

Abstract
Hyperpolarization-activated cyclic nucleotide–regulated cation (HCN) channels generate the hyperpolarization-activated cation current Ih present in many neurons. These channels are directly regulated by the binding of cAMP, which both shifts the voltage dependence of HCN channel opening to more positive potentials and increases maximal Ih at extreme negative voltages where voltage gating is complete. Here we report that the HCN channel brain-specific auxiliary subunit TRIP8b produces opposing actions on these two effects of cAMP. In the first action, TRIP8b inhibits the effect of cAMP to shift voltage gating, decreasing both the sensitivity of the channel to cAMP (K1/2) and the efficacy of cAMP (maximal voltage shift); conversely, cAMP binding inhibits these actions of TRIP8b. These mutually antagonistic actions are well described by a cyclic allosteric mechanism in which TRIP8b binding reduces the affinity of the channel for cAMP, with the affinity of the open state for cAMP being reduced to a greater extent than the cAMP affinity of the closed state. In a second apparently independent action, TRIP8b enhances the action of cAMP to increase maximal Ih. This latter effect cannot be explained by the cyclic allosteric model but results from a previously uncharacterized action of TRIP8b to reduce maximal current through the channel in the absence of cAMP. Because the binding of cAMP also antagonizes this second effect of TRIP8b, application of cAMP produces a larger increase in maximal Ih in the presence of TRIP8b than in its absence. These findings may provide a mechanistic explanation for the wide variability in the effects of modulatory transmitters on the voltage gating and maximal amplitude of Ih reported for different neurons in the brain.
INTRODUCTION
The electrical activity of the nervous system depends on the precise tuning of the electrophysiological properties of neurons through various mechanisms. The interaction of auxiliary subunits of voltage-gated channels with their pore-forming α-subunits provides one powerful means of regulating neural function by controlling channel expression and gating (Arikkath and Campbell, 2003; Vacher and Trimmer, 2011). The actions of neurotransmitters to modulate voltage-gated channel function through various second messenger signaling pathways provide another means for the more dynamic control of neural firing properties. We now find that the cAMP-dependent modulation of hyperpolarization-activated cyclic nucleotide–regulated cation (HCN) channels undergoes a surprising form of regulation by the brain-specific auxiliary HCN channel subunit TRIP8b (Santoro et al., 2004).
HCN channels are composed of four pore-forming α-subunits encoded by members of a small gene family (HCN1–4) that is part of the larger voltage-gated channel superfamily (Robinson and Siegelbaum, 2003; Biel et al., 2009). Unlike most voltage-gated channels, the HCN channels are nonselective cation channels that are activated by membrane hyperpolarization, resulting in the hyperpolarization-activated cation current, Ih. The binding of cAMP to the highly conserved HCN cytoplasmic C-terminal cyclic nucleotide–binding domain (CNBD) enhances channel opening by shifting the voltage dependence of HCN channel gating to more positive potentials (DiFrancesco and Tortora, 1991; Wainger et al., 2001). This effect is sometimes associated with an increase in the maximal current through the population of HCN channels (Imax) at strongly hyperpolarized voltages (Craven and Zagotta, 2004; Shin et al., 2004). These results are consistent with a model in which channel opening consists of a voltage-dependent activation step coupled to a voltage-independent opening step: cAMP binding enhances channel opening and shifts the voltage dependence of gating to more positive potentials by stabilizing the closed to open transition of the channel (Zhou and Siegelbaum, 2007).
One puzzling aspect of the actions of modulatory transmitters on Ih is that the relative extent by which they alter maximal Ih or shift the voltage dependence of channel gating can vary widely among different classes of neurons. In most neurons, neurotransmitters act through second messenger cascades primarily to shift the voltage dependence of Ih gating; in some cells, however, transmitters produce substantial changes in maximal Ih elicited by voltage steps to extreme negative potentials where voltage gating has reached completion, with or without an accompanying shift in voltage gating (Bobker and Williams, 1989; McCormick and Williamson, 1991; Kiehn and Harris-Warrick, 1992; Larkman and Kelly, 1992; Erickson et al., 1993; Gasparini and DiFrancesco, 1999; Bickmeyer et al., 2002; Schweitzer et al., 2003; Frère and Lüthi, 2004; Battefeld et al., 2010; Heys and Hasselmo, 2012).
Here we report that the binding of TRIP8b to the HCN2 α-subunit differentially alters the two major actions of cAMP on HCN channel function. TRIP8b, the major auxiliary subunit of HCN channels in the brain, controls HCN channel membrane trafficking, dendritic localization, and cAMP-dependent voltage gating (Santoro et al., 2004, 2009, 2011; Lewis et al., 2009; Zolles et al., 2009; Han et al., 2011; Piskorowski et al., 2011). TRIP8b undergoes extensive alternative splicing at its N terminus, generating at least 10 splice variants expressed in the brain that produce diverse effects to either enhance or suppress channel plasma membrane expression. However, all isoforms exert an identical action to inhibit the ability of cAMP to shift HCN channel opening to more positive potentials (Santoro et al., 2009; Zolles et al., 2009), an effect which has been mapped to a specific interaction site. TRIP8b interacts with HCN channels at two distinct sites: an upstream site in which a conserved central core region of TRIP8b binds to the HCN CNBD and a downstream site in which the C-terminal tetratricopeptide repeat (TPR) domain of TRIP8b interacts with the Ser-Asn-Leu (SNL) tripeptide at the C terminus of the channel (Lewis et al., 2009; Han et al., 2011; Santoro et al., 2011). It is the interaction of the central core domain of TRIP8b with the CNBD that is responsible for the effect of the auxiliary subunit to antagonize the action of cAMP (Santoro et al., 2011).
The precise mechanism by which TRIP8b binding reduces the response to cAMP is controversial. In a biochemical study, Han et al. (2011) have suggested that TRIP8b directly antagonizes the binding of cAMP by a competitive interaction with the ligand-binding site of the CNBD. In contrast Zolles et al. (2009), using an electrophysiological approach, reported that TRIP8b reduces the maximal voltage shift in response to saturating concentrations of cAMP, ΔVmax, an effect incompatible with direct competition. No study to date has addressed whether TRIP8b binding alters the ability of cAMP to enhance maximal HCN channel current.
Here we have investigated the effects of TRIP8b on HCN channel function using both TRIP8b-HCN2 fusion proteins and direct application of the TRIP8b core region to HCN2 channels in inside-out patches. Our results indicate that TRIP8b acts through an allosteric mechanism to decrease the affinity of the channel for cAMP. Moreover, we report a previously uncharacterized action of TRIP8b to reduce maximal current through the channel in the absence of cAMP. By reversing this action of TRIP8b, cAMP produces an increase in maximal current significantly greater than that seen in the absence of the auxiliary subunit. Thus, TRIP8b exerts opposing influences on the two major actions of cAMP on HCN channel function. The auxiliary subunit reduces the effect of cAMP to shift the voltage dependence of channel gating but enhances the action of cAMP to increase maximal current. These effects have important implications for the physiological actions of cAMP to alter neuronal excitability.
MATERIALS AND METHODS
Constructs and expression
All constructs were cloned in pGHE or pGH19 vectors, linearized, and transcribed into cRNA using T7 polymerase (MessageMachine; Ambion) as described previously (Santoro et al., 2004, 2009). cDNA clones encoding HCN2 and TRIP8b both correspond to the Mus musculus sequence. GFP, TRIP8b, and TRIP8bcore fused to HCN2 channels were created as described previously (Santoro et al., 2011). Site-directed mutagenesis was performed using either the QuikChange Mutagenesis kit (Agilent Technologies) or PCR cloning. Xenopus laevis oocytes were injected with 30–50 nl cRNA solutions at a concentration of 0.5–1 µg/µl.
