

Abstract
Protein ubiquitylation controls many cellular pathways, and timely removal of ubiquitin by de-ubiquitylating enzymes (DUBs) is essential to govern these different functions. To map endogenous expression of individual DUBs as well as that of any interacting proteins, we developed a catch-and-release ubiquitin (Ub) probe. Ub was equipped with an activity-based warhead and a cleavable linker attached to a biotin affinity-handle through tandem site-specific modification, in which we combined intein chemistry with sortase-mediated ligation. The resulting probe is cell-impermeable and was therefore delivered to the cytosol of Perfringolysin-O (PFO) permeabilized cells. This allowed us to retrieve and identify 34 DUBs and their interacting partners. Upon infection with Chlamydia trachomatis, we noted the expression of two additional host DUBs. Furthermore, we retrieved and identified Chlamydial DUB1 (ChlaDUB1) and DUB2 (ChlaDUB2), demonstrating by experiment that ChlaDUB2, the presence and activity of which had not been detected in infected cells, is in fact expressed in the course of infection.
Keywords: Activity-based probe, cleavable linker, deubiquitylating enzymes, ubiquitin, Perfringolysin-O permeabilized cells, Chlamydia trachomatis
Introduction
Protein ubiquitylation is a versatile post-translational modification that regulates a wide array of biological processes, ranging from trafficking of proteins and signaling cascades, to protein stability. Conjugation of ubiquitin is achieved through a cascade of enzymes. The C-terminal carboxylic acid of ubiquitin is activated by an E1 ubiquitin-activating enzyme that uses ATP to yield an ubiquitin-enzyme thioester. Transthioesterification to an E2 ubiquitin-conjugating enzyme is followed by transfer of ubiquitin to protein substrates via an E3 ligase. The vast number of predicted E3 ligases (>600) allow for great specificity. Multiple ubiquitylation reactions can occur on the same protein, either as mono- or polyubiquitin modifications, the latter using structurally different linkages, to produce a highly versatile protein modification system (reviewed in [1]).
Ubiquitylation is reversible. Protein trafficking, where dependent on ubiquitylation, would require removal of ubiquitin once the protein has reached its destination. Similarly, an ubiquitin tag can mark proteins for proteasomal destruction, but ubiquitin itself can be removed to recycle it. De-ubiquitylating enzymes (DUBs), also known as ubiquitin-specific proteases (USPs), cleave ubiquitin from substrate proteins. The human genome encodes ~80 (putative) DUBs, the majority of which have a papain-like active site [2]. DUB activity is tightly regulated by post-translational modifications, protein-protein interactions, and subcellular localization, both to increase specificity and for temporal control of ubiquitin removal [3]. Large-scale efforts have allowed the construction of interaction maps of the individual DUBs to gain insight in their physiological role and regulation.[4]. These efforts have thus far relied mostly on overexpression of individual DUBs modified with affinity handles, and subsequent retrieval of that particular DUB and its interactors. However, this approach has been limited to a small number of tissue culture cell lines and may suffer from artifacts due to overexpression of a DUB or its catalytic domain. Elevated DUB levels can result in strong phenotypes, such as a complete block in proteasomal degradation [5].
Our laboratory has prepared activity-based probes (ABPs) for the (de)ubiquitylation pathway to study in an unbiased manner the enzymes involved in these reactions. This approach samples DUBs at their endogenous expression level, and in their full-length active state. We equipped the C-terminus of HA-tagged ubiquitin with a variety of electrophiles by aminolysis of protein thioesters, prepared by the intein fusion method [6,7]. Recently other labs replaced the HA-tag by fluorophores to create fluorescent analogs of these probes[8,9]. These ubiquitin derivatives act as covalent, cysteine-directed inhibitors of ubiquitin- and ubiquitin-like deconjugating enzymes, as well as of a subset of ubiquitin ligases. The labeled enzymes can be visualized either by immunoblotting or are identified by mass spectrometry after affinity purification in the case of HA-tagged proteins [10] and by in-gel fluorescence scanning in the case of fluorescent probes[8,9]. Of the electrophiles tested, the vinyl methyl ester derivative is probably the most broadly reactive warhead with ubiquitin deconjugating [6] and conjugating [10] enzymes, irrespective of enzyme family or organism of origin.
