

Introduction
Nucleic acids that are selected to bind to a small molecule (1–3), protein (4–7), or whole cells (8) have a variety of applications that range from diagnosing (9,10) and treating disease (11,12) to deciphering the origins of life (13,14). Many methods have been developed to select nucleic acids that bind a small molecule of interest; Systematic Evolution of Ligands by Exponential Enrichment (SELEX) is most frequently used. All of these methods require the isolation of active nucleic acids from inactive ones. These separations are most commonly completed by using affinity chromatography in which a ligand is conjugated onto a resin (4). Capillary electrophoresis (CE) (15) has also been used to perform selections and does not require immobilization of a ligand onto a matrix. CE requires, however, that the mobility of the bound nucleic acid be different from that of the unbound. Though powerful, both methods have disadvantages. For example, in resin-based selections, kinetic biases can be introduced because a high concentration of the bound ligand is washed over a resin to elute the binders (16). This, the resin-based selections, can select against the highest affinity binders since they are the most difficult to compete off. CE SELEX is not affected by kinetic biases but does require specialized equipment. Neither method allows for multiple selections to be completed in parallel.
In an effort to complete multiple selections in parallel, a microarray-based method was developed. Microarrays are advantageous platforms for screening and performing selections because of the small amounts of ligand and analyte required, and the ability of microarrays to probe thousands of interactions in parallel. These advantages are best illustrated in the widespread uses of microarrays, which include studying gene expression (17), and protein–protein (18), carbohydrate–protein (19), cell–ligand (20,21), and small molecule–protein interactions (22–24). In order to develop a microarray-based method to complete selections, a microarray surface that is robust enough for ligand screening and allows bound RNAs to be harvested directly from the array surface is required. We found that the optimal surface is an agarose-coated microarray (25,26). Bound RNAs can be harvested from the array surface by simple excision of the agarose from ligand-functionalized positions (27,28). Additionally, agarose provides an inexpensive and three dimensional surface for high ligand loading. It can also be functionalized to provide a chemical handle for immobilization of a variety of reactive groups. Most importantly, by harvesting all ligand-bound RNAs via gel excision, kinetic biases found in resin-based selections may be mitigated (27).
Not only does completing multiple selections in parallel on a microarray surface increase throughput, but it also allows for the identification of the highest affinity and specific RNA-ligand interactions. Higher affinity RNA-ligand interactions are selected at lower ligand loadings that captured the members of an RNA library (27). In contrast, separate experiments would be required for each ligand loading to identify higher affinity interactions using resin-based SELEX. Specific RNA-ligand interactions are identified using the microarray selection procedure because it may set up a competition experiment between the arrayed ligands for the members of the RNA library (28).
In this protocol, the steps for completion of a microarray-based selection are described (Fig. 1). We specifically focus on ligands that are immobilized onto an agarose array surface using a Huisgen cycloaddition reaction in which the array displays either azide or alkyne groups (29,30), however, other immobilization chemistries are compatible with this platform, including immobilization of amine-containing ligands via reductive amination (25). This approach should be applicable to any nucleic acid-based selection.
Fig. 1.
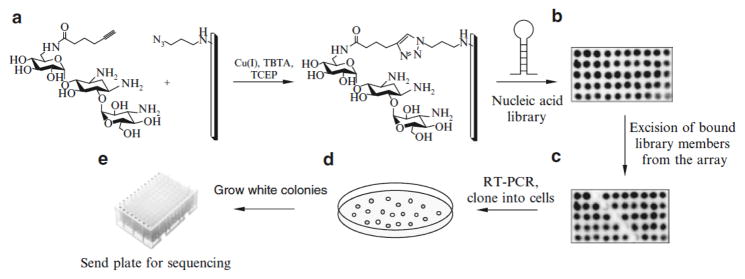
Schematic of the microarray-based selection process: (a) immobilization of small molecules on an activated agarose microarray via a Huisgen 1,3-dipolar cycloaddition reaction. (b) An image of the slide after hybridization with radiolabeled RNA. (c) An image of the same slide after the mechanical removal of bound RNA. (d) A schematic of an agar plate after transformation with a vector containing selected members of the library. (e) Colonies that are white from blue/white screening should be grown in culture and submitted for sequencing to deconvolute the selected members of the library.
2. Materials
2.1 Functionalization of Agarose Slides
31.8 mM solution of NaCNBH3: Dissolve 0.2 g of sodium cyanoborohydride in 80 ml 1× PBS and 20 ml ethanol. Prior to applying this solution to the array surface (preferred vendor is Sigma Aldrich, Silane Prep Slides), ensure that all of the NaCNBH3 is dissolved by stirring the solution for 2–3 min. It is important to prepare this solution fresh every time.
