

Abstract
Ethionamide (ETA) and pyrazinamide (PZA) are considered the drugs of choice for the treatment of multidrug-resistant tuberculosis. Current methods available in the literature for simultaneous determination of ETA and PZA have low sensitivity or involve column modifications with lipophilic cations. The aim of this study was to develop a simple and validated reversed-phase ion-pair HPLC method for simultaneous determination of ETA and PZA for the characterization of polymeric-based porous inhalable microparticles in in vitro and spiked human serum samples. Chromatographic separation was achieved on a Phenomenex C18 column (250 mm × 4.6 mm) using a Shimadzu LC 10 series HPLC. The mobile phase consisted of A: 0.01% trifluoroacetic acid in distilled water and B: ACN/MeOH at 1:1 v/v. Gradient elution was run at a flow rate of 1.5 mL/min and a fixed UV wavelength of 280 nm. The validation characteristics included accuracy, precision, linearity, analytical range, and specificity. Calibration curves at seven levels for ETA and PZA were linear in the analytical range of 0.1–3.0 μg/mL with correlation coefficient of r2 > 0.999. Accuracy for both ETA and PZA ranged from 94 to 106% at all quality control (QC) standards. The method was precise with relative standard deviation less than 2% at all QC levels. Limits of quantitation for ETA and PZA were 50 and 70 ng/mL, respectively. There was no interference from either the polymeric matrix ions or the biological matrix in the analysis of ETA and PZA.
Key words: ethionamide, HPLC, microparticles, pyrazinamide, tuberculosis
INTRODUCTION
Tuberculosis (TB) is one of the most frequent and important infectious diseases that cause morbidity and death (1,2). Current clinical treatment for drug-susceptible tuberculosis which ranges from 6–12 months includes frontline drugs such as rifampicin (RFP), isoniazid (INH), pyrazinamide (PZA), ethambutol, streptomycin, and p-amino salicylic acid. The drugs are initially given in a combination of three to four for a few months since the susceptibility of the organism is not certain, and subsequently, the dose is reduced. Resistance to one or more frontline drugs is a result of incomplete adherence to chemotherapy, errors in the case management, and HIV pandemic (3–5). Mycobacterium TB strains can be resistant to either isoniazid or rifampicin or both isoniazid and rifampicin. Strains that are resistant to both isoniazid and rifampicin are known as multidrug-resistant (MDR) strains and are the causative agents of MDR tuberculosis (6). MDR-TB treatment involves the use of second-line drugs that have limited efficacy and are not suitable for short-course treatment. MDR-TB patients in whom treatment failed have high risk of death (7–10). When the MDR-TB strain is resistant to INH and RFP, initial treatment is the combination of ethionamide (ETA), fluoroquinolone, another bacteriostatic drug such as ethambutol, PZA, and aminoglycoside (kanamycin, amikacin, or capreomycin) for 3 months or until sputum purulence conversion. The combination of ETA, ofloxacin, and another bacteriostatic drug (cycloserine) is recommended for at least 18 months during continuation phase (5,11). PZA, a frontline drug, is recommended for MDR-TB since it is effective in acidic medium (bacilli inside macrophages), and also, resistance to PZA is not easy to acquire nor prove (5). Inhaled therapy is the most promising for the treatment of tuberculosis. However, for an efficient bactericidal activity against the mycobacteria, drugs need to be encapsulated into a suitable drug delivery system to sustain their effects and ensure their rapid uptake into macrophages that harbor mycobacterium TB (12).
Microparticles are particulate systems with diameter ranges from 0.1 to 200 μm that can be formulated with biodegradable or nonbiodegradable polymers (13). Within the microparticles, the drug is uniformly dispersed throughout the polymeric matrix. Polymeric microspheres are widely used in drug delivery due to their ability to encapsulate a wide range of drugs, biocompatibility, high bioavailability, and sustained release characteristics (14). Poly (dl-lactide-co-glycolide) (PLGA) is widely employed for their preparation due to its biodegradation into compatible metabolite monomers, lactic acid and glycolic acid (15). Yang et al. formulated large porous PLGA microparticles with large geometric diameters and small aerodynamic diameters to avoid agglomeration and increase the efficiency of inhalable formulation of porous microparticles. Porous or hollow microparticles aggregate less and disaggregate more easily under shear forces which results in higher aerosolization efficiency (16). Regarding its potential benefits and compliance in anti-TB patients, porous inhalable PLGA microparticles were considered in this study as an efficient inhalable drug delivery system for a binary anti-TB therapy encompassing ETA and PZA. The proposed first part of the study was to develop an efficient chromatographic method for the simultaneous determination of both drugs in in vitro and spiked human serum samples. Another study is in progress that not only focuses on the application of quality by design in the formulation and optimization of their microparticles but also tests their clinical performance in volunteers. Scarce information was found in the literature regarding the simultaneous determination of both drugs. The published chromatographic methods for the quantitation of each drug were based on the mass spectroscopy which is expensive for the routine analysis (17,18).
