

Abstract
This review discusses the synthesis and application of glycosyl thioimidates in chemical glycosylation and oligosaccharide assembly. Although glycosyl thioimidates include a broad range of compounds, the discussion herein centers on S-benzothiazolyl (SBaz), S-benzoxazolyl (SBox), S-thiazolinyl (STaz), and S-benzimidazolyl (SBiz) glycosides. These heterocyclic moieties have recently emerged as excellent anomeric leaving groups that express unique characteristics for highly diastereoselective glycosylation and help to provide the streamlined access to oligosaccharides.
Keywords: carbohydrates, glycoconjugates, oligosaccharides, thioimidates, glycosylation, leaving group
Carbohydrates are the most abundant biomolecules on the planet, having long been known exclusively as an energy source. Recently, researchers have become increasingly aware of the role carbohydrates play aside from energy supply and storage. Carbohydrates are involved in a wide range of biological processes such as fertilization, anti-inflammation, immunoresponse, joint lubrication, cell growth, antigen determination, etc. [1]. Carbohydrates often play important roles during the occurrence of harmful processes including bacterial and viral infections, development of tumors, metastasis, tissue rejection, and congenital disorders [2]. Because many of these cellular processes are directly associated with pathogenesis of deadly diseases including AIDS, cancer, pneumonia, septicemia, hepatitis, and malaria, scientific efforts in the field of modern glycosciences have expanded [3]. While scientists have been able to isolate successfully certain classes of natural carbohydrates, the availability of appreciable quantities of these pure natural isolates is still inadequate to address the challenges offered by modern glycosciences. For this reason, chemical and enzymatic syntheses of complex carbohydrates have been employed to gain access to large quantities of both natural compounds and mimetics thereof. Even with significant advancements, controlled chemical synthesis of complex carbohydrates remains difficult. Hence, further development of methods and strategies for oligosaccharide and glycoconjugate synthesis continues to be a demanding area of research.
The majority of the biologically and therapeutically active carbohydrates are oligosaccharides or complex glycoconjugates (glycolipids, glycoproteins, etc.) in which monomeric sugar units are joined via O-glycosidic bonds. This linkage is formed by a glycosylation reaction, most commonly a promoter-assisted nucleophilic displacement of a leaving group (LG) of the glycosyl donor with a hydroxyl moiety of the glycosyl acceptor (Scheme 1) [4]. Other functional groups on both the donor and acceptor are temporarily masked with protecting groups (PG, T). Since the new glycosidic linkage creates a chirality center, particular care has to be taken with regards to the stereoselectivity. This represents a major challenge in comparison to the synthesis of the intermonomeric linkages in other natural biopolymers, proteins, and nucleosides. Although mechanistic studies of the glycosylation reaction are still scarce, certain conventions have already been established [5]. For instance, 1,2-trans-linked glycosides are often stereoselectively obtained from 2-O-acylated glycosyl donors, as these reactions proceed via an acyloxonium intermediate (Scheme 1) [6]. In the case of ether-type (2-O-benzyl) non-participating substituents, the glycosylation proceeds via the flattened oxacarbenium ion that often leads to anomeric mixtures. Although 1,2-cis-glycosides (for D-gluco/galacto series) are favored by the anomeric effect, the stereoselectivity is often low, which makes their synthesis a notable challenge [7, 8]. In spite of significant progress, chemical O-glycosylation remains among the top challenges of modern synthetic chemistry due to the requirement to achieve complete stereocontrol and due to the necessity to suppress side reactions [4].
Scheme 1.

Aside from glycosyl donor structure, a number of factors can influence the stereoselective outcome of the glycosylation reaction such as structure of glycosyl donor or acceptor, solvent, temperature, and many other factors. Occasionally, the nature of the leaving group may also have an influence on the anomeric stereoselectivity, and these observations led to the development of a large number of different classes of glycosyl donors [4]. Early methods developed by Fischer [9] and Koenigs and Knorr [10] had been the main driving forces in the area of carbohydrate chemistry until the 1970s through the early 1980s, when the traditional approach to glycosylation was revolutionized by the development of a series of new leaving groups. The new generation of glycosyl donors included S-phenyl glycosides by Ferrier [11], Nicolaou [12], Garegg [13], and others [14], cyanoethylidene derivatives by Kochetkov [15], O-imidates by Sinay [16] and Schmidt [17], S-benzothiazolyl derivatives (SBaz) by Mukaiyama [18], S-pyridyl and S-pyrimidin-2-yl derivatives by Hanessian [19] and Woodward [20], and fluorides by Mukaiyama [21] among others. The fate of these methods for chemical glycol-sylation is very different. While the development of trichloroacetimidates [22], thioglycosides [23], and fluorides [24] has produced general glycosylation methodologies, glycosyl thioimidates, broadly defined as compounds equipped with S–C=N leaving group, had been long outshadowed by other glycosyl donors that offered more promise for further development.
For many years, the use of glycosyl thioimidates have been viewed as an insignificant variation of the thioglycoside glycosidation methodology. Because of this, glycosyl thioimidates received very little attention, in part due to marginal stability toward protecting group manipulation that had been assumed. Over the last decade, the thioimidate method has evolved into a very robust methodology for glycosylation [25, 26]. Particularly with the introduction of novel S-benzoxazolyl (SBox) and S-thiazolinyl (STaz) leaving groups, it has become apparent that thioimidates can withstand many reaction conditions associated with protecting group manipulations [27, 28]. In addition, these thioimidates are easily accessible from a variety of simple precursors and can be glycosidated under a range of different and often unique, reaction conditions. Likewise, they are able to provide superior stereoselectivity and could be selectively activated in the presence of other types of leaving groups. All listed are important traits for both glycoside and oligo-saccharide synthesis that are rarely found all in one class of leaving groups.
This is particularly important in the light of the fact that a single-step glycosylation is only one challenge researchers working on oligosaccharide synthesis face wherein additional protecting and/or leaving group manipulations between each glycosylation step are required. Resultantly, the synthesis becomes increasingly inefficient which is particularly evident at the advanced stages of the assembly [29, 30] and often leads to a dramatic drop in yield, and as a conesquence, the availability of oligosaccharides. Further study of glycosyl thioimidates allowed for the design of novel strategies for the synthesis of oligosaccharides including temporary deactivation concept [31, 32], inverse armed-disarmed strategy [33], surface-tethered iterative carbohydrate synthesis (STICS) [34], and the thioimidate-only orthogonal strategy [35]. It also helped to uncover the O-2/O-5 cooperative effect in glycosylation that allows for superarming and superdisarming of building blocks by simple alteration of protecting group pattern [36–39], and to perform basic mechanistic aspects of glycosylation [33, 40], the most common but least understood reaction in glycochemistry [4]. Very recently, S-benzimidazolyl (SBiz) building blocks showed a very good promise to become a new versatile platform for active-latent oligosaccharide synthesis [41]. In addition, a number of medicinally relevant oligosaccharides and glycoconjugates have been obtained using the thioimidate approach [32, 42–47]. This review discusses major aspects of the synthesis and glycosidation of SBaz, SBox, STaz, and SBiz thioimidates and the roles in which they fit into modern strategies for expeditious oligosaccharide synthesis. A particular emphasis has been placed on methods, strategies, and phenomena that have been discovered thanks to unique structural features and reactivity pattern of glycosyl thioimidates.
S-Benzothiazolyl derivatives
Peracetylated S-benzothiazolyl (SBaz) glycosides were first synthesized in 73% yield by Zinner from acetobromoglucose and mercury(II) salt of 2-mercaptobenzothiazole (HSBaz) in the presence of sodium [48]. Mukaiyama was the first to report glycosidation of a glycosyl donor equipped with SBaz leaving group [18]. For this study, glycosyl donor 2 was prepared from perbenzylated chloride 1 and HSBaz in the presence of 1,8-bis(dimethylamino)naphthalene in 80% yield (Scheme 2). Glycosylation studies were performed in the presence of Cu(OTf)2 with various acceptors in diethyl ether, and in most cases glycosylation proceeded with high yields and good stereoselectivity. For example, the reaction of glycosyl donor 2 with acceptor 3 in the presence of Cu(OTf)2 resulted in the formation of disaccharide 4 in an excellent yield of 92% as an anomeric mixture (α/β = 4/1).
Scheme 2.