Biochemical binding assays
The yeast two-hybrid assay and coimmunoprecipitation techniques used here were described in detail in a previous publication (Santoro et al., 2011). In brief, yeast two-hybrid assays were performed using the Grow’N’Glow Two-Hybrid kit (Bio 101) and yeast strain EGY48. Bait constructs representing the indicated HCN1 channel domains were cloned into vector pEG202, and prey constructs representing the indicated TRIP8b domains (or mutants thereof) were cloned in vector pJG4-5. Bait and prey plasmids were cotransformed with reporter plasmid pGNG1, and cells were plated onto glucose-containing medium. Transformants were restreaked (in triplicate) on galactose+/Leu− selective medium and screened for positive GFP expression under a UV light after 3–5 d of growth. For coimmunoprecipitation, Xenopus oocytes were injected with 50 nl of cRNA solution each, at a concentration of 1.0 µg/µl for HCN1 channel constructs and 0.2 µg/µl for TRIP8b or GFP-TRIP8b fusion constructs. Oocytes were collected 3 d after injection, and protein extracts were prepared in ice-cold lysis buffer, followed by coimmunoprecipitation and Western blot analysis as described previously (Santoro et al., 2011). Primary antibodies used were anti-HCN1 (rat monoclonal 7C3; gift of F. Müller [Institute of Complex Systems, Jülich, Germany] and U.B. Kaupp [Center of Advanced European Studies and Research, Bonn, Germany]), anti-TRIP8b (rabbit polyclonal 794; Santoro et al., 2009), and anti-GFP (290; Abcam). HRP-anti–rat conjugate (Jackson ImmunoResearch Laboratories, Inc.) or HRP-anti–rabbit conjugate (Cell Signaling Technology) was used as a secondary antibody. The protein bands were visualized by chemiluminescence using SuperSignal reagent (Thermo Fisher Scientific).
Affinity purification of TRIP8bcore peptide
A cDNA fragment encoding residues 223–303 (TRIP8bcore) of TRIP8b (1a-4) was cloned into vector pET52b (EMD Millipore) downstream of a Strep (II) tag sequence. The plasmid was transformed into Escherichia coli BL21 Rosetta strain (EMD Millipore) under ampicillin selection. Cells were grown at 37°C in Luria broth to 0.6 OD600 and induced with 0.4 mM isopropyl-1-thio-d-galactopyranoside. After 3 h, cells were collected by centrifugation, resuspended in ice-cold lysis buffer (150 mM NaCl, 100 mM Tris-Cl, pH 8, 1 mM EDTA, 1 mM β-mercaptoethanol, 5 mg/ml leupeptin, 1 mg/ml pepstatin, and 100 µM phenylmethylsulfonyl fluoride) with the addition of 10 µg/ml DNase and 0.25 mg/ml lysozyme, and sonicated on ice 12 times for 20 s, and the lysate was cleared by centrifugation for 30 min at 20,000 g. Protein was purified by affinity chromatography using StrepTrap HP columns (GE Healthcare) according to the manufacturer’s instructions and eluted in 150 mM KCl, 30 mM HEPES, pH 7.4, and 10% wt/vol glycerol plus 2.5 mM desthiobiotin. All purification steps were performed at 4°C and monitored using the ÄKTApurifier UPC 10 fast protein liquid chromatography system (GE Healthcare). The eluted protein was then loaded into HiLoad 16/60 Superdex 200 prep grade size exclusion column (GE Healthcare), which was equilibrated with 150 mM KCl, 30 mM HEPES, pH 7.4, and 10% wt/vol glycerol, and the protein purity was confirmed by SDS-PAGE.
Inside-out patch recordings and data analysis
Macroscopic currents were recorded from excised patches 2–3 d after cRNA injection using an EPC-9 amplifier and PULSE acquisition software (HEKA). Patch pipettes had resistances around 1 MΩ after fire polishing. External (pipette) solutions contained (mM): 96 KCl, 1 NaCl, 1 MgCl2, 1.8 CaCl2, and 10 HEPES, pH 7.4 (titrated with 50% KOH). Internal (bath) solutions contained (mM): 96 KCl, 1 NaCl, 10 HEPES, and 5 EGTA, pH 7.4. 3-s voltage steps were applied from a holding potential of −30 mV to a range of test potentials between −70 and −140 mV in 10-mV decrements, followed by a depolarizing step to −40 mV to measure tail currents. All recordings were obtained at room temperature (22–24°C). Peak tail current amplitudes were measured at either 0 mV for two electrode voltage clamp or −40 mV for patch clamp recordings after the decay of the capacitive transient, and tail current–voltage curves were fitted using the Boltzmann equation I(V) = A1 + A2/{1 + exp[(V − V1/2)/s]}, in which A1 is the offset caused by holding current, A2 is the maximal tail current amplitude, V is the test pulse voltage, V1/2 is the midpoint voltage of activation, and s is the slope factor (in mV). Because the 3-s long pulse was not sufficient to reach steady-state activation levels at less negative voltage steps, these represent isochronal activation curves rather than true steady-state curves. Nonetheless, they are likely to provide a good approximation of true V1/2 values, especially in the absence of cAMP or in the presence of relatively high cAMP levels (Wang et al., 2002) and have been routinely used in the literature because of problems with membrane breakdown with longer hyperpolarizations (Wainger et al., 2001; Zhou and Siegelbaum, 2007; Zolles et al., 2009).
The Hill equation was fitted to the cAMP dose–response data (Figs. 1, 3, and 7): ΔV1/2 = ΔVmax/{1 + (K1/2/[cAMP])h}, where ΔV1/2 is the V1/2 shift produced by a given cAMP concentration, ΔVmax is the maximal V1/2 shift produced by saturating cAMP, K1/2 is the concentration of cAMP producing half of the maximal shift, and h is the Hill coefficient. In Fig. 5 F, the Hill equation was fitted to the TRIP8bcore peptide dose–response data. The data analysis and function fitting were performed in PULSE FIT (HEKA) and Igor Pro (WaveMetrics).
Figure 1.

Effect of fusion of TRIP8b to HCN2 on relationship between [cAMP] and voltage dependence of channel gating. (A) Currents elicited by hyperpolarizing voltage steps in inside-out patches from oocytes expressing TRIP8b-HCN2 fusion channels or GFP-HCN2 channels in 0, 0.1, or 100 µM [cAMP]. The membrane was held at −40 mV for 0.5 s and then stepped for 3 s to test potentials from −70 to −140 mV in 10-mV decrements. (B and C) Normalized tail current G-V relationship for GFP-HCN2 (B) or TRIP8b-HCN2 (C) channels in the presence of 0, 0.1, or 100 µM [cAMP]. Fits of Boltzmann relation yield the following values for V1/2 and slope with different [cAMP]. GFP-HCN2 0 cAMP: V1/2 = −116.0 mV, s = 4.98; 0.1 cAMP: V1/2 = −105.7 mV, s = 5.33; and 100 cAMP: V1/2 = −96.9 mV, s = 4.65. TRIP8b-HCN2 0 cAMP: V1/2 = −114.8mV, s = 4.05; 0.1 cAMP: V1/2 = −113.3 mV, s = 4.91; and 100 cAMP: V1/2 = −104.2 mV, s = 4.42. (D) ΔV1/2 as a function of [cAMP] for GFP-HCN2, GFP-HCN2 + TRIP8b expressed as independent proteins, and TRIP8b-HCN2 fusion channels in inside-out patches. Solid lines show fits of Hill equation. Fits of the Hill equation yield GFP-HCN2: ΔVmax = 17.3 mV, K1/2 = 0.08 µM, h = 1.43; GFP-HCN2 + TRIP8b: ΔVmax = 14.2 mV, K1/2 = 0.19 µM, h = 0.83; and TRIP8b-HCN2: ΔVmax = 11.1 mV, K1/2 = 3.42 µM, h = 0.79. Error bars indicate SEM.
Figure 3.

TRIP8b core domain is necessary to antagonize action of cAMP on HCN2. (A) ΔV1/2 as a function of [cAMP] for TRIP8bΔInt-HCN2 and TRIP8bcore-HCN2 channels in inside-out patches, compared with GFP-HCN2 and TRIP8b-HCN2 channels. Solid lines show fits of the Hill equation. Fits of the Hill equation yield TRIP8bΔInt-HCN2: ΔVmax = 18.2 mV, K1/2 = 0.08 µM, h = 1.11; and TRIP8bcore-HCN2: ΔVmax = 13.3 mV, K1/2 = 190 µM, h = 0.69. Note that TRIP8bΔInt, with internal deletion of 22 residues of the core domain, fails to alter cAMP dose–response relation. (B) V1/2 values of various indicated constructs in the absence of cAMP show no significant differences (P > 0.05, ANOVA). Error bars indicate SEM.
Figure 7.