Reversed-phase ion-pair chromatography with perfluorinated carboxylic acids as an ion-pair agent has been used for determination of amino acids and peptides (19–21). Ion-pairing agents such as trifluoroacetic acid (TFA) would increase the affinity of the positively charged solute for the hydrophobic stationary phase as opposed to phosphoric acid. It was reported that TFA could be used as a mobile phase stabilizer and an effective ion-pair reagent to control retention and selectivity (22). The aim of the current study was to develop a validated reversed-phase ion-pair chromatography method for the simultaneous analysis of two anti-TB drugs, namely ETA and PZA (Fig. 1), for the characterization of their porous PLGA microparticles in in vitro and spiked human serum samples. TFA was employed as an ion-pairing agent for the simultaneous separation and further elution of the two drugs.
Fig. 1.
Chemical structures of a ethionamide and b pyrazinamide
METHOD AND MATERIALS
Materials
Ethionamide, pyrazinamide, and perchloric acid were purchased from Sigma-Aldrich (MO, USA). HPLC-grade acetonitrile, methanol, tetrahydrofuran, dimethyl sulfoxide, and polyvinyl alcohol (PVA) (molecular weight (Mwt) of 40 kDa) were purchased from Fisher Scientific (NJ, USA). TFA and acetaminophen USP were purchased from EM Sciences (NJ, USA) and City Chemical Corporation (NJ, USA), respectively. PLGA 50:50 was purchased from Durect Corporation (AL, USA). Millipore™ ultrapure water was used for the preparation of mobile phase and sample working standards. Pooled drug-free human serum type AB was purchased from MP Biomedicals (OH, USA).
Instrumentation and Chromatographic Conditions
Shimadzu HPLC system equipped with dual LC-10ADvp pumps, DGU-14A degasser, SPD-10ADvp UV–vis detector, SIL-10ADvp autosampler, and SCL-10Avp system controller was used. Data were collected using Class VP v5.0 software. The mobile phase was composed of A: 0.01% TFA in water and B: ACN/MeOH (50:50). Gradient elution run was carried out on a Phenomenex Luna C18 column (250 mm × 4.6 mm, 5 μm) with Zorbax C18 (17 × 1 mm, 5 μm) guard column at a flow rate of 1.5 mL/min. The gradient run was started with 100% A and 0% B then 2% B at 1 min, 70% B at 16 min through 18 min, and 2% B at 20 min through 25 min (end of the run). The wavelength was fixed at 280 nm, and injection volume was 20 μL.
Preparation of Standard Solutions
Standard stock solutions of ETA, PZA, and acetaminophen (internal standard) were prepared in amber-colored volumetric flasks by dissolving 100 mg of each drug in 100 mL ACN/MeOH (50:50) with the aid of stirring for 10 min to obtain a final concentration of 1 mg/mL each. Calibration and system suitability standard solutions were prepared by diluting the above standard solution.
System Suitability Standard
System suitability standard solutions were prepared daily by diluting the stock solution with the mobile phase to obtain the working system suitability standard concentration of 1 μg/mL. System suitability was determined from six replicates of the working solution before sample analyses. The USP acceptance criteria was relative standard deviation (RSD) less than 2% for peak area, theoretical plates >3,000, USP tailing factor ≥0.5 and ≤2.0, and capacity factor (k′) greater than 2.0 (23).
Method Validation
Linearity and Range
Standard calibration curves were constructed with seven calibrators over a concentration range of 0.1–3.0 μg/mL for both ETA and PZA using acetaminophen as an internal standard (IS) at a concentration of 1.5 μg/mL. The analytical range was established by demonstrating the accuracy and precision at both the lowest and highest concentrations of the calibration range.
Accuracy and Precision
Accuracy and precision of the method were determined by analyzing the quality control (QC) standard samples at three concentrations of both ETA and PZA (0.1, 1.2, 3.0 μg/mL). The method precision was established by five replicate injections of three QC levels for the intraday precision and on three successive days for the intermediate precision. Precision and accuracy were expressed as RSD and percent recovery, respectively, at three standards for 3 days.