A number of general modifications and ultimately improvements to this approach have emerged. A number of various precursors and reaction conditions have been investigated. For instance, Szeja reported the synthesis of compound 2 from the hemiacetal derivative, 2,3,4,6-tetra-O-benzyl-D-glucopyranose under phase-transfer conditions in the presence of TsCl and Bu4NCl in benzene/50% aq. NaOH [49]. Resultantly, compound 2 was isolated in 95% yield and as a mixture of anomers (α/β = 1/3). Likewise, SBaz mannofuranoside and xylopyranoside were obtained as anomeric mixtures in 92% and 81% yield, respectively [49]. Schmidt et al. reported the synthesis of SBaz mannoside 6 by opening 1,2-ortho-ester 5 in the presence of HgBr2 (Scheme 3) [50]. Bogusiak reported the synthesis of SBaz furanosides of the L-arabino, D-ribo, and D-xylo series from the reducing sugars and HSBaz in the presence of diphenyl phosphoryl chloride [(PhO)2P(=O)Cl] under phase-transfer conditions [51]. Ferrieres and Plusquellec described the synthesis of peracetylated SBaz galactofuranosides from the corresponding β-penta-acetate in the presence of BF3–Et2O in 83% yield as an anomeric mixture [52, 53]. The obtained thioimidates were then deprotected and applied in anomeric phosphorylation. A number of acylated SBaz and analogous 5-methoxy-SBaz derivatives of D-gluco, D-galacto, and D-ribofurano series have been reported by Khodair et al. [54]. While the syntheses of hexoses were accomplished from acetobromosugar and the corresponding sodium thiolates in MeCN, the synthesis of thioribofuranoside derivative was accomplished from the corresponding tetraacetate and trimethylsilylated thiols in the presence of TMSOTf [54]. The synthesis of a range of differently protected SBaz derivatives from the corresponding glycosyl bromides and KSBaz in acetone has also been reported [55].
Scheme 3.

The glycosylation studies of SBaz derivatives have also been significantly expanded. For instance, Gama et al. reported the glycosidation of compound 2 by using methyl iodide as an activator in several solvents under high pressure [56]. It was found that high pressure-assisted glycosidation of compound 2 provided the corresponding disaccharides with an improved stereoselectivity compared to that reported by Mukaiyama [18]. For example, coupling of glycosyl donor 2 with acceptor 3 in dichloromethane at 1.3 GPa produced disaccharide 4 in 71% yield as a mixture of anomers (α/β = 7/1). SBaz mannoside 6 was employed in the elegant intramolecular glycosylation (molecular clamp) approach involving m-xylylene as a rigid spacer (Scheme 3) [50]. For this purpose, the free hydroxyl of mannoside 6 was reacted with α,α′-dibromoxylene resulting in the intermediate 7, which was subsequently linked to acceptor 8. The resulting compound 9 was subjected to NIS/TMSOTf-promoted intramolecular condensation to afford disaccharide 10 in 75% yield with high β-manno stereoselectivity (α/β = 1/9).
It was determined that AgOTf or NIS/TfOH can serve as suitable activators for S-benzothiazolyl furanosides [51]. As an example, AgOTf-promoted glycosylation of diacetone galactose acceptor (6-OH) with glycosyl donors of the L-arabino, D-ribo, and D-xylo series in toluene afforded the corresponding disaccharides in 75% (α/β = 4/3), 98% (α/β = 7/2), and 70% (α/β = 3/10) yield, respectively. It was also reported that the SBaz leaving group can be very rapidly activated for glycosylation in the presence of AgBF4 and is applicable to selective activation over other types of leaving groups involving S-alkyl and O-pentenyl glycosides [55]. It was also observed that the SBaz leaving group can be activated in the presence of MeOTf, but not in the presence of weaker alkylating reagents, which created a basis for selective activation of STaz donors with benzyl bromide or methyl iodide in the presence of glycosyl acceptors equipped with SBaz anomeric group [35].
S-Benzoxazolyl derivatives
Peracetylated S-benzoxazolyl (SBox) derivatives were first synthesized by Zinner from acetobromoglucose and 2-mercaptobenzoxazole (HSBox) in the presence of sodium [57, 58]. Schmidt's group had achieved the synthesis of SBox mannosides via orthoester 5 with HSBox and HgBr2 (Scheme 4) [50]. Shortly thereafter, it was reported the synthesis of SBox glycosyl donors of the D-gluco, D-galacto, and D-manno series from the corresponding anomeric bromides 12 and KSBox in the presence of 18-crown-6 [59]. Alternatively, glycosyl bromides can react directly with HSBox in the presence of K2CO3 in acetone [28]. In addition, SBox glycosides were obtained from anomeric acetates (e.g., 13), thioglycosides 15, or 1,2-anhydro sugars 14 (Scheme 4) [40]. The introduction of the SBox moiety is completely stereoselective, except for the BF3–Et2O-promoted introduction from anomeric acetate 13 that results in the formation of anomeric mixtures of 1,2-cis and 1,2-trans SBox glycosides (α/β = 1/3) [59].
Scheme 4.