ΔV1/2 as a function of [cAMP] and [TRIP8bcore] polypeptide, fitted by the 12-state allosteric model. The TRIP8bcore polypeptide concentrations are indicated. The parameters of the front face of the model (corresponding to the six-state cyclic model) are adopted from Zhou and Siegelbaum (2007): L = 0.43, s = 4.4, = 0.844 µM, = 0.008 µM. The best fit of the model yields the following values for the other parameters: = 0.089 ± 0.027 µM, = 0.064 ± 0.010 µM, = 33.58 ± 8.18 µM, = 3.53 ± 1.33 µM. and are then derived from the other parameters: = = 3.54 µM and = = 28.24 µM. Error bars show SEM. See supplemental text for further details.
Figure 5.

Effects of TRIP8bcore polypeptide on action of cAMP on HCN2 channel voltage gating. (A) Representative experiment showing V1/2 of HCN2 channels in inside-out patches with solutions containing the indicated [cAMP] in the absence or presence of 4 µM TRIP8bcore polypeptide. Open circles indicate V1/2 in the absence of both cAMP and TRIP8bcore; closed circles indicate V1/2 in the presence of 0.1 or 10 µM cAMP, with or without 4 µM TRIP8bcore polypeptide, as indicated. (B) Normalized G-V relationship for HCN2 channel tail currents in 0, 0.1, or 10 µM cAMP. (C) Normalized G-V relationship for HCN2 channel tail currents in the presence of 4 µM TRIP8bcore polypeptide plus 0, 0.1, or 10 µM cAMP. (D) Normalized G-V relationship for HCN2 tail currents in the presence of 4 µM TRIP8bcore-mut polypeptide plus 0, 0.1, or 10 µM cAMP. (E) Shift in V1/2 as a function of [cAMP] in the absence (open circles) or presence (closed circles) of 4 µM TRIP8bcore polypeptide. Solid lines show fits of the Hill equation, which yield HCN2: ΔVmax = 18.1 mV, K1/2 = 0.08 µM, h = 0.86; and HCN2 plus 4 µM TRIP8bcore: ΔVmax = 15.8 mV, K1/2 = 5.64 µM, h = 0.86. (F) Percent decrease in ΔV1/2 produced by the indicated concentrations of cAMP as a function of TRIP8bcore concentration (left ordinate). Fits of the Hill equation (solid lines) yield 0.1 µM cAMP: K1/2 = 0.22 µM, h = 1.35, percent maximal decrease in ΔV1/2 = 100%; 1 µM cAMP: K1/2 = 1.57 µM, h = 1.69, percent maximal decrease in ΔV1/2 = 97%; 10 µM cAMP: K1/2 = 2.34 µM, h = 1.19, percent maximal decrease in ΔV1/2 = 69%; and 100 µM cAMP: K1/2 = 3.96 µM, h = 0.74, percent maximal decrease in ΔV1/2 = 42%. The Hill fit to the TRIP8b dose–response curve for HCN2 maximal current reduction from Fig. 4 B is replicated here for comparison (red dashed line, right ordinate). Error bars indicate SEM.
Model fitting
The modulation of HCN2 channel opening by voltage, cAMP, and TRIP8b was described by a 12-state allosteric model (Fig. 6). Definitions of all terms are described in Fig. 6. The open probability of the channel is determined from the equation
| (1) |
where
Figure 6.

12-state allosteric model for regulation of HCN2 channel opening by voltage, cAMP, and TRIP8b. The vertical and front-back transitions of the cubic scheme represent the cAMP and TRIP8b binding reactions to the channel, respectively. The two horizontal transitions are the voltage-dependent activation step reflecting voltage sensor movement, followed by a voltage-independent opening step. The front face of the cube is the six-state cyclic allosteric model that represents the effects of voltage and cAMP on channel opening in the absence of bound TRIP8b (Zhou and Siegelbaum, 2007); the top face is a six-state cyclic allosteric model that represents the actions of voltage and TRIP8b binding on channel opening in the absence of bound cAMP. TRIP8b and cAMP can both bind to the channel at the same time, as represented by the back and bottom faces of the cube. Definition of states and ligands: CR, unliganded closed channel with voltage sensor in the resting state; CA, unliganded closed channel with voltage sensor in the activated state; O, unliganded channel in the open state; A, cAMP; T, TRIP8b. Definition of parameters: KV, equilibrium constant for transition of closed channel transition between resting state and activated state; L, intrinsic equilibrium constant for channel opening; and dissociation equilibrium constants for cAMP binding to closed and open states, respectively; and dissociation equilibrium constants of TRIP8b binding to channel in closed and open states, respectively; and dissociation equilibrium constants for cAMP binding to TRIP8b-bound channels in closed and open states, respectively; and dissociation equilibrium constants for TRIP8b binding to cAMP-bound channels in closed and open states, respectively.
| (2) |
Solving Eqs. 1 and 2 yields the voltage shift produced by a given concentration of cAMP (A) in the presence of a given concentration of TRIP8b (T)
| (3) |
The relationship in Eq. 3 for ΔV1/2 as a function of cAMP concentration and TRIP8b concentration was used in the model fitting of Fig. 7.
At extreme negative voltages in the absence of cAMP, the maximal open probability is given by
The current reduction caused by the TRIP8bcore polypeptide can be described from the following relationship:
Fitting of the current reduction as a function of TRIP8bcore peptide concentration (data in Fig. 4 B) by the model yields the following values: = 0.34 µM and = 1.90 µM. The fitting of the model was conducted in MATLAB with the fitnlm function.
Figure 4.

Direct application of TRIP8bcore polypeptide to inside-out patches suppresses HCN2 maximal current in the absence of cAMP. (A) Representative experiment showing effects of TRIP8bcore polypeptide on HCN2 currents in inside-out patches elicited by a hyperpolarization to −140 mV, either in the absence or presence of cAMP. The current recording protocol is described in Fig. 3. The internal bath solution contained control solution, 4 µM TRIP8bcore with no cAMP, 4 µM TRIP8bcore plus 100 µM cAMP, or 100 µM cAMP with no TRIP8b. (B) Dose–response curve for the percent reduction in current amplitude at extreme negative voltages as a function of TRIP8bcore polypeptide concentration (in the absence of cAMP). Error bars show SEM. (C) The binding activity of the HCN1 C-linker/CNBD (residues 390–611) with WT and mutant TRIP8b (constant region, exons 5–16) assessed using a yeast two-hybrid assay. For TRIP8b, the yellow square represents the conserved core region. In TRIP8bcore-mut, the WT EEEFE core residues are substituted by RRRAR. The red colors denote the TPR domains. Activity was detected by transactivation of a GFP reporter gene. “+++” indicates very strong fluorescence; “−” indicates no detectable fluorescence (see Santoro et al. [2011]). (D) Representative HCN2 currents before (black trace) and after (blue trace) the application of TRIP8bcore-mut polypeptide to inside-out patches. The current recording protocol is described in Fig. 3.
Online supplemental material
Detailed derivations and fitting procedures are provided in the supplemental text. Online supplemental material is available at http://www.jgp.org/cgi/content/full/jgp.201311013/DC1.
RESULTS
TRIP8b exerts opposing actions on the ability of cAMP to shift voltage-dependent gating and increase maximal current through HCN2 channels
In this study, we examined the biophysical mechanisms that underlie the action of TRIP8b to alter the response of HCN channels to cAMP by addressing two questions: (1) How does the binding of TRIP8b to HCN channels inhibit the effect of cAMP to shift HCN channel voltage gating to more positive voltages, and (2) does TRIP8b alter the action of cAMP to enhance the maximal tail current carried by HCN channels after steps to extreme negative voltages? We focused on the interaction of TRIP8b with HCN2 because this α-subunit forms channels that respond to cAMP with a large depolarizing voltage shift and noticeable enhancement in maximal current.
In a previous study, our laboratory found that the action of TRIP8b to antagonize the cAMP-dependent shift in HCN1 voltage gating observed in intact cells was greatly diminished upon patch excision when TRIP8b and HCN1 were expressed independently, perhaps because of instability of the complex and/or loss of some intracellular modulatory factor (Santoro et al., 2009). In contrast, we found that the regulatory effect of TRIP8b was robustly maintained in cell-free patches with a TRIP8b-HCN1 fusion protein (Santoro et al., 2009). In our present study of the action of TRIP8b on HCN2, we have therefore covalently linked the N terminus of HCN2 to either the C terminus of TRIP8b or to GFP (as a control).