10× phosphate buffered saline (PBS): Dissolve 14.2 g of Na2HPO4, 2.45 g of KH2PO4, 81.8 g of NaCl, 1.86 g of KCl in 900 ml of nanopure water. Adjust the pH to 7.5 using NaOH or H3PO4. Add nanopure water to bring volume to 1 L. Store the solution at room temperature.
0.2% Sodium dodecyl sulfate (SDS): Dissolve 2 g of SDS in 900 ml of nanopure water; add water to make 1 L of solution. Store the solution at room temperature.
2.2 Spotting of Small Molecules onto Microarrays
-
10× phosphate buffer: Prepare the following solutions: 1 M K2HPO4(dissolve 1.74 g of K2HPO4in 10ml of nanopure water). 1 M KH2PO4 (dissolve 1.36 g of KH2PO4in 10 ml of nanopure water). 1 M Na2HPO4 (dissolve 1.42g of Na2HPO4 in 10 ml of nanopure water). 1 M NaH2PO4 (dissolve 1.2 g of NaH2PO4in 10 ml of nanopure water).
Prepare a 100 mM potassium phosphate solution by mixing 940 ml of 1 M K2HPO4, 60 ml of 1 M KH2PO4, and 9 ml of nanopure water. Prepare a 100 mM sodium phosphate solution by mixing 932 ml of 1 M Na2HPO4, 68 ml of 1 M NaH2PO4, and 9 ml of nanopure water. Finally, mix equal volumes of the two 100 mM solutions together; this solution should have a pH of ~8. Store the solution at room temperature.
Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl] amine (TBTA) solution: Stock solutions of TBTA should be made in 4:1 mixture of 2-butanol:DMSO. Store at 4°C.
10% ethylene glycol solution: Mix 20 ml of ethylene glycol with 180 ml of nanopure water. Store the solution at room temperature.
2.3 Hybridization of RNA onto the Slides
10× Hybridization Buffer: Dissolve 11.36 g of Na2HPO4, 3.72 g of Na2EDTA32H2O, 105.2 g of NaCl in 900 ml of nanopure water; adjust the pH to 7.0 with HCl. Add nanopure water to bring volume to 1 L. Store the solution at room temperature.
DEPC-treated water: Add 100 ml of diethylpyrocarbonate (DEPC) to 1 L of distilled water. Mix well and incubate at room temperature for 2 h. Autoclave on the liquid cycle for 20 min at 121 °C to inactivate the DEPC. Store the water at room temperature.
2.4 Cell Cloning
LB medium: Dissolve 10 g of bacto tryptone, 5 g of bacto yeast extract, 10 g of NaCl, and 1 ml of 1 M NaOH in 900 ml of de-ionized water. Adjust the pH to 7.0 with 1 M NaOH. Sterilize by autoclaving on the liquid cycle at 121 °C for 20 min. Store the solution at room temperature.
Terrific broth medium Dissolve 12 g of tryptone, 24 g of yeast extract, and 4 ml of glycerol in 900 ml deionized water. Sterilize by autoclaving on the liquid cycle at 121 °C for 20 min. Add 100 ml of 0.17 M KH2PO4 + 0.72 M K2HPO4 that has been sterile filtered. Store the solution at room temperature.
SOC medium: Dissolve 20 g of bacto tryptone, 5 g of bacto yeast extract, 0.5 g of NaCl, 10 ml of 250 mM KCl, and 3.6 g of glucose in 900 ml of water. Adjust the pH to 7.0 using 1 M NaOH. Sterilize by autoclaving at 121 °C for 20 min on the liquid cycle. Store the solution at room temperature.
100 mM Isopropyl-3-D-thiogalactoside (IPTG): Dissolve 238 mg of IPTG in 10 ml of nanopure water. Store the solution in a brown glass bottle or wrap the container in aluminum foil as IPTG is light sensitive. Store at −20°C.
5-Bromo-4-chloro-3-indolyl-3-D-galactopyranoside (X-gal): Dissolve 200 mg of X-gal in 10 ml of DMF. Store the solution in a brown glass bottle or wrap the container in aluminum foil as X-gal is light sensitive. Store at −20°C.
2.5 Polymerase Chain Reaction and RNA Transcription
10× PCR buffer: Dissolve the following in 45 ml nanopure water: 1.86 g of KCl, 0.6 g of Tris base, 500 μl of Triton X-100. Use KOH to adjust the pH of the solution to 9.0. Bring the total volume to 50 ml. Store the solution at −20°C. A suitable buffer supplied by the manufacturer can also be used.
To transcribe RNA, we use an RNAMaxx transcription kit (Stratagene).