Schmidt and co-workers reported the application of the SBox glycosides to indirect β-mannosylations, in which a glycosyl donor and a glycosyl acceptor were tethered by a rigid spacer [50]. The resulting disaccharide was obtained in 78% yield (α/β = 1/10). Demchenko et al. reported the use of SBox derivatives as glycosyl donors for the synthesis of 1,2-cis glycosides of the D-gluco, D-galacto [28], as well as 1,2-trans glycosides of the D-gluco, D-galacto, and D-manno series [59]. These studies showed that SBox leaving group can be activated using a broad range of promoters including AgOTf, Cu(OTf)2, Bi(OTf)3, AgBF4, NIS/TfOH, TMSOTf, MeOTf, etc. and allowing for stereoselective synthesis of 1,2-trans and 1,2-cis glycosides. For example, the glycosidation of SBox glycosyl donor 16 with acceptor 3 in the presence of MeOTf in dichloroethane afforded disaccharide 17 in 95% yield (Scheme 5). In attempts to improve the stereoselectivity of mannosylation, SBox glycosyl donors bearing remote protecting groups p-methoxy-benzoyl or N,N-diethyl thiocarbamoyl at C-4 atom were investigated [60].
Scheme 5.

Since the SBox moiety can be activated in the presence of AgOTf or Cu(OTf)2, conditions that fail to activate the alkyl/aryl thioglycosides, selective activation of SBox over a range of other leaving groups has been investigated. For example, the SBox donor 18 could be selectively activated over O-pentenyl 19 or S-ethyl acceptor 21 to give the corresponding disaccharides 20 or 22, respectively, in high yields and stereoselectivities (Scheme 5) [28]. Since the resulting disaccharides already bear a suitable leaving group at the anomeric center, no additional protecting/anomeric group manipulations are required for the continuation of the coupling sequence. This is very beneficial because the chain elongation can be conducted by utilizing selective activation principle or a high throughput one-pot strategy (vide infra) [61]. De Meo reported the application of the SBox approach to stereo-selective α-sialylation and to the efficient synthesis of a GM3 analog [62]. In this synthesis, selective activation of the SBox moiety of the sialyl donor over the S-ethyl moiety of the galactosyl acceptor was conveniently achieved in the presence of AgOTf. The obtained disaccharide was used in subsequent coupling directly to afford the desired GM3 trisaccharide sequence in good overall yield. A similar activation principle was also employed in a high output one-pot synthesis [63].
SBox glycosides also showed promising orthogonal combinations with glycosyl alkoxyimidates [64] and STaz glycosides [35]. The orthogonality with other thioimidates is a unique feature of this methodology because conventional orthogonal activation [65, 66] requires two different classes of leaving groups, whereas here both leaving groups are of the same class. The key to this methodology is the choice of activator. In the case of STaz activation, an alkylation agent is used and SBox activation is carried out with Bi(OTf)3 (Scheme 6) [35]. Herein, STaz glycosyl donor 23 and SBox acceptor 24 are coupled in the presence of benzyl bromide. In the subsequent step, disaccharide 25 is activated with Bi(OTf)3 and reacted with STaz acceptor 26 producing trisaccharide 27 in 62% yield. This methodology was largely based on extended mechanistic work, which demonstrated that STaz glycosides are activated via the heterocyclic nitrogen atom (remote activation), whereas SBox glycosides are activated via exocyclic/anomeric sulfur (direct activation) [35, 40].
Scheme 6.

As a directional modification of the traditional orthogonal approach based on the orthogonality of leaving groups, a reverse approach was reported wherein a protecting group-based chain elongation was conducted. This strategy, termed reverse orthogonal, takes advantage of orthogonal stability of protecting groups along with simultaneous activation of the applicable anomeric leaving group [67]. Thus, glycosyl donors employed were SEt glycosides with 6-O-(p-methoxy)benzyl (pMB) protection 28 and SBox glycosides with 6-O-pentenoyl (Pent) protection 29. The first disaccharide was obtained from compounds 28 and 30 in 81% using NIS/TfOH/H2O. Disaccharide 31 was then glycosylated with SBox donor 29 equipped with an O-pentenoyl group. The resulting trisaccharide 32 was obtained in 82% yield and then reacted with glycoside 28 to afford tetrasaccharide 33 in 71% yield. Finally, tetrasaccharide 33 was coupled with glycoside 29 and the resulting pentasaccharide 34 was isolated in 75% yield.
Another general concept for oligosaccharide synthesis makes use of only one class of leaving group for both reaction components, which are either activated (armed donor) or deactivated (disarmed acceptor) by the influence of the protecting groups (PG1, PG2, Scheme 8) [68]. Usually, the protecting groups in both reaction components and careful selection of mild reaction conditions have to be taken into consideration to allow for direct chemoselective activation of the armed glycosyl donor over the disarmed glycosyl acceptor. Many classes of leaving groups can be activated accordingly, and SBox is no exception. For instance, armed donor 35 could be chemoselectively activated over the disarmed acceptor 24. This activation was affected in the presence of Cu(OTf)2 and resulted in chemoselective formation of disaccharide 36 in 64% yield. Further study of chemoselective activation of SBox glycosides revealed other reactivity levels of differently protected building blocks. Thus, it was demonstrated that disarmed disaccharide 36 could be further chemoselectively activated over superdisarmed building block 37. This activation was achieved in the presence of Cu(OTf)2/TfOH to produce trisaccharide 38 in 70% yield [36]. This finding was somewhat surprising because at first it was anticipated that reactivity of building block such as 37 would fall somewhere between that of the armed (perbenzylated) and the disarmed (perbenzoylated) glycosyl donors; similar to the results found in Ley's studies [69] for building blocks of the L-rhamnose series.
Scheme 8.