As shown in Fig. 1, application of cAMP to GFP-HCN2 channels in inside-out patches produces a large, dose-dependent depolarizing shift in the voltage dependence of channel activation and increases the rate of channel opening, actions which are identical to those seen in WT HCN2 channels (Pian et al., 2006). In cell-free patches in the absence of cAMP, TRIP8b-HCN2 channels exhibit a similar voltage dependence and rate of activation compared with GFP-HCN2 channels (Fig. 1, A–C). However, TRIP8b-HCN2 channels show a marked reduction in sensitivity to cAMP compared with GFP-HCN2 channels (Fig. 1, A–C). An examination of the cAMP dose–response curves for GFP-HCN2 and TRIP8b-HCN2 demonstrates that the presence of TRIP8b causes a 40-fold increase in the concentration of cAMP required to produce a half-maximal shift in the voltage dependence of channel activation (K1/2; Fig. 1 D). Moreover, there is a 40% decrease in the maximal shift in the V1/2 in response to saturating concentrations of cAMP (ΔVmax), a measure of ligand efficacy (Fig. 1 D).
We found a qualitatively similar inhibitory action on the modulatory effects of cAMP when TRIP8b is coexpressed with GFP-HCN2 as independent proteins (Fig. 1 D). However, similar to our previous results with HCN1 (Santoro et al., 2009), the quantitative extent of inhibition produced by the independent TRIP8b protein is much less than with the TRIP8b-HCN2 fusion. Thus, expression of TRIP8b with GFP-HCN2 causes only a 2.5-fold increase in the K1/2 for cAMP and a 20% decrease in ΔVmax. Our results differ slightly from those of Zolles et al. (2009), who found that coexpression of TRIP8b with HCN2 caused a somewhat larger decrease in ΔVmax (∼40%) and produced a slight decrease in the K1/2 (∼40%) for cAMP. These discrepancies may reflect small differences in recording conditions or relative levels of protein expression. We also found that the Hill coefficient is reduced by ∼44% by TRIP8b coexpression or fusion to HCN2 (from 1.43 to ∼0.8), suggesting that TRIP8b might inhibit the cooperativity of cAMP binding. Neither TRIP8b coexpression or fusion to HCN2 alters the voltage dependence of HCN2 channel gating in the absence of cAMP, indicating that this regulatory subunit has minimal effect on the basal channel voltage gating (V1/2 values: GFP-HCN2, −116.2 ± 0.86 mV; GFP-HCN2 + TRIP8b, −116.0 ± 0.98 mV; and TRIP8b-HCN2, −114.7 ± 0.74 mV).
Next we asked whether TRIP8b alters the action of cAMP to enhance maximal HCN2 current after voltage steps to extreme hyperpolarized potentials. Unexpectedly, we found that fusion of TRIP8b to HCN2 increases the extent to which cAMP enhances Imax (Fig. 2). Thus, whereas cAMP increases maximal current in GFP-HCN2 channels by only 37 ± 9%, the nucleotide enhances maximal current in TRIP8b-HCN2 channels by 83 ± 9%, more than a twofold increase (P < 0.05; n = 10). Thus, TRIP8b exerts opposing actions on the two modulatory effects of cAMP: TRIP8b inhibits the ability of cAMP to shift voltage gating to more positive potentials, whereas it increases the action of cAMP to enhance maximal current.
Figure 2.

cAMP causes a larger increase in maximal current with TRIP8b-HCN2 and TRIP8bcore-HCN2 channels than with GFP-HCN2 channels. The membrane was held at −40 mV and then hyperpolarized to −140 mV with a 3-s test pulse, followed by a depolarizing pulse to −40 mV to measure the tail current. (A) Representative currents for GFP-HCN2, TRIP8b-HCN2, and TRIP8bcore-HCN2 channels before (black traces) and after (red traces) application of saturating concentrations of cAMP (100 µM for GFP-HCN2 and 1 mM for the other two channels) to inside-out patches. (B) Percent increase in maximal tail current amplitude in response to cAMP for GFP-HCN2, TRIP8b-HCN2, and TRIP8bcore-HCN2; error bars indicate SEM. Mean percent increases in maximal current ± SEM are as follows: GFP-HCN2: 37 ± 9% (n = 9); TRIP8b-HCN2: 83 ± 9% (n = 10); and TRIP8bcore-HCN2: 116 ± 18% (n = 9). Current amplitude increase with GFP-HCN2 by cAMP is significantly less than that seen with TRIP8b-HCN2 and TRIP8bcore-HCN2 (*, P < 0.05, ANOVA). There is no significant difference in current increase between the latter two constructs (P > 0.05, t test).
An 81–amino acid domain in the conserved core of TRIP8b is both necessary and sufficient to produce the opposing actions of TRIP8b on cAMP-dependent modulation of HCN2
Are the two distinct actions of TRIP8b mediated by the same region of the molecule? We previously found that an 81–amino acid core region of TRIP8b, TRIP8bcore corresponding to residues 223–303 in TRIP8b(1a-4), is sufficient to fully reproduce the effect of full-length TRIP8b to antagonize the actions of cAMP on HCN1 channel voltage gating, based on the action of the TRIP8bcore-HCN1 fusion protein in intact oocytes (Santoro et al., 2011). However, these experiments did not quantify the effect of the TRIP8b core region on the relation between cAMP concentration and the voltage shift in HCN channel gating. Moreover, these experiments did not examine whether the core region could mimic the effect of full-length TRIP8b to enhance the ability of cAMP to increase HCN channel maximal current.
We therefore next examined the effect of cAMP on voltage gating and maximal current of TRIP8bcore-HCN2 fusion protein channels. We found that the relatively small core region is necessary and sufficient to both inhibit the ability of cAMP to alter voltage gating and facilitate the action of cAMP to increase maximal current (Figs. 2 and 3 A). Surprisingly, the magnitude of some of the effects of TRIP8bcore on the cAMP-dependent modulation of HCN2 are even greater than those seen with full-length TRIP8b-HCN2. Thus, TRIP8bcore-HCN2 fusion protein channels exhibit a >2,000-fold increase in the K1/2 for cAMP relative to HCN2 alone, a 50-fold larger effect than seen with the full-length TRIP8b-HCN2 fusion (Fig. 3 A). The TRIP8bcore-HCN2 channels also display a decrease in ΔVmax with saturating cAMP (∼27% decrease). cAMP increased the maximal current carried by TRIP8bcore-HCN2 channels by 116 ± 18%, slightly greater than the 83% increase in maximal current seen with cAMP with full-length TRIP8b fusion protein channels (P > 0.05; n = 9; Fig. 2). We next confirmed previous results obtained using HCN1 channels in intact oocytes, that the TRIP8b core is necessary for the action of full-length TRIP8b to inhibit the effects of cAMP on voltage gating (Santoro et al., 2011). Thus, a fusion protein consisting of HCN2 plus full-length TRIP8b lacking only 22 amino acids in the core domain (TRIP8bΔInt-HCN2) generates channels whose response to cAMP is identical to that of HCN2 expressed alone without TRIP8b (Fig. 3 A). These results further suggest that the alterations in the regulatory actions of cAMP with TRIP8b fusion proteins are not the result of some nonspecific effects of the fusion per se, as the effects are completely dependent on the short internal stretch of 22 amino acids. Consistent with results using full-length TRIP8b, neither the TRIP8bΔInt nor the TRIP8bcore fusion proteins show altered voltage-dependent gating in the absence of cAMP (Fig. 3 B), further indicating that basal voltage gating is not affected by this regulatory subunit.
Effects of acute application of a TRIP8bcore peptide to HCN2 channels
How does TRIP8b exert its opposing effects on the cAMP-dependent modulation of HCN2, inhibiting the action of cAMP to shift voltage gating while enhancing the action of cAMP to increase maximal current? Previous studies have suggested that channel opening involves a voltage-dependent activation step followed by a voltage-independent opening reaction. cAMP binding is thought to stabilize the open state of the channel, thereby enhancing maximal open probability and, thereby, maximal current (Shin et al., 2004; Chen et al., 2005; Zhou and Siegelbaum, 2007). The relatively large maximal open probability of HCN2 in the absence of cAMP (>0.5) normally limits the extent to which cAMP can enhance maximal current (Zhou and Siegelbaum, 2007). We therefore hypothesized that TRIP8b might enhance the ability of cAMP to increase maximal current by depressing maximal channel open probability in the absence of cAMP, thereby providing a larger dynamic range by which cAMP can increase channel opening.