Using these ABPs, we identified DUBs in mammalian cells and those encoded by pathogens such as the Herpes viruses, parasites including Plasmodium falciparum and Toxoplasma gondii, and bacteria such as Chlamydia trachomatis [11-13]. Although viral and bacterial pathogens do not possess an intact ubiquitin pathway, they presumably express DUBs to evade detection by the immune system or to otherwise enhance virulence [14] especially for those pathogens with an intracellular lifestyle. Detection of DUBs in the context of host infection can be challenging, as expression levels can vary between infectious agents and with the time of infection. Detection is therefore highly dependent on the sensitivity of the approach used. An example is the bacterium Chlamydia trachomatis, for which no means of genetic manipulation has been established to date. We identified a single Chlamydial DUB (ChlaDUB1) using UbVME probes [11], but the Chlamydia genome also encodes a second putative DUB, termed ChlaDUB2. Even though recombinantly expressed ChlaDUB2 encodes an UbVME-reactive protein, the actual product has escaped detection in the course of a Chlamydial infection.
We attribute this inability to a lack of sensitivity of the activity-based probes available at the time, and to the manner of their application. Furthermore, in the course of affinity-based purification, abundant, poorly soluble, or otherwise nonspecifically interacting proteins invariably accumulate on the affinity matrix. Recovery of samples from the matrix under denaturing conditions will then elute these contaminants together with the specific interactors, resulting in false positive hits during mass spectrometric identification. Moreover, the presence of background peptides adversely affects the bandwidth available to detect true protein targets, in turn decreasing the limit of detection of true positive hits [15]. A second and more specific drawback of many such ABPs is that they are cell-impermeable and therefore can be used only in cell lysates. Lysis of cells usually entails massive dilution of the cytosol, which in turn may lead to dissociation of protein complexes with concomitant loss of activity, and reduced or no binding of the ABP when the affinity of the target for the ABP is low.
To mitigate the first issue, we equipped UbVME derivatives with recently described chemically cleavable linkers [16-18]. These linkers allow cleavage under mild conditions and enable specific retrieval of the tagged proteins while minimizing release of non-specific binders (Figure 1a). The second issue was addressed by developing a method that allows the delivery of the probe to the cytoplasm of permeabilized cells (Figure 1b) with minimal dilution of the target cytosol. The combined methods allowed us to retrieve 34 DUBs −1 of which had not been previously identified by ABP. Two additional host DUBs were detected in HeLa cells upon infection with Chlamydia trachomatis . Furthermore, the covalent modification of DUBs with ABPs in a less dilute cytosol and thus presumably a more physiological setting resulted in retrieval of a large number of interacting proteins, as identified by mass spectrometry. Finally, we were able to detect ChlaDUB2, 24 hours after infection of HeLa cells with Chlamydia trachomatis, showing that this enzyme, the presence of which was previously inferred but never demonstrated in Chlamydia-infected cells, is expressed.
Figure 1.

Schematic overview of the cleavable linker approach.
A) DUBs are labeled with an ABP containing the cleavable linker. The labeled proteins are retrieved on a streptavidin matrix which is then washed before release by selective cleavage of the linker.
B) Schematic overview of probe delivery approach. Cells are incubated on ice with Perfringolysin-O in the presence of the probe. The cells are transferred to 37°C to allow pore formation and are incubated for 30 min to allow labeling to occur. The cytosol is then separated from the cellular compartments by centrifugation.
Results
Preparation and validation of catch-and-release ubiquitin electrophiles
A ubiquitin moiety that carries diverse modifications at the N-terminus such as epitope tags, fluorophores, and biotin can still be recognized by the ubiquitin conjugation and deconjugation machinery [6,9], whereas modification of the ε-amino group of its lysine residues may interfere with activity through elimination of possible conjugation sites. Modifications at the C-terminus may result in altered specificity[19]. We therefore selectively introduced a cleavable linker at the N-terminus of UbVME using the sortase-based transpeptidation reaction. This is a versatile and easily implemented site-specific protein ligation strategy (Scheme 1). Proteins bearing 1-5 glycine residues at the N-terminus can be labeled using appropriately functionalized LPXTG peptides [20,21]. We therefore synthesized peptides 1-3 (Scheme 1), which combine previously reported catch-and-release moieties [16-18] with the above peptide sequence (for synthesis, see SI schemes 1 and 2). Preliminary experiments showed that protein recovery from the streptavidin resin was significantly improved by extending the biotin handle using a polyethylene glycol (PEG)-type spacer (SI, Figure 1a). This scheme is generalizable, in that it can be applied to any protein with a suitably exposed run of Gly residues.
Scheme 1.

Preparation of cleavable linker containing UbVME probes.
G3UbVME was equipped with peptides 1-3 using a transacylation reaction catalyzed the bacterial enzyme sortase A.