2.6 Polyacrylamide Gel Electrophoresis
10× TBE Buffer: Dissolve 108 g of Tris base, 55 g of boric acid, 80 ml of 0.5 M EDTA in 850 ml of nanopure water. Adjust the pH to 8.3 using HCl. Bring volume to 1 L. Store the solution at room temperature.
0.5M EDTA: Dissolve 14.9 g of EDTA in 900 ml of nanopure water. Adjust the pH to 9.0 with NaOH. Bring volume to 1 L with nanopure water. The solution is stored at room temperature.
2× Loading Buffer: Dissolve 24 g of urea, 0.372 g of Na2EDTA32H2O, and 0.0121 g of Tris base in approximately 30 ml of nanopure water. Bring volume to 50 ml with water. Check the pH of solution; it should be approximately 7.5. Do not try to adjust the pH, as this will result in the introduction of excess of salt, which will cause problems during gel electrophoresis. Add 5 mg of Orange G dye and store the solution at room temperature.
2× Loading Buffer with bromophenol blue and xylene cyanol: Make 2× Loading buffer as described above, but do not add Orange G. Instead, add 2 mg of bromophenol blue and 2 mg of xylene cyanol. Store at room temperature.
3 METHODS
3.1 Preparation of Agarose Slides
Prepare 1% agarose solution (w/v) using nanopure water. Melt in a microwave on high for 2–3 min, swirling the solution every 20–30 s.
While the solution is hot, apply ~1.5 ml to the surface of a glass slides using a P-1000 pipette. Ensure that the solution is spread evenly over the slide surface. Allow the agarose to dry to a thin film overnight (Fig. 2a, b).
Fig. 2.

Images of agarose arrays during the preparation and selection of bound members. (a) An agarose slide that has solidified to a gel. (b) An agarose-coated microarray that has dried to a clear film. (c) An image of an agarose array that is placed on top of a paper grid to image use as a guide for spotting ligands; the spots to indicate the four corners of the grid have been put on the array using a hydrophobic marker and help align the grid for precise excision of bound nucleic acid on an array. The same grid is used to align the image of slides after hybridization with an RNA library.
3.2 Functionalization of Agarose Slides
Prepare a solution of 0.02 M NaIO4 in water. Submerge the slides in this solution for 30 min at room temperature, and then wash with nanopure water for 30 min.
Submerge the slides in 10% (v/v) ethylene glycol for 1 h at room temperature to quench residual NaIO4. Wash with water for 1.5 h, changing water every 20 min.
-
To prepare microarrays of amine-displaying ligands, complete the following:
Allow the slides to dry.
Prepare spotting solutions as follows: small molecule at desired concentration (typically serially diluted from 5 mM to 1 mM), 0.1 M NaHCO3, and 10% glycerol. Spot 0.4 ml of the solutions onto aldehyde-agarose slides in duplicate.
Incubate for 3 h at room temperature in a humidity chamber (box containing a saturated solution of NaCl). Wash the slides 3×10 min with 1× Hybridization Buffer, followed by water, 2×10 min. Continue to step 6.
For microarrays of alkyne-displaying ligands, submerge the slides in a solution of 0.1 M NaHCO3 (pH 8.5) and 10 mM 3-azidopropylamine for 3 h at room temperature. Continue to step 6.
For microarrays of azide-displaying ligands, replace 10 mM 3-azidopropylamine with 10 mM propargylamine. Continue to step 6.
-
Submerge slides in solution of 31.8 mM NaBH3 for 3 min to reduce the imine formed on the microarray surface.
Wash the slides with 0.2% SDS, 3×15 min, and then with water, 2×15 min.
Dry the slides under a stream of air. (This is the last step for microarrays of amine-displaying ligands.)
3.3 Immobilization of Aminoglycosides on the Slide Surface
-
Prepare spotting solutions as follows:
For azide-displaying small molecules: the small molecule at the desired concentration (typically two- to three-fold serially diluted from 5 mM to 1 mM), 10 mM Tris-HCl, pH 8.5, 1 mM CuSO4, 100 μM ascorbic acid, 100 μM TBTA (29), and 10% glycerol. Spot 0.4 μl of the solutions onto alkyne-agarose slides in duplicate.
For alkyne-displaying small molecules: the small molecule at the desired concentration (typically serially diluted from 5 mM to 1 mM), 1× phosphate buffer, 1 mM CuSO4, 0.5 mM TCEP, 100 μM TBTA (29), and 10% glycerol. Spot 0.4 μl of the solutions onto azide-agarose slides in duplicate.
Incubate for 3 h at room temperature in a humidity chamber (box containing a saturated solution of NaCl). Wash the slides 3×10 min with 1× Hybridization Buffer, followed by water, 2×10 min.