To directly address this possibility, we examined the effect of acute application of a soluble, purified TRIP8b core peptide (TRIP8b residues 223–303) to HCN2 channels in cell-free inside-out patches (using oocytes where TRIP8b was not coexpressed with HCN2). Consistent with the above hypothesis, application of 4 µM TRIP8b core peptide to HCN2 channels in the absence of cAMP causes a marked, ∼40%, decrease in current amplitude in response to a hyperpolarizing voltage step to −140 mV, a potential at which voltage-dependent activation is complete (Fig. 4 A). In contrast, the core peptide produces no detectable change in the voltage dependence of channel gating (Fig. 3 B). Over a range of concentrations, the core peptide causes a dose-dependent reduction in HCN2 channel maximal current (in the absence of cAMP), resulting in a 60% decrease in current at 40 µM, the maximum concentration of core peptide that was soluble (Fig. 4 B).
To determine whether the effects of TRIP8bcore on maximal current are specific, we examined the action of a mutant core peptide in which a conserved pentapeptide sequence (EEEFE) is replaced with a charge-reversed sequence (RRRAR; Fig. 4, C and D). In a yeast two-hybrid assay, we find that this mutation abolishes the ability of TRIP8b to bind to the HCN1 CNBD (Fig. 4 C). Application of this mutant peptide to HCN2 channels has no effect on maximal current, arguing for a specific action dependent on the conserved core sequence (Fig. 4 D).
Is the effect of the core peptide to reduce maximal current related in any way to the action of cAMP to enhance maximal current? The two processes do indeed interact as the application of 100 µM cAMP fully reverses the inhibitory action of the core peptide on maximal current. Thus, cAMP application is able to increase maximal current to a level similar to that seen when cAMP is applied in the absence of core peptide (Fig. 4 A). As a result, cAMP application in the presence of core peptide produces a larger percentage increase in maximal current amplitude compared with that seen when cAMP is applied in the absence of core peptide. This action therefore accounts for the increased effect of cAMP to enhance maximal current observed above with TRIP8b-HCN2 or TRIP8bcore-HCN2 channels (Fig. 2).
We next asked whether acute application of the core peptide also inhibits the ability of cAMP to shift the voltage dependence of HCN2 opening to more positive potentials, similar to what we observe when the core peptide is fused to HCN2. Indeed, application of the core peptide rapidly antagonizes the ability of cAMP to shift HCN2 gating to more positive potentials (Fig. 5). At a concentration of 4 µM, TRIP8bcore almost fully abolishes the voltage shift in response to 0.1 µM cAMP and significantly reduces the voltage shift with 10 µM cAMP (Fig. 5, A–C). Importantly, the effect of cAMP recovers rapidly after wash-out of the peptide (Fig. 5 A), indicating that the binding between TRIP8b and HCN2 is reversible. This rapid reversibility may explain the finding that the modulatory action of TRIP8b on HCN channel function is diminished after patch excision when TRIP8b and HCN subunits are coexpressed as independent proteins (Santoro et al., 2009, 2011). Importantly, the action of the core peptide to inhibit the effect of cAMP to shift HCN2 voltage gating results from a specific action of the peptide as it is prevented by the EEEFE to RRRAR mutation (Fig. 5 D).
Examination of the action of a range of cAMP concentrations reveals that, at a concentration of 4 µM, the TRIP8bcore peptide shifts the cAMP dose–response curve to higher agonist concentrations, causing a 70-fold increase in the K1/2 for cAMP and a small decrease in ΔVmax (Fig. 5 E). The shift in K1/2 with 4 µM TRIP8bcore is intermediate between the results with TRIP8b-HCN2 and TRIP8bcore-HCN2 fusion protein channels, suggesting that the local effective concentration of TRIP8b in the two fusion proteins may be, respectively, lower and higher than 4 µM. Alternatively, the efficacy or affinity of the core peptide may differ from that observed with the two fusion protein channels.
We next explored the effect of a range of concentrations of TRIP8bcore on the action of cAMP. The core peptide produces a dose-dependent inhibition of the effect of a given concentration of cAMP to shift the V1/2 to more positive potentials (Fig. 5 F). Moreover, as the cAMP concentration is increased, higher concentrations of TRIP8bcore are required to produce a given level of inhibition of the cAMP response. Thus, TRIP8b and cAMP exert reciprocal inhibitory effects on each other’s action. TRIP8b shifts the dose–response curve for cAMP to higher concentrations, and conversely, cAMP produces a concentration-dependent shift in the dose–response curve for TRIP8b. Although these results are consistent with a competition of cAMP and TRIP8b for a single binding site, the finding that TRIP8b reduces the maximal response to saturating concentrations of cAMP is more consistent with an allosteric interaction, as discussed in the next section.
TRIP8b inhibits the effect of cAMP to facilitate HCN2 voltage gating through an allosteric mechanism
To explore the mechanism by which TRIP8b alters the function of HCN2 channels, we examined whether the actions of TRIP8b are consistent with an extension of a six-state cyclic allosteric model previously used to describe the modulatory action of cAMP on HCN2 gating in the absence of TRIP8b (Fig. 6; Zhou and Siegelbaum, 2007). According to this model, in the absence of cAMP, membrane hyperpolarization causes the channel to undergo a voltage-dependent transition from a closed resting state (CR) to a closed active state (CA), from which the channel undergoes a voltage-independent conformational change to the open state (O). cAMP (A) can bind and unbind the CNBD in all three states, yielding three corresponding agonist bound states: ACR, ACA, and AO. The key property of this model is that the opening transition is allosterically coupled to a conformational change in the CNBD that enhances the affinity of the open state for cAMP (dissociation constant ), relative to the affinity of the two closed states (identical dissociation constant ). Thus, cAMP binding to the open state is energetically more favorable than binding to the closed state, causing agonist binding to shift the equilibrium toward the open transition by the factor Because the voltage-independent opening reaction is kinetically coupled to the voltage-dependent activation step, the cAMP-dependent enhancement of channel opening both shifts the apparent voltage dependence of channel gating to more positive potentials and increases maximal open probability.
We incorporated TRIP8b (T) into this model by assuming it binds to the open and closed states of the channel, in both the cAMP-bound or unbound states. This results in an additional six states for the TRIP8b-bound channel, three states with the CNBD unoccupied by cAMP (TCR, TCA, and TO; Fig. 6, top face), and three states with the CNBD occupied by cAMP (ACRT, ACAT, and AOT; Fig. 6, back face). According to the model, TRIP8b binding may, in principle, alter the affinity of both the closed and open states for cAMP. Our finding that TRIP8b decreases ΔVmax implies that TRIP8b must reduce cAMP binding to the open state more than it reduces cAMP binding to the closed state. Finally, as the various states are incorporated in a cyclic reaction scheme, cAMP binding must necessarily reduce the affinity of the channel for TRIP8b. We adjusted the parameters of the model to obtain the best fit to the measured relation between [cAMP] and ΔV1/2, in the presence of 0–12 µM TRIP8bcore peptide (Fig. 7; see supplemental text for details).
The model provides a good fit to the cAMP dose–response curves in both the absence and presence of increasing concentrations of TRIP8b. In particular, the model reproduces the effect in which increasing concentrations of TRIP8b cause both a progressive shift in the cAMP dose–response curve to higher concentrations of cAMP and a progressive decrease in the maximal response to saturating cAMP (Fig. 7). Based on the parameters obtained from the fit (see legend to Fig. 7), we infer that binding of TRIP8b produces a 70-fold decrease in the affinity of the closed channel for cAMP. Conversely, binding of cAMP produces an identical decrease in the affinity of the closed channel for TRIP8b. For the open channel, cAMP or TRIP8b binding produces an even greater 300-fold decrease in affinity for the antagonistic ligand. The finding that the ratio (9.6) is much smaller than the ratio (105) indicates that TRIP8b binding does indeed reduce the affinity of the open state for cAMP to a greater extent than it reduces affinity of the closed state for cAMP. Moreover, this accounts for the action of TRIP8b to inhibit the efficacy with which cAMP shifts the voltage dependence of channel gating, resulting in the decrease in ΔVmax.
In principle, the model could also account for our observation that TRIP8b reduces HCN2 maximal current in the absence of cAMP (as shown in Fig. 4), if TRIP8b were to bind to the cAMP-free closed state more tightly than it were to bind to the cAMP-free open state. However, the best fit of the model yields nearly identical values for (0.089 ± 0.027 µM) and (0.064 ± 0.010 µM). A second discrepancy is seen when we independently adjusted and to fit the observed reduction in Imax (Fig. 4 B). In this case, the model predicts that TRIP8b should produce a −3-mV shift in the voltage dependence of gating in the absence of cAMP, an effect we do not observe (Fig. 3 B). Moreover the estimates of and obtained from the fits in the absence of cAMP are 4- to 30-fold larger ( = 0.34 µM; = 1.90 µM) than those obtained when we fit the relation between TRIP8b core polypeptide concentration and the magnitude of the shift in voltage gating with cAMP (compare Fig. 4 B with Fig. 5 F; see Model fitting subsection of Materials and methods).