Limit of Quantitation and Limit of Detection
Concentration at the signal-to-noise ratio (SN) of 10:1 was the limit of quantitation (LOQ), and SN of 3:1 was the limit of detection (LOD), estimated from the following formula:
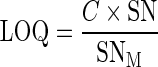 |
where C is the concentration being evaluated, SN is the signal-to-noise ratio of 10:1 for LOQ and 3:1 for LOD, and SNM equals H/ND, where SNM is the calculated signal-to-noise ratio, H is the peak height, and ND is the measured noise height.
Specificity
The ability of the developed method to separate both drugs from the in vitro samples during the development of porous microparticles was investigated. Porous microparticles were prepared by modification of the double emulsion method described by Yang et al. (16). Briefly, a specified amount of PLGA polymer (50:50) was dissolved in a mixture of dichloromethane and methanol as the organic phase. The internal aqueous phase was a solution of the effervescent salt ammonium bicarbonate (NH4HCO3) in distilled water. Two milliliters of the above solution was added dropwise to the organic phase followed by homogenization at 4,000 rpm for 6 min. The resultant w/o emulsion was added dropwise to 1% PVA solution while homogenizing at 5,000 rpm for 8 min. The double emulsion (w/o/w) formed was thus stirred at 900 rpm using a paddle stirrer (R1376, IKA Works Inc., NC, USA) for 3 h followed by centrifugation at 15,000 rpm for 30 min to harvest the formed microparticles. The porous microparticles were then washed with 10 mL of distilled water to remove the adsorbed free drug and PVA then dried under vacuum overnight. Drug loadings were performed by dissolving the specified amount of the two drugs in the primary aqueous phase. The matrix factor was determined by both extracting the two drugs from the formed microparticles and the free drug solutions after harvesting and washings. For efficient extraction of both drugs, the loaded porous microparticles were dissolved in 100 mL of tetrahydrofuran and dimethyl sulfoxide (1:1 v/v) solution by stirring followed by homogenization and vortexing. The above solution was spiked with IS. This solution was diluted to a system suitability working concentration of 1 μg/mL with the mobile phase. Matrix factor percent RSD was determined from six runs of microparticle drug extracts from the microparticles using the following equation:
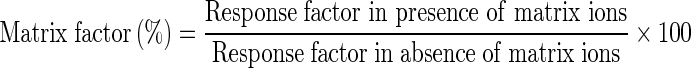 |
The matrix effect for the determination of both drugs in spiked human serum samples was also investigated. Sample preparation to determine the specificity of the method to quantitate both drugs in human serum was performed as described by Watabe et al., with some modifications (24). Briefly, 100 μL of 6% (w/v) perchloric acid was added to 200 μL of human serum spiked with ETA, PZA, and IS. This mixture was vortexed for 1 min followed by addition of 100 μL methanol. The mixture was then centrifuged at 17,000 rpm for 30 min, and the supernatant collected was diluted as required and used for specificity determination as described with the in vitro samples.
Robustness
Robustness examines the effect that operational parameters have on the analysis results (25). Robustness shows the reliability of an analysis with respect to deliberate variations in method parameters. To determine the robustness of the method, the elution parameters were varied individually by adjusting the mobile phase pH (±0.2), detector wavelength (±5 nm), organic phase percentage of the mobile phase (±5%), and HPLC pump flow rate (±0.1 mL/min).
RESULTS AND DISCUSSION
Method Development and Optimization
The available methods for the simultaneous determination of ETA and PZA have some limitations such as the high LOQ (26,27) and the use of lipophilic cations to modify the stationary phase that can deteriorate the HPLC column for other applications (28). The main objective of this study was to develop a simple reversed-phase ion-pair HPLC method for simultaneous analysis of both drugs from the in vitro and spiked human serum samples during characterization of their porous microparticles. Because PZA is more hydrophilic than ETA, gradient elution was preferred over isocratic to avoid the elution of PZA in the void volume or the peak shape deterioration for ETA. High baseline drift was a common problem with high-pressure binary gradient elution method. With a steep gradient, there was either an increase in baseline drift or a decrease in resolution between ETA and PZA. The gradient elution of 2% organic phase at 1 min to 70% organic phase at 16 min resulted in a lower baseline drift and improved resolution between the two compounds. Compared to o-phosphoric acid as an ion-pairing agent, TFA increased not only the resolution between compounds of interest but also the sensitivity to detect both ETA and PZA (Fig. 2, traces A and B). As indicated from the clear chromatogram after sample blank run, there was no significant carryover or overretention of the two compounds (Fig. 2, trace C). The mobile phase was prepared daily as mobile phase contamination was one of the primary reasons for the ghost peaks in gradient elution (29). It was observed that the incidence of ghost peaks decreased significantly with the use of fresh mobile phase. In addition, there was no interference from the ghost peaks in the analysis of compounds of interest (Fig. 3). The detection sensitivity of the developed method to ETA increased significantly by the addition of methanol to the organic phase. The optimum absorptivity was observed with a 1:1 v/v acetonitrile to methanol as the organic compartment of the mobile phase. It is worthy to note that increasing the methanol percentage in the organic phase more than 50% resulted in peak shape deterioration of both compounds.