3.4 Transcription of RNA Using DNA Template
Amplify the randomized DNA template that encodes the RNA library by PCR using a forward primer that includes a T7 RNA polymerase promoter. Set up the reaction as follows: 1× PCR buffer, 0.3 mM dNTPs, 4.25 mM MgCl2, 2 μM forward primer, 2 μM reverse primer, 20 nM template, and one unit of Taq DNA polymerase in a total volume of 50 μl. Alternatively, anneal two strands of complementary DNAs encoding an RNA polymerase promoter and the desired RNA library.
While PCR is running, pour an agarose gel containing ethidium bromide to analyze the PCR reaction. Choose the percentage of agarose that is appropriate for the size of the PCR product. For the RNAs used herein, 2% agarose is sufficient.
Mix 3μl of PCR product with 1 μl 50% glycerol. Load the PCR product and an aliquot of a 100 bp DNA ladder into separate wells of the agarose gel; run the gel at 100 V for 15–30 min, or until proper separation is obtained. Visualize the DNA using a transilluminator or handheld UV lamp. Keep this gel to check the transcription reaction.
Transcribe the PCR product using any transcription protocol that utilizes an RNA polymerase (31). Manufacturers usually provide procedures for transcription of unlabeled RNA or 32P-internally labeled RNA; add 2 μl of α-[32P] ATP, or 20 μCi, to each transcription reaction.
While the RNA is transcribing, pour a 16 cm × 19.7 cm × 0.8 mm denaturing polyacrylamide gel to purify the RNA transcript. The percentage of acrylamide used should be appropriate for the size of the RNA library. For the library described here, 12% acrylamide gels are used (32).
Check that the transcription reaction was successful by agarose gel. Pour a 2% agarose gel and mix about 100 ml of 1× TBE buffer with 5 μl of 10 mg/ml aqueous ethidium bromide solution. Mix 0.5 μl of the transcription reaction with 5 μl of 15% glycerol. Load the sample and an aliquot of a 100 bp DNA ladder on an agarose gel. Run the gel at 100 V for 15–30 min or until proper separation is obtained. Visualize the DNA using a transilluminator or handheld UV lamp.
Once the transcription reaction is complete, add 1 unit of RNase-free DNase and incubate the sample for 30 min at 37°C.
Add an equal volume of 2× Loading Buffer to the transcription reaction. Load on the acrylamide gel prepared in step 5. Also, load running dyes (bromophenol blue and xylene cyanol in 1× Loading Buffer) to determine when proper separation of the RNA has been obtained. Run the gel at 300 V for 1–3 h using the running dyes as a guide.
-
Visualize the RNA transcript:
If the RNA is transcribed without radioactivity, identify the transcript by UV shadowing. Place a TLC plate with a fluorescent indicator under the gel, and excise the product band with a razor blade. (The product will appear purple.)
If the RNA is transcribed in the presence of α-[32P] ATP, the product can be identified by exposing the gel to a phosphorimager screen. Wrap the gel in saran wrap before placing the screen on top of the gel. Use the image as a template to excise the product band with a razor blade.
Place the gel slice into a 15 ml conical tube. Add 3 ml of 0.3 M NaCl in DEPC-treated water and tumble at 4 °C overnight.
Centrifuge the sample to pellet the gel pieces, and transfer the supernatant to a fresh 15 ml conical tube. Use 2-butanol to concentrate the RNA to approximately 0.5 ml, and ethanol precipitate by adding 2.5 volumes of ethanol to the sample, and then place the sample at −20°C for at least 15 min. Pellet the RNA by spinning in a centrifuge for 10 min at 10,000×g at 4 °C (32).
Resuspend the RNA in 100 μl DEPC-treated water. Determine the concentration of RNA using UV absorption at 260 nm and the corresponding extinction coefficient (for example, obtained from HyTher server, HyTher version 1.0, Nicolas Peyret, and John SantaLucia, Jr., Wayne State University) (33,34). Since this is an RNA library, the extinction coefficient is an estimate.
3.5 5′ End Labeling of RNA (If RNA Is Not Internally Labeled)
Remove 5′ phosphate group by incubating 20 pmol RNA, 1× alkaline phosphatase buffer, and three units of calf intestinal alkaline phosphatase (CIAP) in 100 μl total volume at 37°C for 1h.
Remove CIAP from the reaction by phenol/chloroform/isoamyl alcohol extraction and then ethanol precipitate the RNA with 10 μg of glycogen. Place the sample in a vacuum concentrator for 1–2 min to remove residual ethanol.
Kinase the RNA by dissolving the above pellet in 1× kinase buffer containing 3 μl γ–[32P] ATP and five units of T4 polynucleotide kinase. Incubate the mixture at 37 °C for 1 h. Mix with an equal volume of 2× Loading Buffer. Purify the reaction by denaturing polyacrylamide gel electrophoresis (PAGE) and extract RNA as described in Subheading 3.4, step 10.