The above discrepancies suggest that the effects of TRIP8b to inhibit maximal current amplitude in the absence of cAMP are mediated through an interaction with a separate low-affinity binding site on the channel distinct from the high-affinity site responsible for the action of TRIP8b to antagonize the cAMP-dependent shift in HCN2 voltage gating. Consistent with such a dissociation, we find that a point mutation in a conserved arginine, HCN2R591E, which profoundly disrupts the binding to the CNBD of both cAMP (Chen et al., 2001; Zhou and Siegelbaum, 2007) and TRIP8b (Han et al., 2011), has no effect on the ability of the TRIP8b core polypeptide to reduce HCN2 maximal current (Fig. 8). Collectively, these observations strongly argue that TRIP8b reduces maximal current through a kinetic and structural mechanism that is distinct from its action to antagonize the effect of cAMP on HCN2 voltage gating. We next explore in more detail the mechanism for this second action of TRIP8b to suppress maximal current.
Figure 8.

Importance of C-terminal regions of HCN2 for the ability of TRIP8b to reduce maximal current. (A) Representative currents through HCN2 WT and mutant channels before (black traces) and after (blue traces) the application of 4 µM TRIP8bcore polypeptide to inside-out patches (no cAMP present). HCN2ΔCNBD, truncation after residue V526 removing entire CNBD and all downstream residues; HCN2R591E, point mutation of conserved arginine in CNBD required for high-affinity cAMP binding; HCN2FPN, triple point mutation substituting FPN sequence for residues QEK in A′ helix of C-linker (residues 450–452 in HCN2). The recording protocol is described in Fig. 2. (B) Percent reduction in maximal tail current amplitude for HCN2 WT and mutant channels in response to 4 µM TRIP8bcore polypeptide. Error bars indicate SEM. Mean percent reductions ± SEM are as follows: HCN2, 43 ± 4% (n = 9); HCN2ΔCNBD, −1.6 ± 2% (n = 3); HCN2R591E, 48 ± 9% (n = 3); and HCN2FPN, −1 ± 2% (n = 3).
Molecular mechanism underlying the action of TRIP8b to reduce HCN2 channel current
TRIP8b and HCN1 have been previously found to interact at two sites (Lewis et al., 2009; Han et al., 2011; Santoro et al., 2011). At an upstream site the core region of TRIP8b binds to the CNBD of the channel; at a downstream site the C-terminal TPR domain of TRIP8b binds the extreme C-terminal SNL tripeptide of the channel. Which channel domains are required for the action of the TRIP8b core peptide to regulate HCN2 maximal current? We find that truncation of HCN2 at the N terminus of the CNBD blocks the ability of TRIP8b to reduce HCN2 channel current (HCN2ΔCNBD; Fig. 8). In contrast, as stated above, the HCN2R591E point mutation does not alter the action of 4 µM TRIP8b to reduce maximal HCN2 current (Fig. 8). These results suggest that, although a direct interaction of TRIP8b with the CNBD may not be required and some other previously unrecognized low-affinity binding site may be important for the reduction in channel current, the presence of the CNBD is still essential for TRIP8b to exert its effect to reduce maximal current amplitude (either by contributing to the integrity of the low affinity binding site or by acting as an effector domain).
If the action of TRIP8b to reduce HCN2 maximal channel current does not depend directly on its interaction with the CNBD, how does cAMP antagonize this action? A previous study has suggested that cAMP binding enhances HCN channel opening by relieving an inhibitory action of the CNBD on channel gating (Wainger et al., 2001). At a structural level, cAMP is thought to act by promoting assembly of the four CNBDs in a channel into a tetrameric gating ring (Zagotta et al., 2003). We thus hypothesized that the inhibitory action of TRIP8b on channel current is only manifest when the gating ring is disassembled, that is, in the absence of cAMP. To test this hypothesis, we took advantage of a triple point mutation our laboratory previously identified in the first (A′) helix of the cytoplasmic C-linker region of the channel, which connects the S6 transmembrane segment to the CNBD. We found that this mutation promotes gating ring assembly in the absence of cAMP (Zhou et al., 2004). We therefore predicted that this mutation should also block the action of TRIP8b to reduce channel current. As shown in Fig. 8 (HCN2FPN), the triple point mutation does indeed block the effect of TRIP8b to inhibit HCN2 maximal current, supporting the hypothesis outlined above.
DISCUSSION
In this study, we report that TRIP8b exerts multiple effects on HCN2 channel function. The auxiliary subunit shifts the relation between ΔV1/2 and [cAMP] to higher concentrations, decreasing the sensitivity of the channel to cAMP. TRIP8b also decreases the maximal voltage shift in response to a saturating concentration of cAMP (ΔVmax), reflecting a reduction in cAMP efficacy. Finally, in the absence of cAMP, TRIP8b inhibits the maximal current through the channel. Because this latter effect is antagonized by cAMP, TRIP8b enhances the action of cAMP to increase maximal current through the channel.
An allosteric mechanism accounts for the action of TRIP8b to inhibit the effect of cAMP to shift the voltage dependence of HCN2 channel opening
The effect of TRIP8b to inhibit the cAMP-dependent modulation of HCN2 voltage gating can be accounted for by an expansion of a six-state allosteric model that has been previously used to describe the dual actions of voltage and cAMP to promote HCN2 channel opening in the absence of TRIP8b (Zhou and Siegelbaum, 2007). In this model, cAMP facilitates channel opening by binding more tightly to the open than to the closed state of the channel. In the expanded 12-state model, TRIP8b exerts an allosteric action that reduces the affinity of both the closed and open states of the channel for cAMP. Conversely, cAMP binding produces an allosteric effect to reduce the affinity of the closed and open channel for TRIP8b. Because TRIP8b also reduces ΔVmax, its binding must reduce the cAMP affinity of the open state to a greater extent than it reduces the cAMP affinity of the closed state, a prediction which was confirmed by our detailed modeling results. In contrast, unlike cAMP, which binds much more favorably to the open state than the closed state, TRIP8b binds with similar affinity to the open and closed states of the channel.
A recent biochemical study focused on the nature of the interaction between TRIP8b and HCN1 at the upstream interaction site, using a channel in which the C-terminal SNL tripeptide was deleted to prevent the interaction at the downstream site (Han et al., 2011). Under these conditions the binding between TRIP8b and HCN1 is reduced by cAMP in a concentration-dependent manner. Moreover the binding of TRIP8b to the HCN1ΔSNL truncation mutant is abolished when a highly conserved arginine residue in the CNBD, which interacts with the cyclized phosphate of cAMP, is mutated to glutamate (R538E in HCN1 and R591E in HCN2). This led Han et al. (2011) to conclude that the core region of TRIP8b may directly bind to this conserved arginine, thereby competing with cAMP for the CNBD.
However, the finding that TRIP8b reduces the maximal voltage shift with saturating concentrations of cAMP (Zolles et al., 2009) indicates that the auxiliary subunit does not directly compete with cAMP for the channel but acts through an allosteric mechanism to decrease the affinity of the CNBD for ligand. Moreover, several additional lines of evidence make it unlikely that TRIP8b directly interacts with the binding domain arginine (R591 in HCN2). First, the crystal structure of the HCN2 CNBD with cAMP bound shows that the arginine is embedded in the binding pocket and surrounded by the CNBD β-roll (Zagotta et al., 2003). Although cAMP or cGMP are small enough to enter the pocket and interact with the arginine, it is unclear how the internal core domain of TRIP8b would have ready access to this site. Second, we find that the binding of TRIP8b to HCN1 is not altered when the corresponding arginine (R538) is substituted with alanine (HCN1 R538A), although we confirmed that the R538E mutation does weaken the binding of TRIP8b to the channel (Fig. 9). These results can be reconciled if the conserved arginine of the CNBD does not directly interact with TRIP8b, but rather its substitution by glutamate results in a local disruption of the CNBD, thereby weakening the binding of TRIP8b. We further hypothesize that this local structural change in the CNBD may not occur with the less disruptive alanine substitution. More detailed structural studies, including x-ray crystallography, are needed to identify the precise location of the TRIP8b binding site.