As nucleophile we prepared a derivative of ubiquitin vinyl methyl ester (UbVME) with three glycine residues appended at the N-terminus (G3UbVME) [22]. N-terminal sortagging of G3UbVME with catch-and-release probes 1-3 proceeded in 50-70% yield. Purification by RP-HPLC gave highly pure and labeled UbVME (SI, Figure 1b and c). HPLC purification also allowed for recovery from the reaction mixtures of unreacted G3UbVME that could then be used in subsequent reactions, thus minimizing losses of valuable input materials. This synthetic strategy enables the production of a protein reagent bearing two site-specific bio-orthogonal chemical modifications. Examples of this in the literature are scarce [20,23,24].
The activity of the different catch-and-release UbVME probes was evaluated by reacting them with purified recombinant ubiquitin carboxy-terminal hydrolase isozyme L3 (UCH-L3), a well-characterized DUB [25]. Analysis of the reaction products by anti-ubiquitin immunoblot revealed an ubiquitin-bearing product at ~37 kDa, corresponding to the covalent adduct of UCH-L3 (26 kDa) with biotin-labeled ubiquitin (~10 kDa). Binding of the reaction products on streptavidin, followed by exposure of the streptavidin resin to the optimized cleavage conditions for each cleavable linker, yielded the UCH-L3-UbVME adduct in >50% recovered yield for all three derivatives (Figure 2, left and middle lanes). Boiling of the streptavidin resin in Laemmli sample buffer after chemical cleavage released only a minimal amount of residual protein (right lanes), demonstrating the efficiency of cleavage.
Figure 2.
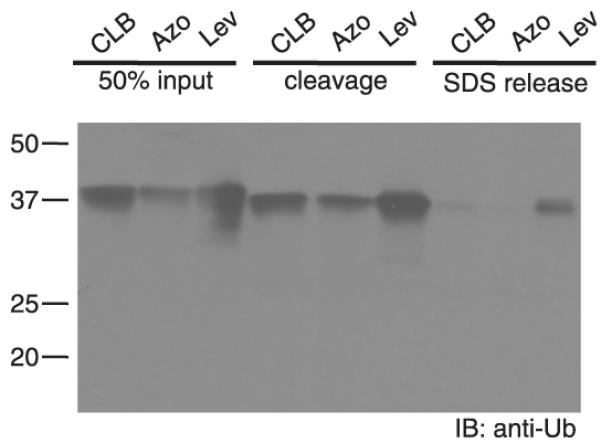
Evaluation of the novel probes using UCH-L3.
UCH-L3 was incubated with the different probes (10 μM) for 2h at 37°C (left lanes). Biotinylated proteins were retrieved on streptavidin agarose and released by chemical cleavage (middle lanes). After cleavage of the linker, beads were boiled to release remaining biotinylated proteins (right lanes).
Probe efficiency in whole cell lysate
The results of the in vitro assay using recombinant UCH-L3 prompted us to assess the efficiency of the catch-and-release probes in whole cell lysates. HEK293T cells were cultured overnight in the presence of [35S]cysteine/methionine to achieve steady state protein labeling. The labeled cells were lysed in NP40 lysis buffer. The lysate was then precleared with streptavidin agarose to remove any endogenously biotinylated proteins prior to incubation with 1 μM of the probe of choice. Modified proteins were retrieved using streptavidin-agarose and were specifically eluted through cleavage of the chemical linker (see material and methods). Finally, the eluted proteins were separated by SDS-PAGE and visualized using autoradiography (Figure 3a).
Figure 3.

Retrieval of DUBs from whole cell lysate using the catch-and-release probe.
A) 293T cells were grown overnight in the presence of [35S]cysteine/ methionine. Cells were lysed in NP40 lysis buffer, precleared with streptavidin beads, followed by incubation with 1 μM of the indicated probe at 4°C. Labeled product was retrieved with streptavidin agarose, eluted with the relevant elution buffer, and analyzed by SDS-PAGE.
B) 293T cells were transiently transfected with YOD1 and treated as in A), though exclusively with the LEV probe. Where indicated, the eluate was redissolved in NP40 buffer, followed by immuno-retrieval of YOD1.
First, we retrieved few proteins in the lanes where the probe was omitted from the reaction (lanes 1, 3 and 5). In contrast, addition of the probe and subsequent chemical cleavage of the linker resulted in the specific retrieval of a large number of proteins (lanes 2, 4 and 6), demonstrating the added benefit of the catch-and-release approach used here. The efficiency of retrieval varied between the three individual probes (Figure 3a), most likely attributable to differences in cleavage efficiencies. Whereas retrieval with the CLB probe proved rather poor, a large number of specific proteins was retrieved with either the LEV, or AZO probe, the latter slightly outperforming the former.