The observed reactivity pattern was rationalized by the occurrence of the so-called “O-2/O-5 Cooperative Effect” [36]. In addition to the “arming/disarming” nature of the protecting group at O-2, stabilization of the glycosyl cation intermediate must also be taken into consideration. If electron withdrawing protecting groups are placed near the O-5 ring oxygen (C-4 and C-6, like in the disarmed donors C or D), the electron density on O-5 will be decreased, effectively suppressing oxacarbenium ion formation (Figure). In this case, the ability of the system to stabilize via the formation of an acyloxonium ion intermediate, like in disarmed glycosyl donor C, may become increasingly important. Crich et al. [70]. emphasized that the anchimeric assistance was particular to the 1,2-trans orientation of the 2-O-acyl and 1-SBox leaving group. If no source of secondary stabilization is available, as in case of 2-O-benzyl substituent in D, this combination will give rise to the overall “superdisarming” protecting group pattern.
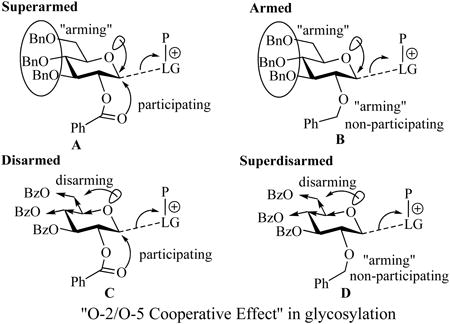
Along these lines, a glycosyl donor containing a participating benzoyl group at C-2 and electron donating groups at the remaining positions was also investigated and these glycosyl donors proved to be even more reactive (superarmed) than their armed perbenzylated counterparts [37, 38]. This observed reactivity pattern was also rationalized by the occurrence of the “O-2/O-5 Cooperative Effect” (see donor A depicted in Figure) [36]. Furthermore, this concept was found to be universal and applicable to glycosidation of O-pentenyl, S-ethyl, S-phenyl, S-tolyl and STaz building blocks [71].
In attempts to employ the SBox moiety with 2-amino-2-deoxy glycoside donors, it was observed that 2-N-trichloroethoxycarbonyl (Troc) derivatives are significantly more reactive than their 2-N-phthaloyl counterparts in MeOTf-promoted glycosylations [72]. This modified armed-disarmed approach allowed for an efficient chemical synthesis of 1,2-trans-linked oligosaccharides (Scheme 9). Taking advantage of the reactivity differences, the 2-N-Phth-protected glycosyl acceptor 40 was glycosylated with 2-N-Troc-protected glycosyl donor 39 in the presence of MeOTf, producing disaccharide 41 in 82% yield. Then, with AgOTf activation, the disaccharide was further subjected to coupling with acceptor 3 affording trisaccharide 42 in 73% yield.
Scheme 9.

It was also demonstrated that SBox-equipped building blocks can be used in both glycosyl acceptor and glycosyl donor bound approaches using polymer supports (Merrifield's resin or Tentagel) [73]. As an extension of this study, a novel approach to solid supported synthesis termed surface-tethered iterative carbohydrate synthesis (STICS) was developed [34]. This methodology employs nanoporous gold (NPG) plates as the support to which the glycosyl acceptor is anchored via a thiol linker (Scheme 10). The process involves the coupling of 6-O-TBDPS-protected glycosyl donor with the anchored acceptor using MeOTf. The tethered disaccharide was then desilylated and subsequently coupled with perbenzoylated SBox donor, cleaved from the NPG, and benzoylated for characterization yielding the trisaccharide in 52% overall on a 7–10 mg scale utilizing ten 8×8×0.2 mm NPG plates assembled in a shelved Teflon mini reactor.
Scheme 10.

S-Thiazolinyl derivatives
The synthesis of peracetylated S-thiazolinyl (STaz) glucopyranosides from acetobromoglucose and 2-mercaptothiazoline (HSTaz) in the presence of DIPEA or directly from glucose pentaacetate 13 and HSTaz in the presence of BF3Et2O in 64 and 69% yield respectively, was first reported by Descotes et al. (Scheme 11) [74]. Ferrieres and Plusquellec described the synthesis of peracetylated thiazolinyl galactofuranosides from β-D-galactofuranose pentaacetate in the presence of BF3 · Et2O in 53% yield [52, 53]. Demchenko et al. reported the synthesis of benzoylated STaz glycosides from the corresponding anomeric bromides 12 and NaSTaz or KSTaz in the presence of a crown ether [27] (Scheme 11). In these syntheses, the STaz glycosides of the D-gluco, D-galacto, and D-manno series were isolated in 60, 90, and 70% yield, respectively. Subsequently, it was determined that direct conversion of the anomeric acetates into STaz glycosides is by far more efficient for the D-gluco and D-galacto series in comparison to the synthesis from glycosyl bromides. Thus, the target compounds were obtained in 91 and 85% respectively [27].
Scheme 11.

Lower yields resulted when glycosyl halide donors were employed in the synthesis of STaz glycosides, this was due to two side reactions, N-glycosylation and β-elimination. Formation of by-products, N-linked “Taz” and 1,2-anhydro derivatives, are probably due to ambient reactivity of HSTaz and its relatively high basicity. Nevertheless, the STaz glycosides were obtained from anomeric chlorides 43 [27], thioglycosides (e.g., 45) [31], 1,2-anhydro sugars 14 [74], or 1,2-orthoesters 44 (Scheme 11) [33]. The formation of 1,2-trans STaz glycosides were found in complete stereoselectivity.
The STaz moiety was found to be stable toward common protecting group manipulations involving basic and acidic conditions including acetylation, benzylation, acetal formation, and cleavage, among others [31]. Moreover, in comparative hydrolytic stability studies, STaz glycosides exhibited even more stability than their 1-S-ethyl and 1-S-phenyl counterparts in the presence of NBS or NIS/TfOH [27, 75]. Studies were conducted to determine whether thiazolinyl 2,3,4,6-tetra-O-benzyl-1-thio-β-D-glucopyranoside 46 could serve as a common precursor toward the synthesis of an array of glycosyl donors [55]. It was found that the STaz glycoside 46 can be directly converted into the following glycosyl donors: acetate 47, chloride 1, bromide 48, fluoride 49, and hemiacetal 50 in excellent yields (Scheme 12). Moreover, glycosyl bromide 48 was then used as a precursor for subsequent synthetic steps to obtain benzoxazolyl, benzothiozolyl, and ethyl thioglycosides (35, 2, and 51), as well as n-pentenyl glycoside 52, all with exclusive β-stereoselectivity. The hemiacetal derivative 50 was converted into the corresponding glycosyl trichloroacetimidate 53.
Scheme 12.