Fig. 2.
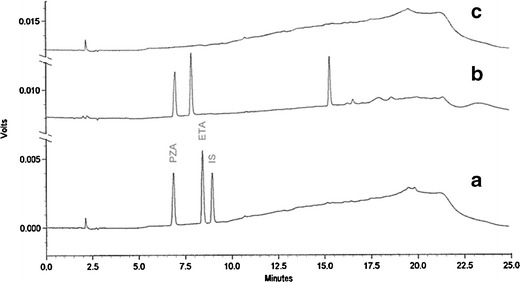
Chromatograms of A system suitability standard of 1 μg/mL ETA and PZA, B system suitability standard of 1 μg/mL ETA and PZA, and C blank run after sample analysis
Fig. 3.
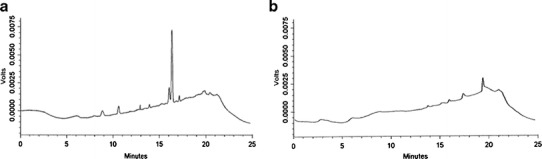
Chromatograms showing a the ghost peaks with a null injection (without injecting sample or blank) due to mobile phase contamination and b a decrease in ghost peaks by optimizing a fresh mobile phase to A: 0.01% TFA in distilled water and B: ACN/MeOH (50:50)
System Suitability
The system suitability test verifies that the resolution and reproducibility of the chromatographic system are adequate for the analysis to be conducted (30,31). Six replicates of system suitability standard 1 μg/mL of both PZA and ETA were injected for three successive days. All critical parameters tested (k′, peak area RSD, retention time RSD, column plates, tailing, etc.) met the USP acceptance criterion on all days (Table I). Area RSD was less than 2% on all 3 days for both drugs (Table I). Both ETA and PZA passed the USP tailing and capacity factor criteria with values <2 and >2, respectively. Adequate resolution of >3 between PZA and ETA peaks ensured the specificity of the method to analyze both compounds.
Table I.
System Suitability of 1 μg/mL of Both ETA and PZA (n = 6)
| Parameters | Specificationsa | Day 1 | Day 2 | Day 3 |
|---|---|---|---|---|
| ETA system suitability standard, 1 μg/mL | ||||
| Capacity factor (k′) | >2.0 | 3.22 | 3.21 | 3.21 |
| Capacity factor (RSD) | ≤2.0 | 0.067 | 0.067 | 0.053 |
| Area (% RSD) | ≤2.0 | 1.25 | 1.27 | 1.54 |
| Plates (column) | >3,000 | 35,236 | 34,456 | 35,962 |
| USP tailing | (0.5–2.0) | 1.11 | 1.11 | 1.11 |
| Pyrazinamide system suitability, 1 μg/mL | ||||
| Capacity factor (k′) | >2.0 | 2.46 | 2.42 | 2.43 |
| Capacity factor (RSD) | ≤2.0 | 0.093 | 0.14 | 0.094 |
| Area (% RSD) | ≤2.0 | 0.69 | 1.03 | 1.45 |
| Plates (column) | >3,000 | 12,982 | 13,037 | 16,995 |
| USP tailing | (0.5–2.0) | 0.87 | 0.90 | 1.02 |
Standard deviations of the listed values did not exceed 2%
ETA ethionamide, RSD relative standard deviation, USP United States Pharmacopeia
aAcceptance criteria per the USP 35/NF 30 specifications
Linearity and Analytical Range
Linearity of the method was established by preparing standard calibration curves over the analytical range of 0.1–3.0 μg/mL for both ETA and PZA (Fig. 4). This narrow but chromatographically necessary analytical range was first a result of ETA sensitivity where, below 0.1 μg/mL, sensitivity was lost, and secondly, above 3 μg/mL results in trace amounts of ETA sticking on the column, causing the excessive need to wash the column with mobile phase. The results in Table II show a linear correlation between analyte peak area and the concentration of the drug over the analytical range with R2 ≥ 0.999.