Figure 9.
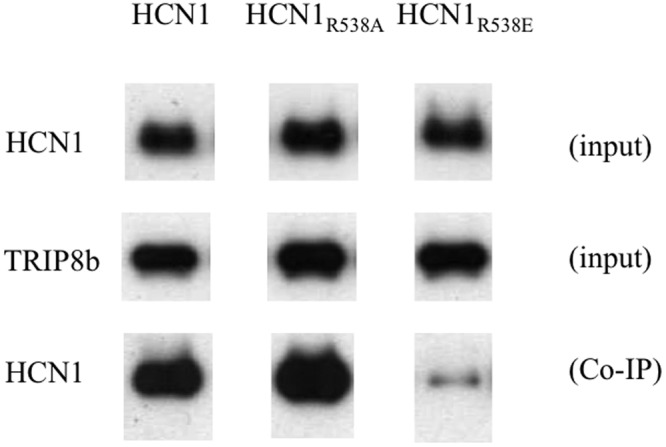
The effect of mutations in the HCN1 key CNBD residue R538 on TRIP8b binding. Western blot analysis shows binding of HCN1, HCN1R538A, and HCN1R538E mutants to WT TRIP8b(1b-2) assessed by coimmunoprecipitation from Xenopus oocyte extracts coinjected with TRIP8b and HCN1 cRNA. The top row shows the HCN1 input signal using an HCN1 antibody. The middle row shows the TRIP8b input signal using an anti-TRIP8b antibody. The bottom row shows the amount of HCN1 protein coimmunoprecipitated with the TRIP8b antibody (Western blot probed using HCN1 antibody). Note that exposure times are directly comparable along each row but not down each column. Individual bands have been cut from intact gel pictures and aligned to allow direct comparison of intensities for WT and mutant constructs.
TRIP8b inhibits maximal current through HCN2 channels through a second action
What is the relation between the action of TRIP8b to reduce maximal Ih in the absence of cAMP and the action of TRIP8b to decrease the affinity of the CNBD for cAMP? As discussed in the Results, although in principle the 12-state allosteric model can account for both of these actions through TRIP8b binding more tightly to the closed than open state of the channel, this single mode of action is not consistent with our experimental and modeling results. Rather, the reduction in maximal macroscopic current appears to be caused by a second, low-affinity action of TRIP8b to reduce maximal channel current. Such an effect could result from a reduction in channel open probability or from a reduction in single channel conductance.
One important caveat with our modeling results is that the 12-state allosteric reaction scheme does not take into account the tetrameric nature of the channel and its interaction with cAMP (Ulens and Siegelbaum, 2003) and TRIP8b (Bankston et al., 2012). Furthermore, a recent study suggests that the gating of HCN2 channels is much more complex than our model, involving a profound cooperative interaction (Kusch et al., 2012). Although our initial attempts to fit a model with multiple equivalent cAMP and TRIP8b binding sites did not yield a significant improvement in our ability to describe the two effects of TRIP8b through a single mechanistic action, some more complex model with nonequivalent binding sites (Ulens and Siegelbaum, 2003; Kusch et al., 2012) may prove adequate to explain our results based on a single underlying action of TRIP8b.
If TRIP8b does indeed exert separate high- and low-affinity actions at two distinct binding sites that, respectively, antagonize the action of cAMP on voltage gating and reduce maximal current in the absence of cAMP, where are these sites located? One possibility is that TRIP8b binds to two separate sites within the CNBD. However it is unclear as to how the relatively compact CNBD could accommodate multiple TRIP8b proteins. Also, any interaction of TRIP8b with a second CNBD-binding site that reduces maximal current must be impervious to the R591E mutation, which disrupts high-affinity binding of TRIP8b to the CNBD (Fig. 9; Han et al., 2011). Rather, we favor a model in which the core region of TRIP8b binds to two separate sites on the channel, a high-affinity site on the CNBD that allosterically reduces cAMP binding and a low-affinity site located elsewhere in the channel that reduces maximal current, perhaps by decreasing apparent single channel conductance. As we previously failed to detect an interaction between the TRIP8b core peptide and the C-terminal downstream HCN1 binding site using a yeast two-hybrid–based assay (Santoro et al., 2011), this second site is likely to be located either within the C-linker or within the channel’s intracellular loops.
Our finding that cAMP antagonizes the ability of TRIP8b to reduce maximal current indicates that the low-affinity TRIP8b binding site must be allosterically coupled to the CNBD. We further postulate that this allosteric coupling may be mediated by the action of cAMP to promote assembly of the four CNBDs of the tetrameric HCN2 channel into a fourfold symmetric gating ring (Zagotta et al., 2003). Some support for this idea comes from our finding that a triple point mutation in the A′ helix of the HCN2 C-linker, previously shown to promote gating ring assembly in the absence of cAMP (Zhou et al., 2004), blocks the action of TRIP8b to inhibit HCN2 maximal current (Fig. 8). However, this result is also consistent with the possibility that the A′ helix of the C-linker may help form the TRIP8b low-affinity binding site.
Implications of the dual actions of TRIP8b for the physiological effects of cAMP to modulate Ih
Our finding that TRIP8b exerts opposing actions on the modulatory effects of cAMP, enhancing the action of cAMP to increase maximal current while inhibiting the action of cAMP to shift voltage gating to more positive potentials, has interesting potential implications for the physiological consequences of the diverse modulatory actions of cAMP on Ih as previously reported in different neurons. Our results suggest that in neurons with high levels of TRIP8b expression, cAMP will exert a larger action to enhance maximal current and a smaller action to alter the voltage dependence of channel gating compared with neurons in which TRIP8b expression is low. Differences in TRIP8b expression among different neuronal types could help explain the varied results reported in the literature for the modulatory effects of neurotransmitters on the hyperpolarization-activated Ih (Bobker and Williams, 1989; McCormick and Williamson, 1991; Kiehn and Harris-Warrick, 1992; Larkman and Kelly, 1992; Erickson et al., 1993; Gasparini and DiFrancesco, 1999; Bickmeyer et al., 2002; Schweitzer et al., 2003; Heys and Hasselmo, 2012). Whereas in most neurons the predominant effect of neurotransmitter actions results from a cAMP-dependent alteration in the voltage-dependent gating of Ih, the extent to which transmitters also regulate maximal current amplitude varies greatly. In some instances, the predominant effect of transmitter is to enhance maximal current, with little or no change in channel voltage-dependent gating (e.g., Bickmeyer et al., 2002). Further studies using manipulations of TRIP8b expression in intact neurons may help in characterizing the role of this auxiliary subunit in the modulatory control of Ih function under more physiological conditions.
Supplementary Material
Acknowledgments
We thank John Riley and Claudia Corso for their excellent technical support.
This work was partially supported by grant NS36658 from the National Institutes of Health (to S. Siegelbaum) and by grant SAL-49 ASTIL Regione Lombardia (to A. Moroni).
Sharona E. Gordon served as editor.