3.6 Hybridization of RNA to the Slide
Prepare a solution of 32P-labeled RNA in 1× Hybridization Buffer. We generally use the amount of RNA that gives ≥2,000 cpm by Geiger counter for every slide. Refold the RNA as appropriate. (For example, heat the sample at 60 °C for 5 min and allow to slowly cool to room temperature on bench top.) If the isolation of specific RNA-ligand interactions in which the RNAs are small secondary structures is desired, then competitors – oligonucleotide mimics of the constant regions in the library – should be added to the hybridization solution (Fig. 3). If only the isolation of aptamers that bind a ligand is desired, then it is not necessary to use competitor oligonucleotides. Add 1× Hybridization Buffer and competitor oligonucleotides if appropriate to a total volume of 500 μl.
While the RNA is cooling to room temperature, prehybridize ligand-functionalized slides with 500 μl of 0.1% BSA (w/v) in 1× Hybridization Buffer for 5 min. Distribute the buffer evenly over the surface of the slide by placing a piece of parafilm over the applied solution. After prehybridization, remove parafilm and shake off the buffer.
Apply RNA solution to the slide, distributing it evenly over the surface using custom-cut 1 in. × 3 in. piece of parafilm. Incubate at room temperature for at least 20 min.
Wash the slides with 50 ml of 1× Hybridization Buffer with slight agitation 3×10 min. Gently blow air over the slide for 1 min to remove excess of buffer, and dry at room temperature by sitting on bench. Wrap the slide in saran wrap and expose to a phosphorimager screen at −20°C. Exposure time depends on how radioactive the slides are (3 h – overnight).
Scan the screen on a phosphorimager and print the image as its actual size to use as a template for excision (Fig. 4a) (see Notes 1 and 2).
Fig. 3.
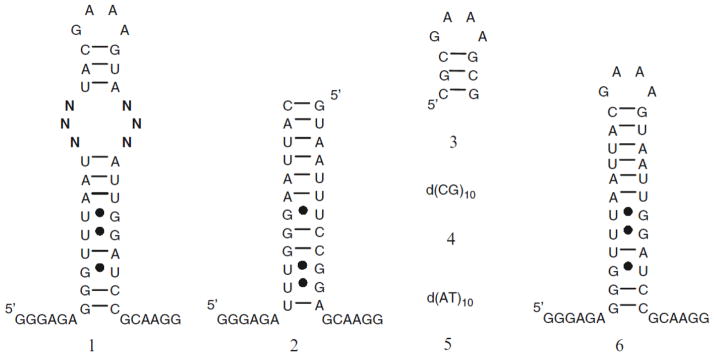
An example of oligonucleotides that may be used to identify RNA internal loop motifs that bind ligands. Oligonucleotide 1 is an internal loop library with six randomized positions (N) that contains 4,096 unique members; 2–5 are examples of competitors used to ensure that the interaction between a small molecule and RNA is specific; and 6 is the cassette in which the internal loop library is embedded.
Fig. 4.
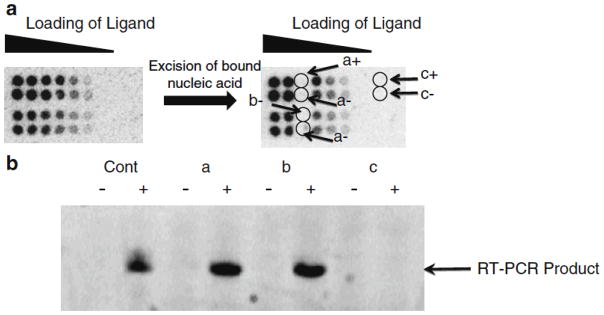
Selection of RNA-ligand interactions on an array. A left, image of an array after hybridization with an RNA motif library. (a, right ) Image of the array after the six indicated positions are excised. (b) Gel analysis of RT-PCR reactions completed with the samples that were excised. The plus indicates RT-PCR reactions in the presence of RT while the minus indicates RT-PCR reactions in the absence of RT.
3.7 Excision of Bound RNAs
Place an image of the array under the slide and use it as a template to identify captured RNA. Add 0.4 μl of nanopure water to each position to be excised. Incubate for 30 s at room temperature, and then remove the unabsorbed water using a pipette. Using a toothpick, trace the circumference of the hydrated spot and remove it; once loosened, the gel slice will stick to the toothpick. Place the gel slice in a PCR tube.
Re-expose the slide to a phosphorimager screen overnight to ensure that the agarose containing bound RNAs was removed cleanly (without affecting neighboring spots) (Fig. 4a).