Fig. 4.
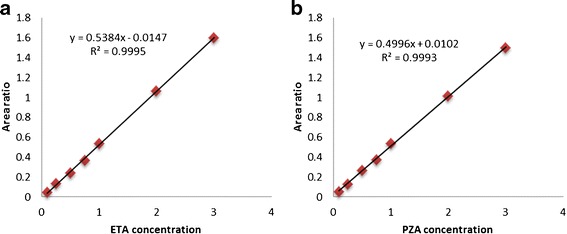
Calibration curves of a ETA and b PZA for analytical range of 0.1–3.0 μg/mL
Table II.
Parameters and Linearity Data of Both ETA and PZA Calibration Curves
| Standard curves | Range, μg/mL | Calibrators | Slope | Y-intercept | R 2 |
|---|---|---|---|---|---|
| ETA calibration | |||||
| Day 1 | 0.1–3.0 | 7 | 0.5384 | −0.0147 | 0.9995 |
| Day 2 | 0.1–3.0 | 7 | 0.5394 | −0.0134 | 0.9994 |
| Day 3 | 0.1–3.0 | 7 | 0.5391 | −0.0142 | 0.9995 |
| PZA calibration | |||||
| Day 1 | 0.1–3.0 | 7 | 0.4996 | 0.0102 | 0.9993 |
| Day 2 | 0.1–3.0 | 7 | 0.4989 | 0.0103 | 0.9995 |
| Day 3 | 0.1–3.0 | 7 | 0.4992 | 0.0104 | 0.9994 |
ETA ethionamide, PZA pyrazinamide
Accuracy and Precision
Accuracy and precision were established across the analytical range for both compounds. The intra-and interday accuracy and precision were calculated using multiple injections of the QC samples. Results for the intraday accuracy and precision of PZA and ETA are summarized in Tables III and IV, respectively. The accuracy across the analytical ranged from 94.6 to 107% for all QC samples. Despite being within the acceptance limits, the matrix effect resulted in accuracy of more than 100% in the higher QC samples.
Table III.
Accuracy of Both ETA and PZA (n = 5)
| Sample | 0.1 μg/mL | 1.2 μg/mL | 3.0 μg/mL |
|---|---|---|---|
| Ethionamide | |||
| Day 1 | 106.97% | 102.48% | 101.09% |
| Day 2 | 101.21% | 106.45% | 100.44% |
| Day 3 | 98.82% | 103.57% | 97.44% |
| Pyrazinamide | |||
| Day 1 | 104.71% | 100.43% | 99.11% |
| Day 2 | 106.45% | 104.67% | 99.66% |
| Day 3 | 94.61% | 102.42% | 97.44% |
Accuracy was expressed as the percent recovery of QC samples. Standard deviations of the listed values did not exceed 2%
Table IV.
Precision of Both ETA and PZA (Percent RSD, n = 5)
| Sample | 0.1 μg/mL | 1.2 μg/mL | 3.0 μg/mL |
|---|---|---|---|
| Ethionamide | |||
| Day 1 | 0.83 | 1.04 | 0.72 |
| Day 2 | 1.48 | 1.33 | 0.41 |
| Day 3 | 1.41 | 0.41 | 0.33 |
| Pyrazinamide | |||
| Day 1 | 0.39 | 0.92 | 1.08 |
| Day 2 | 1.55 | 1.17 | 0.97 |
| Day 3 | 1.76 | 1.12 | 1.11 |
Standard deviations of the listed values did not exceed 2%
Limit of Quantitation and Limit of Detection
LOQs determined by estimating signal-to-noise ratio of 10:1 for ETA and PZA were 50 and 70 ng/mL, respectively (Fig. 5). Area RSD for six runs of both the compounds at the LOQ concentrations was <3%. LODs determined by estimating signal-to-noise ratio of 3:1 for ETA and PZA were 15 and 21 ng/mL, respectively.
Fig. 5.