Investigation into the properties of the STaz glycosyl donor brought about the development of a general methodology for 1,2-cis and 1,2-trans glycosylation. Early studies by Descotes and co-workers involved the displacement of the STaz functionality of peracetylated derivatives of the D-gluco series using MeOH in the presence of HgNO3 [74]. Further investigation into the glycosylation protocol toward disaccharide and oligosaccharide synthesis was reported by Demchenko et al. Activators such as AgOTf, MeOTf, NIS/TfOH, benzyl bromide, methyl iodide, AgBF4 and Cu(OTf)2 have been found to be suitable for efficient STaz activation for glycosylation [27]. STaz glycosyl donors were shown to react smoothly with common glycosyl acceptors. As shown in Scheme 13, STaz donor 46 was coupled with acceptors 54, 56, and 58 in the presence of AgOTf to afford disaccharides 55 in 96% yield (α/β = 2.9/1), 57 in 97% yield (α/β = 4.5/1), and 59 in 95% yield (α/β = 3.5/1). De Meo et al. reported application of the STaz approach to stereo-selective α-sialylation [62].
Scheme 13.

The applicability of the STaz method to selective glycosylation strategies for expeditious oligosaccharide synthesis was also evaluated [27]. As aforementioned, the STaz leaving group remains inert in the presence of NIS in combination with catalytic amount of TfOH, common conditions for alkyl/aryl thioglycoside activation. This finding led to the development of a promising combination of orthogonal leaving groups, wherein STaz is activated selectively with AgOTf, whereas ethyl thioglycoside are activated with NIS and catalytic TfOH [27]. This finding was applied to the synthesis of pentasaccharide 64, as depicted in Scheme 14 [76]. Thioglycoside donor 51 was first activated over the STaz acceptor 26 in the presence of NIS/cat. TfOH. The resulting disaccharide 60, isolated in 98% yield, was in turn activated over thioglycoside acceptor 61 with AgOTf to give trisaccharide 62 in 93% yield. The sequence was then reiterated as follows. Thioglycoside donor 62 was activated over STaz acceptor 26 in the presence of NIS/cat. TfOH. The resulting tetrasaccharide 63 was selectively activated over thioglycoside acceptor 61 in the presence of AgOTf to give pentasaccharide 64 in 59% yield. It should be noted that NIS and stoichiometric TfOH are capable of effective activation of the STaz leaving group [27].
Scheme 14.

The selective activation methodology was further applied in a five-step synthesis of a hexasaccharide, wherein two classes of glycosyl thioimidates, STaz and SBox, were used as building blocks (Scheme 15) [77]. First, thiocyanate glycoside donor 65 was activated with Cu(OTf)2 over STaz acceptor 26. Subsequently, disaccharide 66 was coupled with SBox glycosyl donor 24 in the presence of benzyl bromide to achieve the trisaccharide 67 in 67% yield. The latter was coupled with fluoride acceptor 68 in the presence of MeOTf to produce tetrasaccharide 69 in 87% yield. Tetrasaccharide 69 was then reacted with SEt acceptor 70 in the presence of AgClO4/Cp2ZrCl2 to afford the pentasaccharide 71 in 84% yield. Lastly, the coupling of O-pentenyl acceptor 72 with the pentasaccharide 71 using MeOTf as an activator produced hexasaccharide 73 in 72% yield. Selective activation principle was found beneficial for the synthesis of oligosaccharides of Streptococcus pneumoniae serotypes 6A, B and C and mimetics thereof for the development of synthetic vaccine components [43, 45, 46] and for the synthesis of a tumor associated glycosphingolipid SSEA-3 analog [47].
Scheme 15.

One-pot glycosylation methods, wherein a few sequential glycosylation steps are executed in one reaction vessel, are of further advantage because there is no need for purification of the intermediates. Many different leaving groups and methods fit into this general concept [61], and thioimidate-based one-pot glycosylation procedures have also emerged [55, 78]. For instance, SBox glycoside 74 was activated over SEt acceptor 21 with AgOTf (Scheme 16) [78]. Upon completion of the coupling, NIS and catalytic TfOH were added to the reaction mixture along with the STaz glycosyl acceptor 26. In the final step, glycosyl acceptor 3 and AgOTf were added in order to glycoside the STaz moiety of the trisaccharide intermediate. Upon completion of the one-pot sequence and purification by column chromatography, the tetrasaccharide 75 was isolated in 73% yield over three steps [78].
Scheme 16.

Additionally, the armed and disarmed properties of differently protected STaz glycosides were evaluated in the context of chemoselective oligosaccharide synthesis. It was determined that STaz glycosides follow the general concept of the armed-disarmed [79] approach. The activated (benzylated) STaz derivatives could be promoted over electronically disarmed (acylated) STaz glycosyl acceptors in the presence of either AgOTf or Cu(OTf)2 [33]. The scope of this methodology was expanded by the invention of a novel arming participating group, which allowed to expand chemoselective activation principle to the synthesis of trans-trans sequenced oligosaccharides [33]. Thus, it was observed that when 2-O-picolinyl-protected glycosyl donor 76 was activated, the corresponding disaccharides were obtained with exclusive β-stereoselectivity. Extended mechanistic study demonstrated that these reactions proceed via the formal intermediacy of the α-pyridinium salt intermediate that can be stereoselectively opened to provide β-linked products. This finding gave rise to the development of a new strategy for oligosaccharide synthesis that was called “inverse armed-disarmed strategy” [33]. When STaz donor 76 was activated over the disarmed acceptor 26 disaccharide 77 was obtained in 74% yield with complete 1,2-trans stereoselectivity (Scheme 17), which is the inverse outcome in comparison to the classical armed-disarmed approach resulting in 1,2-cis bond formation in the first step (vide supra, see Scheme 8). Subsequent activation of disaccharide 77 with AgOTf yielded the trans-trans-linked trisaccharide 78 in 91% yield [80].
Scheme 17.

