme. J Biol Chem. 1996;271(29):17124–31. doi: 10.1074/jbc.271.29.17124. [DOI] [PubMed] [Google Scholar]
- 94.Holopainen JM, et al. Activation of matrix metalloproteinase-8 by membrane type 1-MMP and their expression in human tears after photorefractive keratectomy. Invest Ophthalmol Vis Sci. 2003;44(6):2550–6. doi: 10.1167/iovs.02-1190. [DOI] [PubMed] [Google Scholar]
- 95.Kostoulas G, et al. Stimulation of angiogenesis through cathepsin B inactivation of the tissue inhibitors of matrix metalloproteinases. FEBS Lett. 1999;455(3):286–90. doi: 10.1016/s0014-5793(99)00897-2. [DOI] [PubMed] [Google Scholar]
- 96.Taggart CC, et al. Cathepsin B, L, and S cleave and inactivate secretory leucoprotease inhibitor. J Biol Chem. 2001;276(36):33345–52. doi: 10.1074/jbc.M103220200. [DOI] [PubMed] [Google Scholar]
- 97.Welss T, et al. Hurpin is a selective inhibitor of lysosomal cathepsin L and protects keratinocytes from ultraviolet-induced apoptosis. Biochemistry. 2003;42(24):7381–9. doi: 10.1021/bi027307q. [DOI] [PubMed] [Google Scholar]
- 98.Higgins WJ, et al. Heparin enhances serpin inhibition of the cysteine protease cathepsin L. J Biol Chem. 2010;285(6):3722–9. doi: 10.1074/jbc.M109.037358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Johnson DA, Barrett AJ, Mason RW. Cathepsin L inactivates alpha 1-proteinase inhibitor by cleavage in the reactive site region. J Biol Chem. 1986;261(31):14748–51. [PubMed] [Google Scholar]