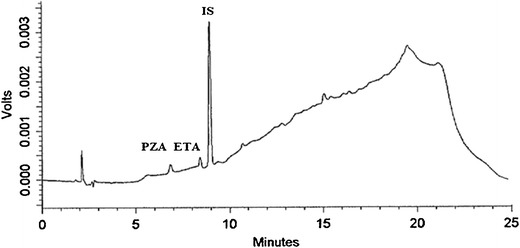
Chromatogram of simultaneous ETA and PZA elution at their LOQ concentrations
Specificity
Specificity was established by observing no peaks in the blank chromatograms and no coeluting peaks in the drugs’ loaded samples shown in Fig. 6. Additionally, the Shimadzu Class VP peak purity software was used to evaluate diode array spectral data of the chromatographic peaks and determined that no impurities were present or coeluting with both drugs. The validated method was used to assess its potential utility not only to characterize the porous microparticles loaded with both drugs but also to analyze spiked human serum samples for the determination of both drugs in human blood. Direct determination of the drugs’ loadings was done by extraction from their formulation in biodegradable PLGA-based microparticles. The extract was diluted with mobile phase and injected onto the HPLC using the described validated method. Representative chromatograms for ETA and PZA are shown in Fig. 6 (trace A and B). The chromatogram showed that there was no interference from the microparticle components with the peaks of both drugs. The mean matrix factors for six runs of the drugs’ extract from destruction of drug-loaded PLGA porous microparticles with ETA and PZA were 0.95 and 0.99, respectively. The matrix factor percent RSD for six runs of ETA and PZA were 1.28% and 1.21%, respectively. The drug-loaded blood sample was utilized to assess the potential of the analytical method to qualitatively separate both drugs from the biological matrix as a surrogate for evaluating the bioavailability of both drugs after extravascular administration. For this purpose, drug-loaded human serum was diluted with mobile phase and injected on the HPLC using the described method. Representative chromatograms for both drugs are shown in Fig. 6 (traces C and D). The chromatograms show that there was no coelution of both drugs with any of the biological matrix components. Moreover, the mean matrix factors for six runs of the drugs’ extract from human serum loaded with both ETA and PZA at concentrations of 1 μg/mL were 1.02 and 0.97 with RSD values of 1.02% and 2.27%, respectively. Further formulation screening in vitro and in vivo studies are in progress for optimizing the formulation parameters for higher drug loadings and bioavailability using the validated analytical method.
Fig. 6.
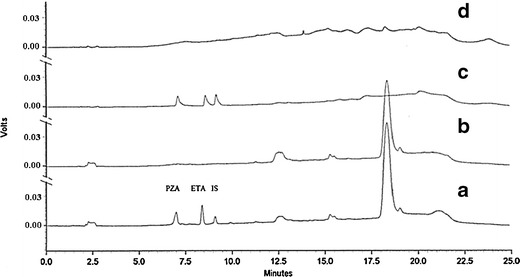
Chromatograms of A drugs’ extract from destruction of drug-loaded PLGA porous microparticles with ETA, PZA, and IS; B extract of drug-free PLGA porous microparticles; C drugs’ extract from human serum loaded with both ETA and PZA at a concentration of 1 μg/mL; and D extract of drug-free human serum
Robustness
The robustness of the method was evaluated by making small adjustments to the operating parameters of the system suitability procedure. The obtained data (not shown) indicated that the method could handle small deliberate changes to the operating conditions while maintaining its accuracy, precision, and reproducibility. All robustness data remained <5% RSD for each adjusted parameter with the exception of increased pH from nominal to high. For example, the capacity factor (k′) of ETA elution changed from 3.2 ± 0.06 to 3.1 ± 0.04 by varying the pH of the mobile phase from 2.8 to 3.2, respectively. Variability increased to 3.1% RSD due to sensitivity to increasing pH. As a result, none of the adjustments caused a significant change (p < 0.05) in resolution between ETA system suitability standard and PZA, peak area, theoretical plates, and/or USP tailing factor.
CONCLUSION
Insufficient therapeutic levels of ETA and PZA in patients diagnosed with MDR-TB can lead to death or cases of extensive drug-resistant tuberculosis. A validated analytical method would be helpful for the therapeutic drug monitoring and can be also applied for simultaneous pharmacokinetic studies of ETA and PZA. In this prospect, a simple and efficient gradient reverse-phase ion-pair HPLC method has been developed and validated according to USP category I. The proposed method was accurate, precise, sensitive, specific, and linear in the range specified. Moreover, it can be successfully used for determination of ETA and PZA from both the in vitro and spiked human serum samples of their porous PLGA microparticles and similar formulations with PLGA matrix.