An interesting observation that STaz glycosides can participate in stable nonionizing transition metal complexes led to the development of a temporary deactivation concept [31]. The outline of this unprecedented strategy that involves the temporary disarming of a leaving group by external deactivation of its active sites is depicted in Scheme 18. Thus, the deactivation of the would-be-armed glycosyl acceptor 79 was accomplished by coordinating its STaz functionality into a stable, non-ionizing metal complex with PdBr2 80. This allowed for the activation of the “free” STaz moiety of the disarmed glycosyl donor 81 over the temporarily deactivated (capped) STaz moiety of the complexed acceptor 80 in the presence of MeOTf. Upon glycosylation, disaccharide 82 was released from the complex by treatment with NaCN to permit a “free” disaccharide 83, which could be used in subsequent transformations.
Scheme 18.

The temporary deactivation concept was further expanded by the development of a new generation of leaving groups capable of bidentate, and therefore more stable, complexation mode with the deactivating reagent PdBr2. This approach was applied to the synthesis of medicinally relevant oligosaccharides of Streptococcus pneumoniae serotype 14 (Scheme 19) [32]. To undergo this synthesis, STaz donor 84 was then reacted with the complexed 4-(pyridin-2-yl)thiazol-2-yl thioglycoside acceptor 85 in the presence of AgOTf followed by desilylation with TBAF to allow the disaccharide acceptor 86.
Scheme 19.

The latter was then coupled with SBox lactose donor 87 to afford the pneumococcal tetrasaccharide repeating unit 88. For the subsequent convergent elongation, one portion of tetrasaccharide 88 was treated with NaCN to afford thiazolyl donor 89, whereas another portion of compound 88 was treated with 10% TFA to afford complexed glycosyl acceptor 90. The coupling of compounds 89 and 90 followed by decomplexation under the standard reaction conditions afforded octasaccharide 91 [32].
S-Benzimidazolyl derivatives
Peracetylated S-Benzimidazolyl (SBiz) glycosides were first reported by Zinner [81]. The SBiz moiety was then investigated as a leaving group for phosphorylation of unprotected glycosyl donors [52, 53]. It was recently reported the use of the SBiz moiety as a new platform for active-latent-like glycosylations [41]. For this study, the synthesis SBiz glycosyl donors of the D-gluco and D-galacto series 12, as well as the 2-amino-2-deoxy-D-gluco series 92 was accomplished from the corresponding anomeric bromides and potassium salt of HSBiz (KSBiz). The synthesis of SBiz building blocks from the anomeric acetate 13, thioglycoside 93, and 1,2-orthoester 94 were also reported (Scheme 20) [41].
Scheme 20.

It was determined that activation of the SBiz leaving group can be readily achieved with the use of AgOTf, Cu(OTf)2, and DMTST. For example SBiz glycosyl donor 95 was coupled with glycosyl acceptor 3 in the presence of AgOTf to produce disaccharide 96 in 98% yield (Scheme 21).
Scheme 21.

Similarly, the activation of N-anisoylated derivative of SBiz (SBizAn) 97, obtained by simple acylation of compound 95, could be also achieved with AgOTf. Thus, disaccharide 96 was obtained in 98% yield. Alkylating reagents, such as methyl iodide, benzyl bromide, and allyl bromide are also able to promote SBiz leaving group departure (Scheme 21). For instance, the reaction between SBiz glycosyl donor 95 and acceptor 3 in the presence of methyl iodide produced disaccharide 96 in 89% yield. Further investigation of the alkylation activation pathway led to the discovery that when the SBiz moiety is N-anisoylated its reactivity in the presence of methyl iodide was essentially turned off. As an example, in the attempt to react SBiz donor 97 with acceptor 3 using methyl iodide, no product 96 was formed.
This differential reactivity of the SBiz vs. SBizAn moieties allowed to execute the active-latent-like oligosaccharide synthesis, a strategy pioneered by Roy et al. [82, 83]. In ordinance with this methodology, the active SBiz donor 95 was glycosidated with the latent SBizAn acceptor 98 in the presence of methyl iodide to afford disaccharide 99 in 75% yield. After subsequent deprotection of the anisoyl group, using NaOMe, the resulting active disaccharide 100 was further coupled with acceptor 3, methyl iodide, promoted, in 71% yield. Since both SBiz and SBizAn donors are activated smoothly in the presence of AgOTf, further activation of SBizAn disaccharide 99 can be also conducted to afford trisaccharide 101 directly. Extended mechanistic study led to a conclusion that the deactivation effect of N-anisoyl moiety in SBizAn donor 98 is of electronic nature similarly to that described for the classical disarming effect in glycosylation [79]. Herein, the disarming is achieved by acylation of the leaving group, not by introducing the neighboring acyl substituents in the sugar moiety.
Glycosyl thioimidates represent a broad class of leaving groups used in carbohydrate syntheses. While the discussion of these compounds was very limited, it should be noted that generalization of synthesis, reactivity and application should be avoided. Thioimidates can be prepared from a broad range of precursors including acetates, halides, anhydrosugars, hemiacetals, orthoesters, thioglycosides. Singly step activation of thioimidates for glycosylation is typically a simple and fast process that leads to disaccharides in very high yields and often superior stereoselectivities. Due to the polyfunctional character of the thioimidoyl leaving group, the activation can be achieved via a variety of modes including the direct and remote pathways with the use of AgOTf, Cu(OTf)2, NIS/TfOH, TfOH, MeOTf, Bi(OTf)3, AgBF4, and with alkylating reagents such as benzyl bromide, methyl iodide, and allyl bromide. This ultimately allows for fine tuning of the activation conditions. The high stability of glycosyl thioimidates toward other glycosyl donor activation conditions permits their use for the temporary protection of the anomeric center of the glycosyl acceptor. This feature is what made possible to fit thioimidates into many existing strategies for oligosaccharide assembly and to develop conceptually new approaches and methods.
Scheme 7.

Scheme 22.