3.8 RT-PCR of Selected RNAs
Add 16 μl water, 2 μl 10× DNase buffer, and four units of DNase to each tube. Incubate the solution at 37 °C for 2 h. Quench the reaction by the addition of 2 μl of 10× DNase stop solution (provided by Promega). Incubate at 65 °C for 10 min to completely inactivate the DNase. Note: for a positive control, use approximately 100 fmoles of the original RNA library and set up the reaction under the same conditions.
Add 2 μl of 100 μM reverse primer (5′-CCT TGC GGA TCC AAT) to the tube. Anneal the RNA and DNA primer at 70 °C for 10 min, and then incubate on ice for 10 min.
To complete reverse transcription, add 1.6 μl of 25 mM dNTPs, 0.8 μl of 10 mg/ml BSA, 4 μl of 10× RT buffer (provided by RT supplier), and two units of RT to +RT samples and the same volume of water for no RT controls. Incubate the reaction for 1h at 60°C, and then inactivate the RT by heating at 95°C for 3 min.
To the above samples add 4 μl of 100 μM of forward primer (5′-GGC CGA ATT CTA ATA CGA CTC ACT ATA GGG AGA GGG TTT AAT), 2 μl of 100 μM of reverse primer, 0.6 μl of 250 mM MgCl2, one unit of Taq DNA polymerase, 13 μl of H2O, and 6 μl of 10× PCR buffer. Run 20–25 PCR cycles.
Set up a gel to determine if the RT-PCR was successful. Depending on the size of the PCR product, either an agarose or an acrylamide gel can be used to determine if the RT-PCR was successful. For acrylamide gels, a much smaller gel can be used: for example, 8.6 cm × 6.8 cm × 1 mm gel.
Mix 1μl of RT-PCR reactions with 1μl of 2× Loading Buffer. Load onto gel and run at 200–250 V for 30–45 min for acrylamide gels or at 100 V for 15–30 min for agarose gels.
Mix about 100 ml of 1× TBE buffer with 5 μl of 10 mg/ml aqueous ethidium bromide solution. (Alternatively, ethidium bromide can be added to agarose gels prior to pouring.) Soak the gel in this solution for 10 min and then image (Fig. 4b). Only experiments in which the +RT background samples (Fig. 4, position c) contain no product should be carried on toward cloning (see Notes 3 and 4).
3.9 Cell Culture and Cloning (32)
Steps 1–3 should be completed the day before transformation of cells (steps 4–6). Prepare LB broth as described above but add 15 g of agar before autoclaving. Autoclave on the liquid cycle at 121 °C for 20 min. Remove the flask from autoclave, and cool the solution to about 60 °C ensuring that the agar does not settle to the bottom (by swirling every 20 min). Add 0.05 g of Ampicillin dissolved in 1 ml of water; mix by swirling the flask.
Pour a thin layer (5 mm) of LB agar into a petri dish. Let each plate cool until the agar is solidified (about 20 min). Incubate the plates upside down at 37°C overnight. Store plates in plastic bags at 4°C.
Ligate PCR products into pGEM T Vector following the manufacturer’s protocol (Promega).
DH5-α cells stored at −80°C should be thawed on ice for 5 min prior to use. Mix 100 μl DH5-α cells with 3 μl of ligation reaction in a 15 ml conical tube and incubate on ice for 30 min. Heat shock the cells at 42°C for 45 s, and then immediately place the tube on ice for 2 min. Add 1 ml of SOC medium and incubate at 37 °C with shaking (220rpm) for 1 h.
While the transformed cells are shaking prepare the LB-agar plates for blue/white screening. Add 40 μl of 100 mM IPTG onto agar plate and spread evenly using a spreader bar. Close the lid and wait for 2 min for the solution to absorb into the plates. Add 40 μl of 20 mg/ml X-gal; spread using the spreader. Place the plates in the 37°C incubator.
Plate 100 μl of transformed cells onto the plates prepared in step 2. Incubate plates at 37°C overnight (see Notes 5 and 6).
Transfer white colonies into separate wells of a deep 96-well plate containing 1 ml Terrific broth supplemented with 50 mg/l Ampicillin. (If it is difficult to determine if a colony is blue or white, place plates at 4 °C for 30–60 min) Cover the 96-deep well plate with a piece of foil and grow the cells at 37 °C with shaking (220 rpm) until they reach an OD600>4 (24–48 h). Wrap the LB-agar plates with parafilm and store at 4 °C.
Prepare glycerol stocks of the cells cultured in step 7. It is easiest to use a multichannel pipette and a 96-well plate to prepare glycerol stocks. Add 100 μl of 50% glycerol (sterilized) to each well of a 96-well plate. Aseptically transfer 100 μl of the cells to the plate with glycerol. Mix by pipetting up and down; cover the plate and store at −80 °C.
