

Abstract
Purpose
Targeting multiple anti-apoptotic proteins is now possible with the small molecule BH3 domain mimetics such as ABT-737. Given recent studies demonstrating that autophagy is a resistance mechanism to multiple therapeutic agents in the setting of apoptotic inhibition, we hypothesized that hydroxychloroquine (HCQ), an anti-malarial drug that inhibits autophagy, will increase cytotoxicity of ABT-737.
Experimental Design
Cytotoxicity of ABT-737 and HCQ was assessed in vitro in PC-3 and LNCaP cells, and in vivo in a xenograft mouse model. The role of autophagy as a resistance mechanism was assessed by siRNA knockdown of the essential autophagy gene beclin1. ROS was measured by flow cytometry, and mitophagy assessed by the mCherry-Parkin reporter.
Results
Induction of autophagy by ABT-737 was a mechanism of resistance in prostate cancer cell lines. Therapeutic inhibition of autophagy with HCQ increased cytotoxicity of ABT-737 both in vitro and in vivo. ABT-737 induced LC-3 and decreased p62 expression by immunoblot in cell lines and by immunohistochemistry in tumors in vivo. Assessment of ROS and mitochondria demonstrated that ROS production by ABT-737 and HCQ was a mechanism of cytotoxicity.
Conclusions
We demonstrated that autophagy inhibition with HCQ enhances ABT-737 cytotoxicity in vitro and in vivo, that LC-3 and p62 represent assessable markers in human tissue for future clinical trials, and that ROS induction is a mechanism of cytotoxicity. These results support a new paradigm of dual targeting of apoptosis and autophagy in future clinical studies.
Keywords: Prostate Cancer, Autophagy, Metabolism, Bcl-2, BH3, ABT-737, ABT-263
Introduction
Advanced prostate cancer is incurable with current approaches to therapy including androgen ablation therapy and chemotherapy, due to the development of mechanisms of drug resistance. Efforts to improve therapeutic effectiveness against prostate cancer include drug development of agents capable of inhibiting multiple Bcl-2 family protein members based on prior studies demonstrating the importance of Bcl-2 and Bcl-xL expression as a mechanism of drug resistance, and increased expression of Bcl-2 with castration in prostate cancer (1–3). As a potent inhibitor of apoptosis, the BH3 mimetic ABT-737 is a small molecule inhibitor of the anti-apoptotic proteins Bcl-2, Bcl-xL and Bcl-w, and an analogue with oral formulation (ABT-263) is currently in clinical trials (4–6). Our prior studies demonstrated the activity of ABT-737 in a mouse model of prostate cancer (7). Although promising, preclinical development in an effort to understand mechanisms of drug resistance are warranted to better guide future clinical studies (8, 9).
As an emerging, and common, mechanism of tumor cell survival and drug resistance, autophagy appears to account for decreased therapeutic effectiveness for a variety of agents under drug development (10). In fact, as a common cellular pathway used for degradation of cytoplasmic organelles, autophagy has been shown to protect cells by decreasing proteotoxic, metabolic and oxidative stress (10). Studies by our group and other groups demonstrated that autophagy is particularly important as a survival mechanism in tumors with defects in the apoptotic pathway, supporting a paradigm of dual apoptotic and autophagic inhibtion (11–13).
Therefore, in the current study we determined whether ABT-737 is effective in prostate cancer, if autophagy is a mechanism of resistance, and if combination with hydroxychloroquine (HCQ), a known inhibitor of autophagy, improves cytotoxicity. We found that the combination of ABT-737 and HCQ resulted in enhanced cytotoxicity of prostate cancer in vitro and in vivo; demonstrated that modulation of ROS was a potential mechanism of enhanced cytotoxicity with autophagy inhibition; and developed autophagy biomarkers for future studies. These results support a new paradigm of dual targeting of apoptosis and autophagy in clinical studies.
Materials and Methods
Cell Culture and Cytotoxicity
PC-3 (human androgen insensitive prostate cancer cell line), LNCaP (human androgen sensitive prostate cancer cell line) were obtained from ATCC. LNCaP and PC-3 cells were maintained in RPMI-1640 media with 2 g/L glucose and 10% FBS. ABT- 737 was obtained from Abbott Laboratories and HCQ was obtained from Sigma (St. Louis, MO). Cells were plated initially in 96-well microtiter plates and cells were treated after 24 hrs with different concentrations of ABT-737, HCQ or combination of both as indicated. After 72 hrs of incubation in the presence or absence of drug, viability studies were performed by an MTT assay as previously described (14). Clonogenic viability assays were performed on 6 well plates using PC-3 cells after treatment with ABT-737, HCQ or a combination of both in the presence or absence of N-acetylcysteine (NAC), (Sigma, St. Louis, MO). After 14 days of treatment, clones were stained (0.1% methylene blue, 50% ethanol, 50% water) and counted. Experiments were performed in triplicates to analyze the statistical significance.