References
- 1.Tomioka H, Tatano Y, Sano C, Shimizu T. Development of new antituberculous drugs based on bacterial virulence factors interfering with host cytokine networks. J Infect Chemother. 2011;17(3):302–317. doi: 10.1007/s10156-010-0177-y. [DOI] [PubMed] [Google Scholar]
- 2.Tomioka H, Namba K. Development of antituberculous drugs: current status and future prospects. Kekkaku Tuberc. 2006;81(12):753–774. [PubMed] [Google Scholar]
- 3.Chang KC, Yew WW. Management of difficult multidrug-resistant tuberculosis and extensively drug-resistant tuberculosis: update 2012. Respirology. 2013;18(1):8–21. doi: 10.1111/j.1440-1843.2012.02257.x. [DOI] [PubMed] [Google Scholar]
- 4.Chiang CY, Schaaf HS. Management of drug-resistant tuberculosis. Int J Tuberc Lung Dis. 2010;14(6):672–682. [PubMed] [Google Scholar]
- 5.Falzon D, Jaramillo E, Schunemann HJ, Arentz M, Bauer M, Bayona J, et al. WHO guidelines for the programmatic management of drug-resistant tuberculosis: 2011 update. Eur Respir J. 2011;38(3):516–528. doi: 10.1183/09031936.00073611. [DOI] [PubMed] [Google Scholar]
- 6.Nardell EA. Programmatic management of multidrug-resistant tuberculosis—15 years later. Int J Tuberc Lung Dis. 2011;15(10):1280. doi: 10.5588/ijtld.11.0604. [DOI] [PubMed] [Google Scholar]
- 7.Nathanson E, Lambregts-van Weezenbeek C, Rich ML, Gupta R, Bayona J, Blondal K, et al. Multidrug-resistant tuberculosis management in resource-limited settings. Emerg Infect Dis. 2006;12(9):1389–1397. doi: 10.3201/eid1209.051618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quelapio MI, Mira NR, Orillaza-Chi RB, Belen V, Munez N, Belchez R, et al. Responding to the multidrug-resistant tuberculosis crisis: mainstreaming programmatic management to the Philippine National Tuberculosis Programme. Int J Tuberc Lung Dis. 2010;14(6):751–757. [PubMed] [Google Scholar]
- 9.Schaaf HS, Marais BJ. Management of multidrug-resistant tuberculosis in children: a survival guide for paediatricians. Paediatr Respir Rev. 2011;12(1):31–38. doi: 10.1016/j.prrv.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 10.Yew WW. Management of multidrug-resistant tuberculosis and extensively drug-resistant tuberculosis: current status and future prospects. Kekkaku Tuberc. 2011;86(1):9–16. [PubMed] [Google Scholar]
- 11.Blumberg HM, Burman WJ, Chaisson RE, Daley CL, Etkind SC, Friedman LN, et al. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167(4):603–662. doi: 10.1164/rccm.167.4.603. [DOI] [PubMed] [Google Scholar]
- 12.Pandey R, Khuller GK. Antitubercular inhaled therapy: opportunities, progress and challenges. J Antimicrob Chemother. 2005;55(4):430–435. doi: 10.1093/jac/dki027. [DOI] [PubMed] [Google Scholar]
- 13.Sundaram S, Trivedi R, Durairaj C, Ramesh R, Ambati BK, Kompella UB. Targeted drug and gene delivery systems for lung cancer therapy. Clin Cancer Res. 2009;15(23):7299–7308. doi: 10.1158/1078-0432.CCR-09-1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varde NK, Pack DW. Microspheres for controlled release drug delivery. Expert Opin Biol Ther. 2004;4(1):35–51. doi: 10.1517/14712598.4.1.35. [DOI] [PubMed] [Google Scholar]
- 15.Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010;75(1):1–18. doi: 10.1016/j.colsurfb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 16.Yang Y, Bajaj N, Xu P, Ohn K, Tsifansky MD, Yeo Y. Development of highly porous large PLGA microparticles for pulmonary drug delivery. Biomaterials. 2009;30(10):1947–1953. doi: 10.1016/j.biomaterials.2008.12.044. [DOI] [PubMed] [Google Scholar]
- 17.Conte JE, Jr, Wang G, Lin ET, Zurlinden E. High-performance liquid chromatographic–tandem mass spectrometric method for the determination of ethionamide in human plasma, bronchoalveolar lavage fluid and alveolar cells. J Chromatogr B Biomed Sci Appl. 2001;753(2):343–353. doi: 10.1016/S0378-4347(00)00572-7. [DOI] [PubMed] [Google Scholar]