Acknowledgments
The authors are indebted to the National Science Foundation (Award CHE-1058112) for providing generous support for the development of new glycosylation methodologies and National Institute of General Medical Studies (Award GM077170) for providing generous support for the development of new strategies for expeditious oligosaccharide synthesis.
References
- 1.Varki A, Cummings RD, Esko JD, Freeze HH, Bertozzi CR, Stanley P, Hart GW, Etzler ME. Essentials of Glycobiology. 2nd. CSH Laboratory Press; New York: 2009. [PubMed] [Google Scholar]
- 2.Cipolla L, Arajo AC, Bini D, Gabrielli L, Russo L, Shaikh N. Expert Opin Drug Discovery. 2010;5:721. doi: 10.1517/17460441.2010.497811. [DOI] [PubMed] [Google Scholar]
- 3.Seeberger PH, Werz DB. Nature. 2007;446:1046. doi: 10.1038/nature05819. [DOI] [PubMed] [Google Scholar]
- 4.Demchenko AV. Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance. Wiley-VCH; Weinheim: 2008. [Google Scholar]
- 5.Mydock LK, Demchenko AV. Org Biomol Chem. 2010;8:497. doi: 10.1039/b916088d. [DOI] [PubMed] [Google Scholar]
- 6.Goodman L. Adv Carbohydr Chem Biochem. 1967;22:109. doi: 10.1016/s0096-5332(08)60152-6. [DOI] [PubMed] [Google Scholar]
- 7.Demchenko AV. Synlett. 2003:1225. [Google Scholar]
- 8.Demchenko AV. Curr Org Chem. 2003;7:35. [Google Scholar]
- 9.Fischer E. Ber Dtsch Chem Ges. 1893;26:2400. [Google Scholar]
- 10.Koenigs W, Knorr E. Ber Dtsch Chem Ges. 1901;34:957. [Google Scholar]
- 11.Ferrier RJ, Hay RW, Vethaviyasar N. Carbohydr Res. 1973;27:55. [Google Scholar]
- 12.Nicolaou KC, Seitz SP, Papahatjis DP. J Am Chem Soc. 1983;105:2430. [Google Scholar]
- 13.Garegg PJ, Henrichson C, Norberg T. Carbohydr Res. 1983;116:162. [Google Scholar]
- 14.Oscarson S. In: Glycoscience: Chemistry and Chemical Biology. Fraser-Reid B, Tatsuta K, Thiem J, editors. Vol. 1. Springer; Berlin–Heidelberg–New York: 2001. p. 643. [Google Scholar]
- 15.Bochkov AF, Kochetkov NK. Carbohydr Res. 1975;39:355. [Google Scholar]
- 16.Pougny JR, Jacquinet JC, Nassr M, Duchet D, Milat ML, Sinay P. J Am Chem Soc. 1977;99:6762. doi: 10.1021/ja00462a052. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt RR, Michel J. Angew Chem, Int Ed Engl. 1980;19:731. [Google Scholar]
- 18.Mukaiyama T, Nakatsuka T, Shoda SI. Chem Lett. 1979;8:487. [Google Scholar]
- 19.Hanessian S, Bacquet C, Lehong N. Carbohydr Res. 1980;80:17. [Google Scholar]
- 20.Woodward RB, Logusch E, Nambiar KP, Sakan K, Ward DE, Au-Yeung BW, Balaram P, Browne LJ, Card PJ, Chen CH. J Am Chem Soc. 1981;103:3215. [Google Scholar]
- 21.Mukaiyama T, Murai Y, Shoda S. Chem Lett. 1981;10:431. [Google Scholar]
- 22.Zhu X, Schmidt RR. In: Handbook of Chemical Glycosylation. Demchenko AV, editor. Wiley-VCH; Weinheim: 2008. p. 143. [Google Scholar]
- 23.Zhong W, Boons GJ. In: Handbook of Chemical Glycosylation. Demchenko AV, editor. Wiley-VCH; Weinheim: 2008. p. 261. [Google Scholar]
- 24.Shoda Si. In: Handbook of Chemical Glycosylation. Demchenko AV, editor. Wiley-VCH; Weinheim: 2008. p. 29. [Google Scholar]
- 25.Pornsuriyasak P, Kamat MN, Demchenko AV. ACS Sym Ser. 2007;960:165. [Google Scholar]
- 26.Szeja W, Grynkiewicz G. In: Handbook of Chemical Glycosylation. Demchenko AV, editor. Wiley-VCH; Weinhein: 2008. p. 329. [Google Scholar]
- 27.Demchenko AV, Pornsuriyasak P, De Meo C, Malysheva NN. Angew Chem, Int Ed. 2004;43:3069. doi: 10.1002/anie.200454047. [DOI] [PubMed] [Google Scholar]
- 28.Demchenko AV, Malysheva NN, De Meo C. Org Lett. 2003;5:455. doi: 10.1021/ol0273452. [DOI] [PubMed] [Google Scholar]
- 29.Demchenko AV. Lett Org Chem. 2005;2:580. [Google Scholar]
- 30.Smoot JT, Demchenko AV. Adv Carbohydr Chem Biochem. 2009;62:161. doi: 10.1016/S0065-2318(09)00005-5. [DOI] [PubMed] [Google Scholar]
- 31.Pornsuriyasak P, Gangadharmath UB, Rath NP, Demchenko AV. Org Lett. 2004;6:4515. doi: 10.1021/ol048043y. [DOI] [PubMed] [Google Scholar]
- 32.Pornsuriyasak P, Rath NP, Demchenko AV. Chem Commun. 2008:5633. doi: 10.1039/b810569c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smoot JT, Pornsuriyasak P, Demchenko AV. Angew Chem, Int Ed. 2005;44:7123. doi: 10.1002/anie.200502694. [DOI] [PubMed] [Google Scholar]
- 34.Pornsuriyasak P, Ranade SC, Li A, Parlato MC, Sims CR, Shulga OV, Stine KJ, Demchenko AV. Chem Commun. 2009:1834. doi: 10.1039/b817684a. [DOI] [PubMed] [Google Scholar]
- 35.Kaeothip S, Pornsuriyasak P, Rath NP, Demchenko AV. Org Lett. 2009;11:799. doi: 10.1021/ol802740b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kamat MN, Demchenko AV. Org Lett. 2005;7:3215. doi: 10.1021/ol050969y. [DOI] [PubMed] [Google Scholar]