Immunoblot Analysis
Cells treated for 72 hours were lysed, and equivalent amounts of protein from each sample were subjected to electrophoresis on 15% gel SDS–PAGE. LC3 was assessed using polyclonal rabbit (Novus Biologicals, Littleton, CO) primary antibody. p62 was assessed with p62 polyclonal guinea pig (Progen Biotechnik, Heidelberg Germany) primary antibody. Densitometry was performed using ImageJ v1.45 software (NIH, Bethesda, MA).
RNA interference studies
To modulate autophagy, LNCaP and PC-3 cells were transfected with annealed, purified, and desalted double-stranded beclin1 siRNA (30 mg/ 3*106 cells) using the Amaxa nucleofection system (Gaithersburg, MD) (kit V, program G-16) as performed previously (14). siRNA targeted against beclin1 and LaminA/C control were obtained from Dharmacon Research (Lafayette, CO). Cytotoxicity assays (MTT) were performed as described above.
Measurement of ROS production
ROS levels were determined using 10 μM 2′-7′-dichlorodihydrofluorescene diacetate (DCF- DA, Molecular Probes) as described previously (13). Briefly, PC-3 cells were treated with ABT-737, HCQ or the combination of both for 16 hours followed by incubation with 10 μM 2′-7′-dichlorodihydrofluorescene diacetate (DCF- DA, Molecular Probes) in HBSS (GIBCO) for 10 min, trypsinized, washed in and reconstituted in PBS. Samples were analyzed by Flow Cytometry (FC500; Beckman Coulter, Fullerton, CA).
Mouse Xenograft studies
NCR NUNU mice (Taconic) were inoculated subcutaneously with 1 × 107 PC3 prostate cancer cells. Drug injections were made once the tumors reached an average volume of about 200 mm3. Mice were divided randomly into 4 groups of 7 each and subjected to intraperitoneal (i.p) injection of vehicle, ABT-737 (50mg/kg), HCQ (50mg/kg) or the combination of ABT-737 (50mg/kg) and HCQ (50mg/kg) for 15 days. Tumor volume was measured by caliper measurements (V = L × W2/2). For i.p injection, ABT-737 was prepared in 30% propylene glycol, 5% tween 80, and 65% D5W (5% dextrose in water), pH 4–5. HCQ was prepared in PBS.
Immunohistochemistry
Xenograft tumor tissues from mice treated with ABT-737, HCQ or the combination of both were fixed in formalin and embedded in paraffin. Paraffin sections were deparaffinized, boiled in citrate buffer (pH 6) at 95°C for 30 minutes, blocked in 10% goat serum for 30 minutes, and incubated in LC3 5F10 mAb (nanoTools; 1:100) for 15 minutes. Intermediate washes were performed with 0.1% tween-20 in PBS. Further processing was conducted using LSAB2 System-HRP kit from DAKO. Nuclei counterstaining was done with hematoxylin (Ricca Chemical Company) for 1 minute, and the slides were washed with water, dehydrated and mounted. For the other antibodies, deparaffinized, boiled slides, were blocked in 10% goat serum for 1 hour, and incubated overnight in p62 (SQSTM1), rabbit polyclonal antibody (Biomol International; 1:1000) at 4°C, in a humid chamber.
Mitophagy assessment
For quantification of mitophagy induction, PC3 cells were transiently transfected (Amaxa Nucleofector Reagent V) with the mCherry-Parkin reporter, plated on Lab-Tek® Chamber Slide™ (Nalge Nunc, Naperville, IL) and allowed to recover overnight. Cells were treated with ABT-737, HCQ or combination of ABT-737 and HCQ for 16 hours. Cells were fixed with 4% paraformaldehyde for 15 minutes at room temperature and imaged at 600× magnification.
Results
Effect of ABT-737 on cultured cells with and without autophagic modulation
Prostate cancer cell lines were treated with ABT-737 in a cell culture model to assess for autophagy and the effect of autophagic modulation. As shown in Figure 1A, transfection of PC-3 cells with EGFP-LC3 reporter demonstrated LC-3 localization in autophagosomes by fluorescence microscopy with HCQ, representing inhibition of the distal autophagy pathway, or with ABT-737, representing induction of autophagy. As confirmed in Figure 1B by immunoblot, LC3-II expression increased compared to LC3-I with HCQ and ABT-737 in both LNCaP and PC-3 cells, representing cleavage to LC3. Specifically, in figure 1B, ABT-737 and HCQ increase LC3-II/LC3-I ratios, as expected with ABT-737 inducing autophagy, and as expected with HCQ inhibiting autophagy late in the pathway, which increases autophagosome accumulation. Also shown in Figure 1B, p62 decreased with ABT-737, as would be expected with autophagic clearance of p62. As shown in Figure 1C (all four bars of which represent treatment of ABT-737 compared to untreated control), sensitivity to treatment with ABT-737 increased in PC-3 and LNCaP prostate cancer cell lines by decreasing autophagy in a model developed previously in which the autophagy regulator beclin1 is decreased by beclin1 siRNA (14). Specifically, Beclin1 siRNA decreased viability of ABT-737 treated cells (−Beclin1) compared to Lamin siRNA (+Beclin1) in both PC-3 (P< 0.001) and LNCaP (p=0.035) cells, demonstrating the importance of autophagy as a resistance mechanism of ABT-737 induced cytotoxicity.