Centrifuge the 96-well deep well plate with the remaining culture to pellet the cells. Discard the supernatant by tipping the plate upside down and tapping gently on the bench top. Freeze cells at −80°C, and then send to a sequencing company.
Acknowledgments
We thank Professor Jessica Disney for careful proofreading of the manuscript. This work was supported by funding from the University at Buffalo, the NYS Center of Excellence and Bioinformatics and Life Sciences, a New Investigator Award from the Camille and Henry Dreyfus Foundation, a Cottrell Scholar Award from the Research Corporation, a NYSTAR J. D. Watson Young Investigator Award, and the National Institutes of Health (GM079235).
Footnotes
- Arrays might not be properly functionalized. Make new slides and try again.
- Spotting solutions might be old. Make new spotting solution.
- The hybridization buffer might be wrong. Check that the hybridization buffer was made properly.
- Too few counts of the radiolabeled library might have been used. Radiolabel the RNA with a new stock of α-[32P] ATP.
- Reactive groups were not quenched as NaBH3CN is a hygroscopic substance. If there are clumps in the solid NaBH3CN, obtain a new stock.
- If slides were not prehybridized with BSA, repeat the experiment and prehybridize the slides with hybridization buffer containing 0.1% BSA.
- Bound RNA positions were not properly excised from the array. In this case, reimage the slide after excision to ensure that the proper spots were excised.
- Too little RNA was harvested from the array. Excise a spot that captured more RNA (higher concentration of ligand loaded)
- RT-PCR buffers are contaminated with DNA or RNA. To resolve this problem, obtain new stock solutions for RT-PCR.
- Poor transformation efficiency might be the cause. Ethanol precipitates the RT-PCR product and then ligates into vector.
- Vector ligation was inefficient. Use more RT-PCR product in the vector ligation or use a new stock of DNA ligase to ligate into the vector.
- Vector ligation was inefficient. Try using more RT-PCR product in the vector ligation.
- pGEM T or TOPO TA Vector is old. Obtain a fresh stock of vector and try again.
- T4 DNA ligase is no longer active. Obtain a fresh stock of ligase and repeat the experiment.
References
- 1.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 2.Ellington AD, Szostak JW. Selection invitro of single-stranded DNA molecules that fold into specific ligand-binding structures. Nature. 1992;355:850–852. doi: 10.1038/355850a0. [DOI] [PubMed] [Google Scholar]
- 3.Osborne SE, Ellington AD. Nucleic acid selection and the challenge of combinatorial chemistry. Chem Rev. 1997;97:349–370. doi: 10.1021/cr960009c. [DOI] [PubMed] [Google Scholar]
- 4.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 5.Cox JC, Ellington AD. Automated selection of anti-protein aptamers. Bioorg Med Chem. 2001;9:2525–2531. doi: 10.1016/s0968-0896(01)00028-1. [DOI] [PubMed] [Google Scholar]
- 6.Giver L, Bartel DP, Zapp ML, Green MR, Ellington AD. Selection and design of high-affinity RNA ligands for HIV-1 REV. Gene. 1993;137:19–24. doi: 10.1016/0378-1119(93)90246-y. [DOI] [PubMed] [Google Scholar]
- 7.Hesselberth JR, Miller D, Robertus J, Ellington AD. In vitro selection of RNA molecules that inhibit the activity of ricin A-chain. J Biol Chem. 2000;275:4937–4942. doi: 10.1074/jbc.275.7.4937. [DOI] [PubMed] [Google Scholar]
- 8.Daniels DA, Chen H, Hicke BJ, Swiderek KM, Gold L. A tenascin-C aptamer identified by tumor cell SELEX: Systematic evolution of ligands by exponential enrichment. Proc Natl Acad Sci USA. 2003;100:15416–15421. doi: 10.1073/pnas.2136683100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minunni M, Scarano S, Mascini M. Affinity-based biosensors as promising tools for gene doping detection. Trends Biotechnol. 2008;26:236–243. doi: 10.1016/j.tibtech.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 10.Famulok M, Hartig JS, Mayer G. Functional aptamers and aptazymes in biotechnology, diagnostics, and therapy. Chem Rev. 2007;107:3715–3743. doi: 10.1021/cr0306743. [DOI] [PubMed] [Google Scholar]