- 37.Mydock LK, Demchenko AV. Org Lett. 2008;10:2103. doi: 10.1021/ol800345j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mydock LK, Demchenko AV. Org Lett. 2008;10:2107. doi: 10.1021/ol800648d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamkhachorn T, Parameswar AR, Demchenko AV. Org Lett. 2010;12:3078. doi: 10.1021/ol101089u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamat MN, Rath NP, Demchenko AV. J Org Chem. 2007;72:6938. doi: 10.1021/jo0711844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hasty SJ, Kleine MA, Demchenko AV. Angew Chem, Int Ed. 2011;50:4197. doi: 10.1002/anie.201007212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park S, Parameswar AR, Demchenko AV, Nahm MH. Infect Immun. 2009;77:3374. doi: 10.1128/IAI.00319-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parameswar AR, Park IH, Saksena R, Kovac P, Nahm MH, Demchenko AV. ChemBioChem. 2009;10:2893. doi: 10.1002/cbic.200900587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shulga OV, Zhou D, Demchenko AV, Stine KJ. Analyst. 2008;133:319. doi: 10.1039/b712760j. [DOI] [PubMed] [Google Scholar]
- 45.Parameswar AR, Hasty SJ, Demchenko AV. Carbohydr Res. 2008;343:1707. doi: 10.1016/j.carres.2008.03.035. [DOI] [PubMed] [Google Scholar]
- 46.Parameswar AR, Pornsuriyasak P, Lubanowski NA, Demchenko AV. Tetrahedron. 2007;63:10083. [Google Scholar]
- 47.Pornsuriyasak P, Demchenko AV. Carbohydr Res. 2006;341:1458. doi: 10.1016/j.carres.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 48.Zinner H, Peseke K. Chem Ber. 1965;98:3508. [Google Scholar]
- 49.Bogusiak J, Szeja W. Chem Lett. 1988;17:1975. [Google Scholar]
- 50.Abdel-Rahman AAH, El Ashry ESH, Schmidt RR. Carbohydr Res. 2002;337:195. doi: 10.1016/s0008-6215(01)00306-8. [DOI] [PubMed] [Google Scholar]
- 51.Bogusiak J. Lett Org Chem. 2005;2:271. [Google Scholar]
- 52.Ferrieres V, Blanchard S, Fischer D, Plusquellec D. Bioorg Med Chem Lett. 2002;12:3515. doi: 10.1016/s0960-894x(02)00822-3. [DOI] [PubMed] [Google Scholar]
- 53.Euzen R, Ferrieres V, Plusquellec D. J Org Chem. 2005;70:847. doi: 10.1021/jo0484934. [DOI] [PubMed] [Google Scholar]
- 54.Khodair AI, Al-Masoudi NA, Gesson JP. Nucleosides, Nucleotides Nucleic Acids. 2003;22:2061. doi: 10.1081/NCN-120026407. [DOI] [PubMed] [Google Scholar]
- 55.Kaeothip S, Pornsuriyasak P, Demchenko AV. Tetrahedron Lett. 2008;49:1542. doi: 10.1016/j.tetlet.2007.12.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gama Y, Yasumoto M. Chem Lett. 1993;22:319. [Google Scholar]
- 57.Zinner H, Pfeifer M. Chem Ber. 1963;96:432. [Google Scholar]
- 58.Zinner H, Peseke K. Chem Ber. 1965;98:3515. [Google Scholar]
- 59.Demchenko AV, Kamat MN, De Meo C. Synlett. 2003:1287. [Google Scholar]
- 60.De Meo C, Kamat MN, Demchenko AV. Eur J Org Chem. 2005:706. [Google Scholar]
- 61.Parameswar AR, Demchenko AV. In: Progress in the Synthesis of Complex Carbohydrate Chains of Plant and Microbial Polysaccharides. Nifantiev NE, editor. Transworld Res Network; Kerala: 2009. p. 463. [Google Scholar]
- 62.De Meo C, Parker O. Tetrahedron: Asymmetry. 2005;16:303. [Google Scholar]
- 63.Wang CC, Lee JC, Luo SY, Kulkarni SS, Huang YW, Lee CC, Chang KL, Hung SC. Nature. 2007;446:896. doi: 10.1038/nature05730. [DOI] [PubMed] [Google Scholar]
- 64.Ranade SC, Kaeothip S, Demchenko AV. Org Lett. 2010;12:5628. doi: 10.1021/ol1023079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kanie O, Ito Y, Ogawa T. J Am Chem Soc. 1994;116:12073. [Google Scholar]
- 66.Kaeothip S, Demchenko AV. Carbohydr Res. 2011;346:1371. doi: 10.1016/j.carres.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fujikawa K, Vijaya Ganesh N, Tan YH, Stine KJ, Demchenko AV. Chem Commun. 2011:10602. doi: 10.1039/c1cc13409d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mootoo DR, Konradsson P, Udodong U, Fraser-Reid B. J Am Chem Soc. 1988;110:5583. [Google Scholar]
- 69.Douglas NL, Ley SV, Lucking U, Warriner SL. J Chem Soc, Perkin Trans. 1998;1:51. [Google Scholar]
- 70.Crich D, Li M. Org Lett. 2007;9:4115. doi: 10.1021/ol701466u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Premathilake HD, Mydock LK, Demchenko AV. J Org Chem. 2010;75:1095. doi: 10.1021/jo9021474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bongat AFG, Kamat MN, Demchenko AV. J Org Chem. 2007;72:1480. doi: 10.1021/jo062171d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Parlato MC, Kamat MN, Wang H, Stine KJ, Demchenko AV. J Org Chem. 2008;73:1716. doi: 10.1021/jo701902f. [DOI] [PubMed] [Google Scholar]
- 74.Cottier L, Descotes G, Kudelska W, Seances CR. Acad Sci, Ser II. 1992;314:657. [Google Scholar]
- 75.Ramakrishnan A, Pornsuriyasak P, Demchenko AV. J Carbohydr Chem. 2005;24:649. [Google Scholar]
- 76.Pornsuriyasak P, Demchenko AV. Chem Eur J. 2006;12:6630. doi: 10.1002/chem.200600262. [DOI] [PubMed] [Google Scholar]
- 77.Kaeothip S, Demchenko AV. J Org Chem. 2011;76:7388. doi: 10.1021/jo201117s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pornsuriyasak P, Demchenko AV. Tetrahedron: Asymmetry. 2005;16:433. [Google Scholar]
- 79.Fraser-Reid B, Wu Z, Udodong UE, Ottosson H. J Org Chem. 1990;55:6068. [Google Scholar]
- 80.Smoot JT, Demchenko AV. J Org Chem. 2008;73:8838. doi: 10.1021/jo801551r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zinner H, Peseke K. J Prakt Chem. 1969;311:997. [Google Scholar]
- 82.Roy R, Andersson FO, Letellier M. Tetrahedron Lett. 1992;33:6053. [Google Scholar]
- 83.Cao S, Hernandez-Mateo F, Roy R. J Carbohydr Chem. 1998;17:609. [Google Scholar]