Figure 1.
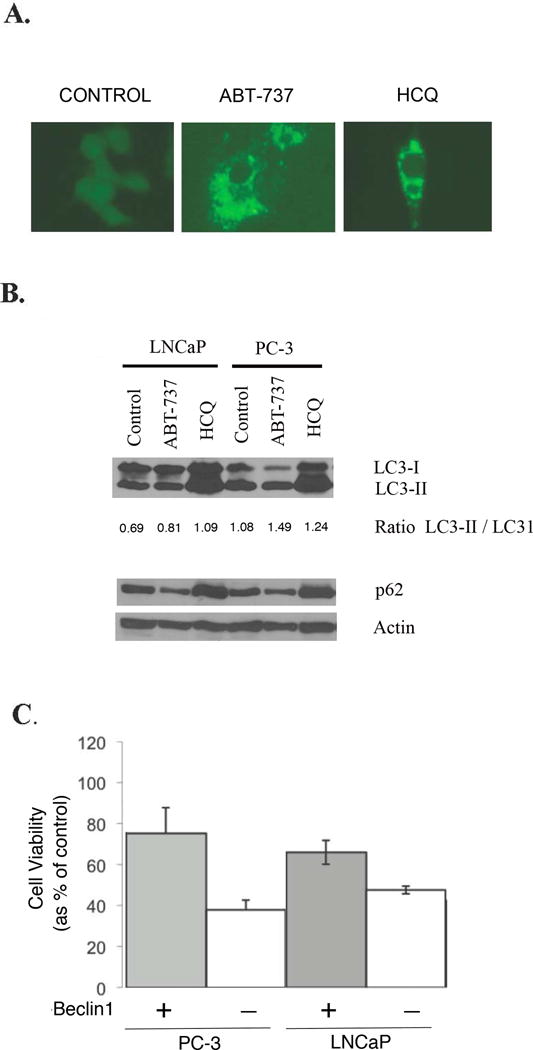
Cell viability assay with and without inhibition of autophagy. 1A: PC-3 cells transfected with EGFP-LC3 reporter treated with 10uM of Hydroxychloroquine (HCQ), or 5uM of ABT-737 for 72 hours demonstrating LC-3 localized to autophagosomes by fluorescence microscopy. 1B: Immunoblot of LC3-I, LC3-II, and p62 expression in LNCaP and PC3 cells treated with HCQ or ABT-737. The ratio of LC3-II/I represents an assessment of cleavage of LC3 with autophagosome formation. 1C: PC-3 and LNCaP cells were treated for 72 hours with 2.5uM of ABT-737 in cells transfected with beclin1 siRNA or Lamin siRNA control. The X axis of Figure 1C represents treatment of cells with ABT-737 with either Beclin siRNA to decrease Beclin1 (shown as negative (−) for Beclin1) or with Lamin siRNA (shown as positive (+) for Beclin1) in PC-3 and LNCaP cells respectively. The effect of drug treatment on cell viability was determined by MTT assay. The percentage of cell viability as a percentage of untreated cells is shown to decrease with treatment of ABT-737 in both PC-3 and LNCaP cells, with a greater decrease in cell viability with Beclin1 inhibition. All experiments were conducted in triplicate.
Cytotoxic effect of combinations of ABT-737 and HCQ in vitro
Given these data demonstrating that ABT-737 induces autophagy, and that autophagy is a mechanism of cell survival, efforts to modulate autophagy through a therapeutic approach would be important for future drug development. Therefore, to determine the effect of therapeutic inhibition of autophagy on the cytotoxic effect of ABT-737, cells were treated with ABT-737 with and without HCQ, a known inhibitor of autophagy. As shown in Figure 2, PC-3 (2A) and LNCaP (2B) cells were treated with ABT-737 and cell viability was assessed. As shown, ABT-737 induced cytotoxicity was dose dependent in both PC-3 and LNCaP cells, whereas HCQ alone had only a moderate cytotoxic effect. However, HCQ enhanced the cytotoxicity of ABT-737 compared to each agent alone at multiple ABT-737 concentrations (P<0.01) supporting plans for further assessment in an in vivo model (which is described below).