- 11.Wu CC, Sabet M, Hayashi T, Tawatao R, Fierer J, Carson DA, Guiney DG, Corr M. In vivo efficacy of a phosphodiester TLR-9 aptamer and its beneficial effect in a pulmonary anthrax infection model. Cell Immunol. 2008;251:78–85. doi: 10.1016/j.cellimm.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keefe AD, Schaub RG. Aptamers as candidate therapeutics for cardiovascular indications. Curr Opin Pharmacol. 2008;8:147–152. doi: 10.1016/j.coph.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 13.Lorsch JR, Szostak JW. In vitro evolution of new ribozymes with polynucleotide kinase activity. Nature. 1994;371:31–36. doi: 10.1038/371031a0. [DOI] [PubMed] [Google Scholar]
- 14.Bartel DP, Szostak JW. Isolation of new ribozymes from a large pool of random sequences. Science. 1993;261:1411–1418. doi: 10.1126/science.7690155. [DOI] [PubMed] [Google Scholar]
- 15.Mendonsa SD, Bowser MT. In vitro evolution of functional DNA using capillary electrophoresis. J Am Chem Soc. 2004;126:20–21. doi: 10.1021/ja037832s. [DOI] [PubMed] [Google Scholar]
- 16.Klug SJ, Famulok M. All you wanted to know about SELEX. Mol Biol Rep. 1994;20:97–107. doi: 10.1007/BF00996358. [DOI] [PubMed] [Google Scholar]
- 17.Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 18.MacBeath G, Schreiber SL. Printing proteins as microarrays for high-throughput function determination. Science. 2000;289:1760–1763. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 19.Houseman BT, Mrksich M. Carbohydrate arrays for the evaluation of protein binding and enzymatic modification. Chem Biol. 2002;9:400–401. doi: 10.1016/s1074-5521(02)00124-2. [DOI] [PubMed] [Google Scholar]
- 20.Barrett OJ, Childs JL, Disney MD. Chemical microarrays to identify ligands that bind pathogenic cells. Chembiochem. 2006;7:1882–1885. doi: 10.1002/cbic.200600260. [DOI] [PubMed] [Google Scholar]
- 21.Disney MD, Magnet S, Blanchard JS, Seeberger PH. Aminoglycoside microarrays to study antibiotic resistance. Angew Chem Int Ed. 2004;43:1591–1594. doi: 10.1002/anie.200353236. [DOI] [PubMed] [Google Scholar]
- 22.Bradner JE, McPherson OM, Koehler AN. A method for the covalent capture and screening of diverse small molecules in a microarray format. Nat Prot. 2006;1:2344–2352. doi: 10.1038/nprot.2006.282. [DOI] [PubMed] [Google Scholar]
- 23.Duffner JL, Clemons PA, Koehler AN. A pipeline for ligand discovery using small-molecule microarrays. Curr Opin Chem Biol. 2007;11:74–82. doi: 10.1016/j.cbpa.2006.11.031. [DOI] [PubMed] [Google Scholar]
- 24.MacBeath G, Koehler AN, Schreiber SL. Printing small molecules as microarrays and detecting protein-ligand interactions en masse. J Am Chem Soc. 1999;121:7967–7968. [Google Scholar]
- 25.Afanassiev V, Hannemann V, Wolfl S. Preparation of DNA and protein microarrays on glass slides coated with an agarose film. Nucleic Acids Res. 2000;28:E66. doi: 10.1093/nar/28.12.e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dufva M, Petronis S, Jensen LB, Krag C, Christensen CB. Characterization of an inexpensive, nontoxic, and highly sensitive microarray substrate. Biotechniques. 2004;37:286–292. 294, 296. doi: 10.2144/04372MT02. [DOI] [PubMed] [Google Scholar]
- 27.Childs-Disney JL, Wu M, Pushechnikov A, Aminova O, Disney MD. A small molecule microarray platform to select RNA internal loop-ligand interactions. ACS Chem Biol. 2007;2:745–754. doi: 10.1021/cb700174r. [DOI] [PubMed] [Google Scholar]
- 28.Disney MD, Labuda LP, Paul DJ, Poplawski SG, Pushechnikov A, Tran T, Velagapudi SP, Wu M, Childs-Disney JL. Two-dimensional combinatorial screening identifies specific aminoglycoside-RNA internal loop partners. J Am Chem Soc. 2008;130:11185–11194. doi: 10.1021/ja803234t. [DOI] [PubMed] [Google Scholar]
- 29.Chan TR, Hilgraf R, Sharpless B, Fokin VV. Polytriazoles as copper(I)-stabilizing ligand in catalysis. Org Lett. 2004;6:2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 30.Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 31.Milligan JF, Uhlenbeck OC. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- 32.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning. Vol. 1. Cold Spring Harbor, NY: 1989. [Google Scholar]
- 33.Peyret N, Seneviratne PA, Allawi HT, SantaLucia J. Nearest-neighbor thermodynamics and NMR of DNA sequences with internal A-A, C-C, G-G, and T-T mismatches. Biochemistry. 1999;38:3468–3477. doi: 10.1021/bi9825091. [DOI] [PubMed] [Google Scholar]
- 34.SantaLucia J., Jr A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc Natl Acad Sci USA. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]