- 18.Wu J-W, Shih H-H, Wang S-C, Tsai T-H. Determination and pharmacokinetic profile of pyrazinamide in rat blood, brain and bile using microdialysis coupled with high-performance liquid chromatography and verified by tandem mass spectrometry. Anal Chim Acta. 2004;522(2):231–239. doi: 10.1016/j.aca.2004.06.004. [DOI] [Google Scholar]
- 19.Quintana JB, Reemtsma T. Rapid and sensitive determination of ethylenediaminetetraacetic acid and diethylenetriaminepentaacetic acid in water samples by ion-pair reversed-phase liquid chromatography–electrospray tandem mass spectrometry. J Chromatogr A. 2007;1145(1–2):110–117. doi: 10.1016/j.chroma.2007.01.044. [DOI] [PubMed] [Google Scholar]
- 20.Xi H, Han G, Lu L, Zhang D. Simultaneous determination of exogenous phosphocreatine and its metabolite creatine in rabbit plasma using ion-pair reversed-phase high performance liquid chromatography. Se pu = Chin J Chromatogr/Zhongguo hua xue hui. 2011;29(10):1000–1004. [PubMed] [Google Scholar]
- 21.Yau WP, Vathsala A, Lou HX, Chan E. Simple reversed-phase ion-pair liquid chromatography assay for the simultaneous determination of mycophenolic acid and its glucuronide metabolite in human plasma and urine. J Chromatogr B Anal Technol Biomed Life Sci. 2004;805(1):101–112. doi: 10.1016/j.jchromb.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 22.Bock MJ, Neilson KL, Dudley A. Use of trifluoroacetic acid to quantify small, polar compounds in rat plasma during discovery-phase pharmacokinetic evaluation. J Chromatogr B Anal Technol Biomed Life Sci. 2007;856(1–2):165–170. doi: 10.1016/j.jchromb.2007.05.024. [DOI] [PubMed] [Google Scholar]
- 23.United States Pharmacopeia. Convention USP, Usp. USP35 NF30, 2012: United States Pharmacopoeia National Formulary: United States Pharmacopeia; 2011
- 24.Watabe S, Yokoyama Y, Nakazawa K, Shinozaki K, Hiraoka R, Takeshita K, et al. Simultaneous measurement of pazufloxacin, ciprofloxacin, and levofloxacin in human serum by high-performance liquid chromatography with fluorescence detection. J Chromatogr B Anal Technol Biomed Life Sci. 2010;878(19):1555–1561. doi: 10.1016/j.jchromb.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 25.Danaher M, O’Keeffe M, Glennon JD. Validation and robustness testing of a HPLC method for the determination of avermectins and moxidectin in animal liver samples using an alumina column clean-up. Analyst. 2000;125(10):1741–1744. doi: 10.1039/b004836o. [DOI] [PubMed] [Google Scholar]
- 26.Seifart HI, Kruger PB, Parkin DP, van Jaarsveld PP, Donald PR. Therapeutic monitoring of antituberculosis drugs by direct in-line extraction on a high-performance liquid chromatography system. J Chromatogr. 1993;619(2):285–290. doi: 10.1016/0378-4347(93)80118-n. [DOI] [PubMed] [Google Scholar]
- 27.Peloquin CA. Therapeutic drug monitoring in antituberculosis chemotherapy. Ther Drug Monit. 1999;21(4):426–427. doi: 10.1097/00007691-199908000-00008. [DOI] [PubMed] [Google Scholar]
- 28.Gennaro MC, Calvino R, Abrigo C. Ion interaction reagent reversed-phase high-performance liquid chromatography determination of anti-tuberculosis drugs and metabolites in biological fluids. J Chromatogr B Biomed Sci Appl. 2001;754(2):477–486. doi: 10.1016/S0378-4347(01)00037-8. [DOI] [PubMed] [Google Scholar]
- 29.Williams S. Ghost peaks in reversed-phase gradient HPLC: a review and update. J Chromatogr A. 2004;1052(1–2):1–11. doi: 10.1016/j.chroma.2004.07.110. [DOI] [PubMed] [Google Scholar]
- 30.Kohn A. Automating method validation and system suitability testing in HPLC and CE. Am Biotechnol Lab. 1994;12(11):44. [PubMed] [Google Scholar]
- 31.Wilson TD, Fogarty DF. The effect of column age on system suitability parameters for an HPLC assay of bitolterol mesylate aerosols. J Chromatogr Sci. 1988;26(2):60–66. doi: 10.1093/chromsci/26.2.60. [DOI] [PubMed] [Google Scholar]