Figure 2.
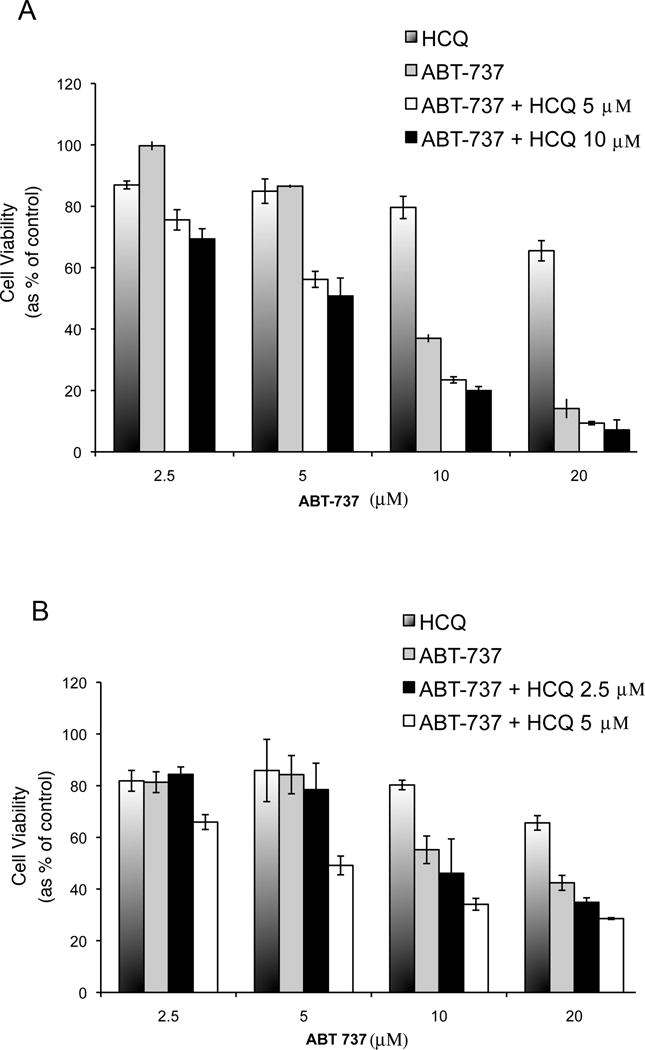
Cell viability assay of PC-3 (A) and LNCaP cells (B) in culture treated with HCQ alone, ABT-263 alone at indicated concentrations, and the combination at differing ABT-263 concentrations. Cells were seeded in 96-well plates 24 hours before treatment. MTT assays were performed 48 hours after treatment. The percentage of cell viability is shown for each treatment group compared to the viability of untreated cells, which were considered as 100%. Experiments were conducted in triplicate.
Effect of ABT-737 and Hydroxychloroquine on tumor growth in vivo
To confirm in vitro findings and provide pre-clinical support for future clinical trials that test this new paradigm of dual apoptotic and autophagic targeting, the effect of ABT-737, HCQ and the combination of both agents were assessed on tumor growth in a mouse PC-3 xenograft model. As shown in Figure 3, growth of PC-3 tumor xenografts was rapid and uninhibited by ABT-737 alone. In contrast, tumor growth was significantly inhibited in mice treated with the combination of ABT-737 and HCQ compared to ABT-737 (p=0.003) or HCQ (p=0.001) alone.
Figure 3.
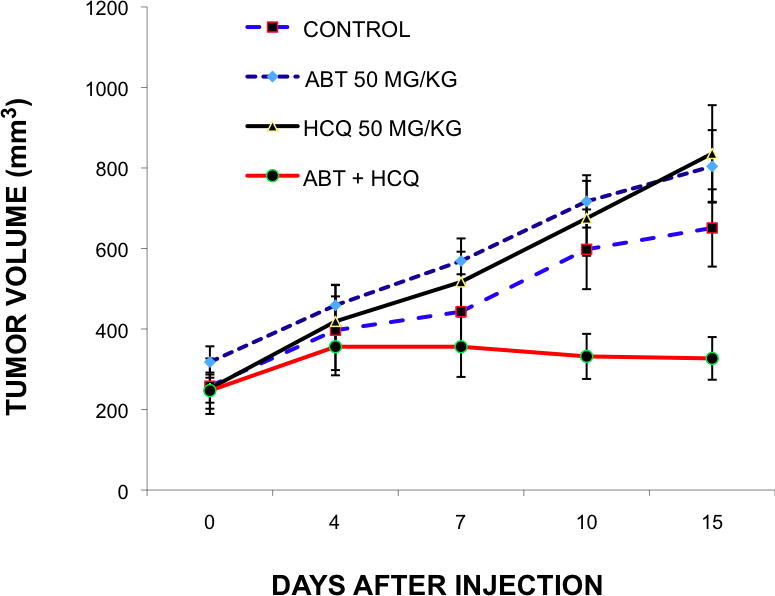
Effect of ABT-737 (ABT), Hydroxychloroquine (HCQ), and the combination in vivo. In vivo tumor growth is shown with combined ABT-737 and HCQ treatment in mouse xenograft model. PC-3 prostate cancer cells were injected subcutaneously in nude mice and grown for 10 days. Mice were treated daily for 14 days with vehicle control, ABT-737 (50mgm/kg), HCQ (50mgm/kg) or both in combination. Each treatment was performed in quadruplicate.