Footnotes
Abbreviations used in this paper:
- CNBD
- cyclic nucleotide–binding domain
- HCN
- hyperpolarization-activated cyclic nucleotide–regulated cation
- TPR
- tetratricopeptide repeat
References
- Arikkath J., Campbell K.P. 2003. Auxiliary subunits: essential components of the voltage-gated calcium channel complex. Curr. Opin. Neurobiol. 13:298–307 10.1016/S0959-4388(03)00066-7 [DOI] [PubMed] [Google Scholar]
- Bankston J.R., Camp S.S., DiMaio F., Lewis A.S., Chetkovich D.M., Zagotta W.N. 2012. Structure and stoichiometry of an accessory subunit TRIP8b interaction with hyperpolarization-activated cyclic nucleotide-gated channels. Proc. Natl. Acad. Sci. USA. 109:7899–7904 10.1073/pnas.1201997109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battefeld A., Bierwirth C., Li Y.C., Barthel L., Velmans T., Strauss U. 2010. I(h) “run-up” in rat neocortical neurons and transiently rat or human HCN1-expressing HEK293 cells. J. Neurosci. Res. 88:3067–3078 10.1002/jnr.22475 [DOI] [PubMed] [Google Scholar]
- Bickmeyer U., Heine M., Manzke T., Richter D.W. 2002. Differential modulation of I(h) by 5-HT receptors in mouse CA1 hippocampal neurons. Eur. J. Neurosci. 16:209–218 10.1046/j.1460-9568.2002.02072.x [DOI] [PubMed] [Google Scholar]
- Biel M., Wahl-Schott C., Michalakis S., Zong X. 2009. Hyperpolarization-activated cation channels: from genes to function. Physiol. Rev. 89:847–885 10.1152/physrev.00029.2008 [DOI] [PubMed] [Google Scholar]
- Bobker D.H., Williams J.T. 1989. Serotonin augments the cationic current Ih in central neurons. Neuron. 2:1535–1540 10.1016/0896-6273(89)90041-X [DOI] [PubMed] [Google Scholar]
- Chen S., Wang J., Siegelbaum S.A. 2001. Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J. Gen. Physiol. 117:491–504 10.1085/jgp.117.5.491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Sirois J.E., Lei Q., Talley E.M., Lynch C., III, Bayliss D.A. 2005. HCN subunit-specific and cAMP-modulated effects of anesthetics on neuronal pacemaker currents. J. Neurosci. 25:5803–5814 10.1523/JNEUROSCI.1153-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven K.B., Zagotta W.N. 2004. Salt bridges and gating in the COOH-terminal region of HCN2 and CNGA1 channels. J. Gen. Physiol. 124:663–677 10.1085/jgp.200409178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco D., Tortora P. 1991. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature. 351:145–147 10.1038/351145a0 [DOI] [PubMed] [Google Scholar]
- Erickson K.R., Ronnekleiv O.K., Kelly M.J. 1993. Electrophysiology of guinea-pig supraoptic neurones: role of a hyperpolarization-activated cation current in phasic firing. J. Physiol. 460:407–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frère S.G.A., Lüthi A. 2004. Pacemaker channels in mouse thalamocortical neurones are regulated by distinct pathways of cAMP synthesis. J. Physiol. 554:111–125 10.1113/jphysiol.2003.050989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini S., DiFrancesco D. 1999. Action of serotonin on the hyperpolarization-activated cation current (Ih) in rat CA1 hippocampal neurons. Eur. J. Neurosci. 11:3093–3100 10.1046/j.1460-9568.1999.00728.x [DOI] [PubMed] [Google Scholar]
- Han Y., Noam Y., Lewis A.S., Gallagher J.J., Wadman W.J., Baram T.Z., Chetkovich D.M. 2011. Trafficking and gating of hyperpolarization-activated cyclic nucleotide-gated channels are regulated by interaction with tetratricopeptide repeat-containing Rab8b-interacting protein (TRIP8b) and cyclic AMP at distinct sites. J. Biol. Chem. 286:20823–20834 10.1074/jbc.M111.236125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heys J.G., Hasselmo M.E. 2012. Neuromodulation of I(h) in layer II medial entorhinal cortex stellate cells: a voltage-clamp study. J. Neurosci. 32:9066–9072 10.1523/JNEUROSCI.0868-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiehn O., Harris-Warrick R.M. 1992. 5-HT modulation of hyperpolarization-activated inward current and calcium-dependent outward current in a crustacean motor neuron. J. Neurophysiol. 68:496–508 [DOI] [PubMed] [Google Scholar]
- Kusch J., Thon S., Schulz E., Biskup C., Nache V., Zimmer T., Seifert R., Schwede F., Benndorf K. 2012. How subunits cooperate in cAMP-induced activation of homotetrameric HCN2 channels. Nat. Chem. Biol. 8:162–169 [DOI] [PubMed] [Google Scholar]
- Larkman P.M., Kelly J.S. 1992. Ionic mechanisms mediating 5-hydroxytryptamine- and noradrenaline-evoked depolarization of adult rat facial motoneurones. J. Physiol. 456:473–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis A.S., Schwartz E., Chan C.S., Noam Y., Shin M., Wadman W.J., Surmeier D.J., Baram T.Z., Macdonald R.L., Chetkovich D.M. 2009. Alternatively spliced isoforms of TRIP8b differentially control h channel trafficking and function. J. Neurosci. 29:6250–6265 10.1523/JNEUROSCI.0856-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick D.A., Williamson A. 1991. Modulation of neuronal firing mode in cat and guinea pig LGNd by histamine: possible cellular mechanisms of histaminergic control of arousal. J. Neurosci. 11:3188–3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pian P., Bucchi A., Robinson R.B., Siegelbaum S.A. 2006. Regulation of gating and rundown of HCN hyperpolarization-activated channels by exogenous and endogenous PIP2. J. Gen. Physiol. 128:593–604 10.1085/jgp.200609648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskorowski R., Santoro B., Siegelbaum S.A. 2011. TRIP8b splice forms act in concert to regulate the localization and expression of HCN1 channels in CA1 pyramidal neurons. Neuron. 70:495–509 10.1016/j.neuron.2011.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson R.B., Siegelbaum S.A. 2003. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu. Rev. Physiol. 65:453–480 10.1146/annurev.physiol.65.092101.142734 [DOI] [PubMed] [Google Scholar]
- Santoro B., Wainger B.J., Siegelbaum S.A. 2004. Regulation of HCN channel surface expression by a novel C-terminal protein-protein interaction. J. Neurosci. 24:10750–10762 10.1523/JNEUROSCI.3300-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B., Piskorowski R.A., Pian P., Hu L., Liu H., Siegelbaum S.A. 2009. TRIP8b splice variants form a family of auxiliary subunits that regulate gating and trafficking of HCN channels in the brain. Neuron. 62:802–813 10.1016/j.neuron.2009.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B., Hu L., Liu H., Saponaro A., Pian P., Piskorowski R.A., Moroni A., Siegelbaum S.A. 2011. TRIP8b regulates HCN1 channel trafficking and gating through two distinct C-terminal interaction sites. J. Neurosci. 31:4074–4086 10.1523/JNEUROSCI.5707-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer P., Madamba S.G., Siggins G.R. 2003. The sleep-modulating peptide cortistatin augments the h-current in hippocampal neurons. J. Neurosci. 23:10884–10891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin K.S., Maertens C., Proenza C., Rothberg B.S., Yellen G. 2004. Inactivation in HCN channels results from reclosure of the activation gate: desensitization to voltage. Neuron. 41:737–744 10.1016/S0896-6273(04)00083-2 [DOI] [PubMed] [Google Scholar]
- Ulens C., Siegelbaum S.A. 2003. Regulation of hyperpolarization-activated HCN channels by cAMP through a gating switch in binding domain symmetry. Neuron. 40:959–970 10.1016/S0896-6273(03)00753-0 [DOI] [PubMed] [Google Scholar]
- Vacher H., Trimmer J.S. 2011. Diverse roles for auxiliary subunits in phosphorylation-dependent regulation of mammalian brain voltage-gated potassium channels. Pflugers Arch. 462:631–643 10.1007/s00424-011-1004-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainger B.J., DeGennaro M., Santoro B., Siegelbaum S.A., Tibbs G.R. 2001. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature. 411:805–810 10.1038/35081088 [DOI] [PubMed] [Google Scholar]
- Wang J., Chen S., Nolan M.F., Siegelbaum S.A. 2002. Activity-dependent regulation of HCN pacemaker channels by cyclic AMP: signaling through dynamic allosteric coupling. Neuron. 36:451–461 10.1016/S0896-6273(02)00968-6 [DOI] [PubMed] [Google Scholar]
- Zagotta W.N., Olivier N.B., Black K.D., Young E.C., Olson R., Gouaux E. 2003. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 425:200–205 10.1038/nature01922 [DOI] [PubMed] [Google Scholar]
- Zhou L., Siegelbaum S.A. 2007. Gating of HCN channels by cyclic nucleotides: residue contacts that underlie ligand binding, selectivity, and efficacy. Structure. 15:655–670 10.1016/j.str.2007.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., Olivier N.B., Yao H., Young E.C., Siegelbaum S.A. 2004. A conserved tripeptide in CNG and HCN channels regulates ligand gating by controlling C-terminal oligomerization. Neuron. 44:823–834 10.1016/j.neuron.2004.11.012 [DOI] [PubMed] [Google Scholar]
- Zolles G., Wenzel D., Bildl W., Schulte U., Hofmann A., Müller C.S., Thumfart J.-O., Vlachos A., Deller T., Pfeifer A., et al. 2009. Association with the auxiliary subunit PEX5R/Trip8b controls responsiveness of HCN channels to cAMP and adrenergic stimulation. Neuron. 62:814–825 10.1016/j.neuron.2009.05.008 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.