Effect of ABT-737 on biomarkers of autophagy
To effectively develop future clinical trials with therapeutics targeting Bcl-2 and autophagy, the development of autophagy specific markers will be important. To establish such potential markers that can be assessed in the future for validation, we assessed the feasibility of candidate markers of autophagy in tumor tissue. Based on studies by our laboratories and other groups demonstrating the importance of p62 and LC3 in the autophagy pathway, we assessed tissue from the tumor xenografts in our in vivo studies following treatment with vehicle control, ABT-737, HCQ, and the combination of agents. As shown in Figure 4, ABT-737 and the combination of HCQ and ABT-737 induced LC-3 localization, as expected based on induction of autophagy by ABT-737 in vitro, as was shown in Figure 1A. Also in agreement with our prior studies that demonstrated the effect of autophagy to clear p62 from autophagosomes, p62 decreased with treatment of ABT-737 in xenograft tumor tissue, as assessed by immunohistochemistry (IHC). To further assess the feasibility of IHC assessment in human tissue for future studies, we assessed LC3 and p62 in human samples of low (Gleason Score 7) and high (Gleason Score 9) grade tumors. As shown in Figure 4I–L, LC-3 and p62 staining by IHC was feasible in human tumor, supporting future studies that assess the ability of IHC to predict outcome and distinguish low from high grade tumors.
Figure 4.
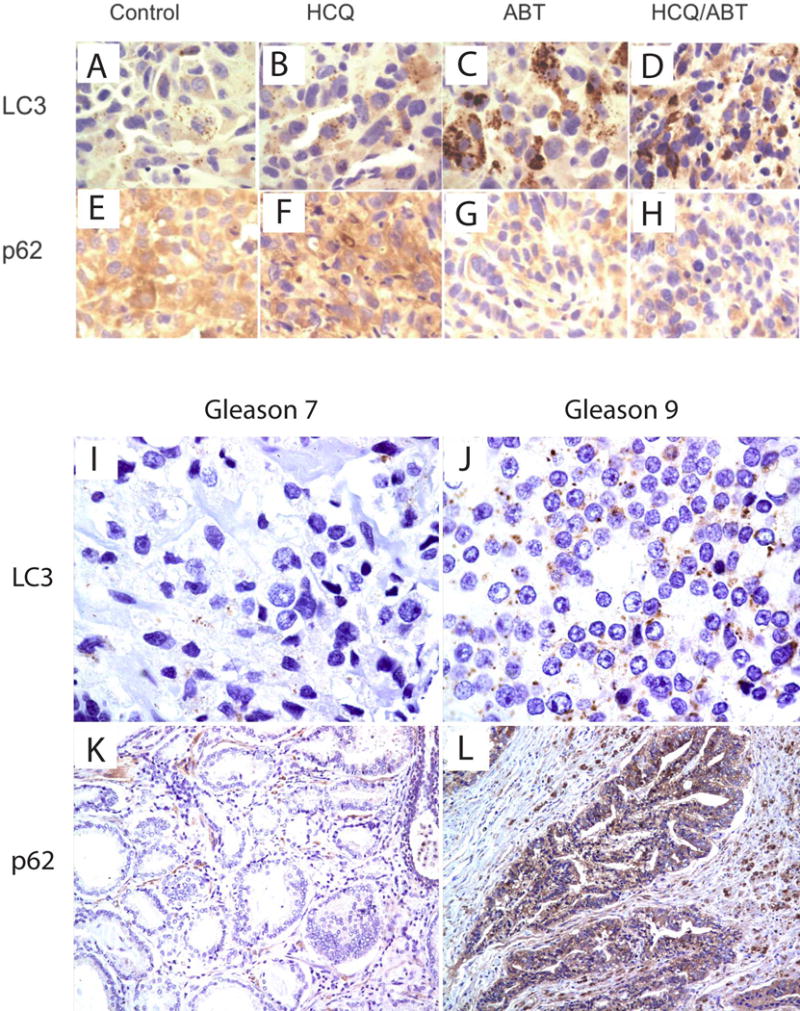
Assessment of LC-3 and p62 in human tumor xenograft and human tissue. 4A-H: Histologic sections were obtained from the tumors of the mice in each treatment group from xenograft studies. LC3 and p62 antibody staining was performed in the tumor tissue sections from Control (A and E), ABT-737 (C and G), HCQ (B and F) and combination of ABT-737 and HCQ (D and H) by immunohistochemistry at 14 days of treatment. 4I–L: Assessment of LC3 and p62 in human cancers with low (Gleason Score 7) and high (Gleason Score 9) grade tumors.
Assessment of ROS and mitochondrial function as mechanisms of cytotoxicity
To further assess the specific mechanisms of autophagy induced resistance, with prior evidence that autophagy reduces ROS and overall oxidative stress related to dysfunctional mitochondria (see Figure 5D), we assessed the role of ROS in HCQ and ABT-737 induced cytotoxicity. As shown in Figure 5A, colony forming units (CFU) increased with the addition of N-acetylcysteine (NAC) in cells treated with ABT-737, HCQ, and with the combination of ABT-737 and NAC, suggesting that ROS is increased with combination therapy and is a mechanism of cytotoxicity. Further evidence of a role of ROS is shown in Figure 5B, which shows increased ROS in cells treated with the ABT-737 combined with HCQ (green) compared to ABT-737 alone (blue), HCQ alone (yellow), and untreated control (red), by flow cytometry. As a measure of dysfunctional mitochondria and mitophagy, mCherry-Parkin expressing cells demonstrated increased activity in ABT-737 and combination therapy with HCQ and ABT-737, as shown in Figure 5C. Figure 5D: These data support the hypothesis that autophagy controls cellular stress including the abrogation of ROS production from dysfunctional mitochondria. Inhibition of autophagy with agents such as HCQ, therefore, hinder the autophagic protective effect and increase dysfunctional mitochondria (as shown in 5C) and ROS production (as shown in 5A and B).
Figure 5.
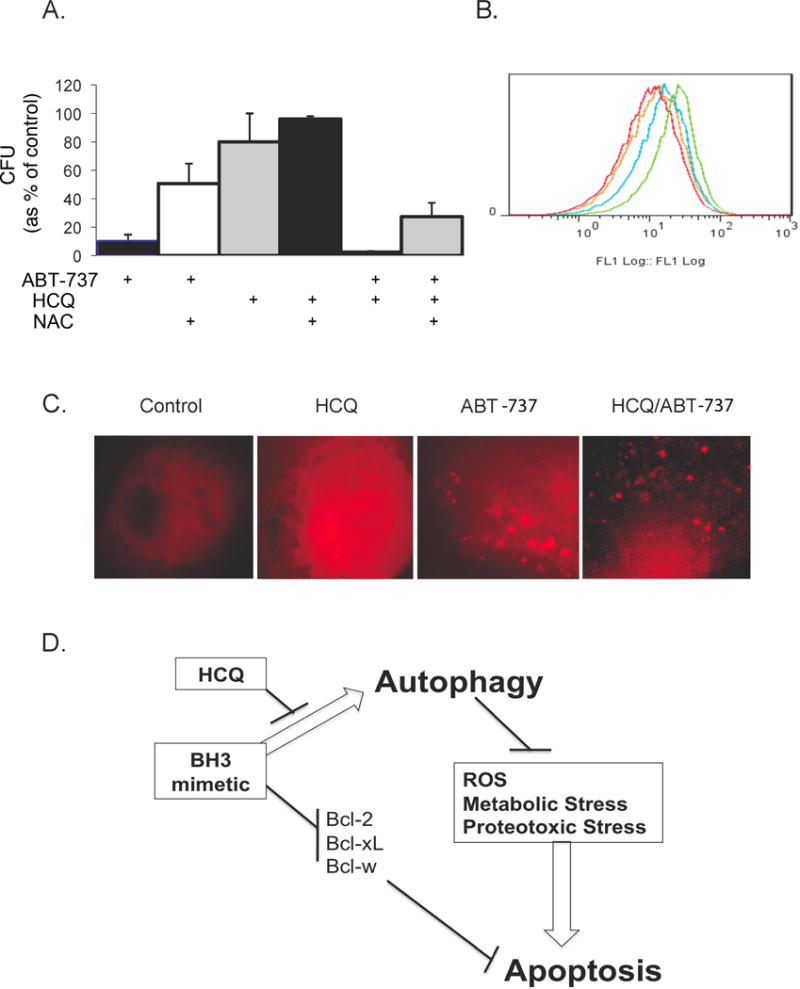
Effect of ABT-737 and HCQ on ROS and mitochondrial function. 5A: PC-3 cells were treated with 10uM of ABT-737 alone and in combination with 1mM N-acetylcysteine (first and second bars on the left) and assessed by clonogenic assay; treated with HCQ alone and in combination with N-acetylcysteine (middle two bars); or treated with the combination of both ABT-737 and HCQ with and without N-acetylcysteine (last two bars on right of figure 5A). 5B: ROS levels were determined using 10uM 2′-7′-dichlorodihydrofluorescene diacetate (DCF-DA, Molecular Probes) in cells treated with the combination of ABT-737 and HCQ (green), ABT-737 (blue), HCQ (yellow), or untreated control (red). 5C: mCherry-Parkin expressing PC-3 cells treated with 20uM of HCQ, 10uM of ABT-737 or the combination of ABT-737 and HCQ to identify dysfunctional mitochondria. 5D: a depiction of autophagy as a cell survival and resistance mechanism through abrogation of oxidative (ROS), metabolic and proteotoxic stress. Figure 5D shows a potential paradigm of optimal tumor targeting by dual inhibition of apoptosis and autophagy.
Discussion
We found in the current study that autophagy was a mechanism of resistance to ABT-737; that the combination of ABT-737 and HCQ resulted in enhanced cytotoxicity of prostate cancer cells and reduction of xenograft growth in a mouse model; developed the feasibility of measuring LC3 and p62 in xenograft and human tissue; and demonstrated that modulation of ROS is a mechanism of enhanced cytotoxicity with autophagy inhibition.
As a catabolic process by which cells degrade intracellular components in vesicles that fuse with lysosomes, autophagy degrades toxic protein aggregates, removes damaged organelles, and provides metabolic and biosynthetic substrates under stress conditions (10). As a pro-survival response, autophagy has been shown to be induced by metabolic stress and multiple cytotoxic therapies (12). Although multiple groups have demonstrated that autophagy represents a pro-survival response to stress, autophagy may also induce cell death in specific conditions, including prolonged periods of autophagic induction, supporting the need to assess the effect of autophagy with a particular condition or novel agent, as was done in this assessment of ABT-737. In a prior study, we demonstrated that metabolic stress and cytotoxicity induced by deoxyglucose in prostate cancer cells was attenuated by autophagy, as a pro-survival response, and increased cytotoxicity by inhibition of the autophagy regulator beclin1 (14). In our current study, the BH3 mimetic ABT-737 induced autophagy as a mechanism of drug resistance both in vitro (Figure 1A and B) and in vivo (Figure 4C and 4D), and cytotoxicity was increased by inhibition of beclin1 by siRNA (Figure 1C). These findings were consistent with recent studies demonstrating autophagy induction with the BH3 mimetic AT-101, and with ABT-737 in combination with celecoxib (15, 16). In an effort to develop a therapeutic regimen for future clinical studies, our results further demonstrated that HCQ, as an inhibitor of autophagy, increased cytotoxicity of ABT-737 in vitro (Figure 2) and in vivo (Figure 3).
We also demonstrated that ABT-737 induced LC3 autophagosome localization and decreased expression of p62 in tumor xenograft tissue (Figure 4A–H). This is consistent with in vitro results (Figure 1A) and prior results demonstrating increased LC3 localization and decreased p62 with induction of autophagy (10, 17). As a potential biomarker for future clinical trials, measurement of p62 may be complimentary to LC3. Both autophagy induction or inhibition distal in the autophagy pathway, such as lysosomal inhibition with HCQ, results in increased autophagosome accumulation with increased LC3 localization. In contrast, it has been demonstrated that p62 has a role to clear protein aggregates through autophagy, and that p62 expression decreases with autophagy induction through protein aggregate degradation (13) (18) (19). Although further clinical studies are warranted to confirm our findings, our initial results support these prior findings in vivo in an animal model demonstrating that induction with an agent such as ABT-737 results in increased LC3 and reciprocal decrease in p62 expression (Figure 4A–H).
In an effort to further understand the mechanism of enhanced cytotoxicity with autophagy inhibition, we found increased ROS with the combination of ABT-737 and HCQ, and dependence of cytotoxicity on ROS (Figure 5A and B). Prior studies supported the hypothesis that one mechanism of autophagy drug and tumor stress resistance is by modulation of oxidative damage through consumption of dysfunctional mitochondria, resulting in decreased ROS (11, 20). In agreement with these data, our current study demonstrated that use of N-acetylcysteine to suppress ROS restored tumor cell growth (Figure 5A) with ABT-737, and ABT-737 in combination with HCQ. Additionally, because prior studies suggest that a mechanism of decreased ROS by autophagy is through consumption of dysfunctional mitochondria (11), we assessed mitochondria using transient transfection with the mCherry-Parkin reporter, which is recruited to dysfunctional mitochondria prior to mitophagy (Figure 5C). Results in Figure 5C demonstrated that ABT-737 and the combination of ABT-737 with hydroxychloroquine had increased dysfunctional mitochondria consistent with the expectation, and demonstration, of increased ROS (Figure 5B).
In summary, taken together our data supports a new therapeutic paradigm to abrogate autophagy in combination with efforts to enhance apoptosis through targeting Bcl-2 or the Bcl-2 family of proteins. Our studies also support the assessment of LC3 and p62 as potential complimentary biomarkers of autophagy modulation in future clinical trials, as well as further mechanistic studies focused on mitochondrial health and function.
References
- 1.Castilla C, Congregado B, Chinchon D, Torrubia FJ, Japon MA, Saez C. Bcl-xL is overexpressed in hormone-resistant prostate cancer and promotes survival of LNCaP cells via interaction with proapoptotic Bak. Endocrinology. 2006;147:4960–7. doi: 10.1210/en.2006-0502. [DOI] [PubMed] [Google Scholar]
- 2.Yoshino T, Shiina H, Urakami S, et al. Bcl-2 expression as a predictive marker of hormone-refractory prostate cancer treated with taxane-based chemotherapy. Clin Cancer Res. 2006;12:6116–24. doi: 10.1158/1078-0432.CCR-06-0147. [DOI] [PubMed] [Google Scholar]
- 3.Lin Y, Fukuchi J, Hiipakka RA, Kokontis JM, Xiang J. Up-regulation of Bcl-2 is required for the progression of prostate cancer cells from an androgen-dependent to an androgen-independent growth stage. Cell Res. 2007;17:531–6. doi: 10.1038/cr.2007.12. [DOI] [PubMed] [Google Scholar]
- 4.Cragg MS, Harris C, Strasser A, Scott CL. Unleashing the power of inhibitors of oncogenic kinases through BH3 mimetics. Nat Rev Cancer. 2009;9:321–6. doi: 10.1038/nrc2615. [DOI] [PubMed] [Google Scholar]
- 5.Chonghaile TN, Letai A. Mimicking the BH3 domain to kill cancer cells. Oncogene. 2008;27(Suppl 1):S149–57. doi: 10.1038/onc.2009.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 7.Bray K, Chen HY, Karp CM, et al. Bcl-2 modulation to activate apoptosis in prostate cancer. Mol Cancer Res. 2009;7:1487–96. doi: 10.1158/1541-7786.MCR-09-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hikita H, Takehara T, Shimizu S, et al. The Bcl-xL inhibitor, ABT-737, efficiently induces apoptosis and suppresses growth of hepatoma cells in combination with sorafenib. Hepatology. 52:1310–21. doi: 10.1002/hep.23836. [DOI] [PubMed] [Google Scholar]
- 9.Kim KW, Moretti L, Mitchell LR, Jung DK, Lu B. Combined Bcl-2/mammalian target of rapamycin inhibition leads to enhanced radiosensitization via induction of apoptosis and autophagy in non-small cell lung tumor xenograft model. Clin Cancer Res. 2009;15:6096–105. doi: 10.1158/1078-0432.CCR-09-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amaravadi RK, Lippincott-Schwartz J, Yin XM, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 17:654–66. doi: 10.1158/1078-0432.CCR-10-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo JY, Chen HY, Mathew R, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25:460–70. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–16. doi: 10.1158/1078-0432.CCR-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–75. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DiPaola RS, Dvorzhinski D, Thalasila A, et al. Therapeutic starvation and autophagy in prostate cancer: a new paradigm for targeting metabolism in cancer therapy. Prostate. 2008;68:1743–52. doi: 10.1002/pros.20837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang S, Sinicrope FA. Celecoxib-induced apoptosis is enhanced by ABT-737 and by inhibition of autophagy in human colorectal cancer cells. Autophagy. 6:256–69. doi: 10.4161/auto.6.2.11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao P, Bauvy C, Souquere S, et al. The Bcl-2 homology domain 3 mimetic gossypol induces both Beclin 1-dependent and Beclin 1-independent cytoprotective autophagy in cancer cells. J Biol Chem. 285:25570–81. doi: 10.1074/jbc.M110.118125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanida I, Waguri S. Measurement of autophagy in cells and tissues. Methods Mol Biol. 648:193–214. doi: 10.1007/978-1-60761-756-3_13. [DOI] [PubMed] [Google Scholar]
- 18.Stein M, Lin H, Jeyamohan C, et al. Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate. 70:1388–94. doi: 10.1002/pros.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watanabe Y, Tanaka M. p62/SQSTM1 in autophagic clearance of a non-ubiquitylated substrate. J Cell Sci. doi: 10.1242/jcs.081232. [DOI] [PubMed] [Google Scholar]
- 20.Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A. 2009;106:2770–5. doi: 10.1073/pnas.0807694106. [DOI] [PMC free article] [PubMed] [Google Scholar]