The complex mixture of labeled DUBs and the interacting partners that are recovered in this approach can complicate data-analysis. The mild chemical cleavage conditions are expected to yield, upon cleavage, native protein complexes with the possibility of performing an immunoprecipitation (IP) for a particular DUB of choice (and its interactors). To demonstrate the feasibility of this approach, 293T cells were transiently transfected with YOD1 – a DUB involved in glycoprotein dislocation from the endoplasmic reticulum – and treated as described above [26]. After chemical cleavage, the eluted proteins were diluted in NP40 buffer and subjected to IP with an antibody against YOD1. When we analyzed the chemical eluate by SDS-PAGE, we observed a complex banding pattern with a prominent polypeptide at a molecular weight corresponding to YOD1 modified with the ubiquitin probe (Figure 3b). The same polypeptide was observed also after a second immunoprecipitation for YOD1, confirming its identity. We found that the LEV probe, although slightly less efficient in the first set of experiments (Figure 3a), performed better in this setting. Far less protein was retrieved in a consecutive immunoprecipitation when an eluate from an AZO-UbVME labeled cell lysate was used. Dithionite concentrations of >10 mM can denature proteins [27]. The reducing conditions used to cleave the azobenzene linker may thus have reduced protein complexes and/or denatured the antibody in the course of the second immunoprecipitation. Addition of oxidized glutathione to quench the excess of sodium dithionite did not alter the outcome of the experiment (data not shown), and this issue was not explored further.
Probe delivery in semi-intact cells
In order to efficiently retrieve the protein complexes in which individual DUBs participate, we devised a strategy that allows labeling of DUBs in a more physiological setting. To this end, we chose to deliver the probe in a permeabilized cell system, instead of incubating a whole cell lysate with an ABP. Cells were permeabilized through incubation with Perfringolysin-O (PFO), a pore-forming toxin that binds cholesterol in the plasma membrane. We previously documented that after pore formation, purified compounds can be delivered to the cytosol by exposure to mild hypotonic conditions, equivalent to 0.5 HBSS (Figure 1b) [28]. 293T cells (5×106) were labeled overnight with [35S]cysteine/methionine, pelleted, transferred on ice, and resuspended in 100 μL 0.5 HBSS, 0.1 μM PFO, in the presence or absence of 10 μM AZO probe (the probe is diluted ~2 times upon addition to the cells). The cells were then transferred to 37°C, which allows pore formation to occur, concomitant with an influx of buffer and delivery of the probe to the intracellular milieu. After an incubation of 30 minutes, cells were again transferred to 4°C, and the cytosol was separated from the membrane fraction by mild centrifugation (5 min at 1150 × g).
This approach offers three major advantages. First, it allows labeling of DUBs under more physiological conditions, that is, with minimal dilution of cytosol and - excepting the plasma membrane- with retention of organellar integrity, thus increasing the probability of retrieving meaningful interaction partners. Second, as the reaction is performed in a small volume, it also allows for higher concentrations of probe to be used. Finally, separation of the soluble from the membrane fractions provides useful information on the localization of any retrieved DUBs and their associated proteins.
The experiment was performed as described above, and the recovered fractions were subjected to denaturation in 1% SDS, followed by retrieval with streptavidin agarose (Figure 4a). Denaturation results in release from the recovered DUBs of any interacting proteins, thus visualizing only labeled DUBs. We retrieved a large number of specific proteins -with minimal background- that distribute over the membrane and soluble fractions. To assess the integrity of intracellular compartments such as the ER, the ER-luminal protein PDI was retrieved with minimal spill-over in the soluble fraction (Figure 4c). To test whether we can recover DUB complexes in this fashion, the experiment was repeated, but now the fractions were not exposed to an SDS-containing buffer (Figure 4b). To minimize labeling of DUBs post-lysis, a protease inhibitor cocktail (Roche complete, EDTA free) was added to the NP40 lysis buffer. Using these conditions, we indeed retrieved a larger number of proteins, but at the expense of increased background.
Figure 4.

Retrieving DUBs from semi-intact cells
A) 293T cells were grown overnight in the presence of [35S]methionine/cysteine. The cells were collected and resuspended in 100 μL of 0.5 HBSS, 0.1 μM PFO, in the presence or absence of 10 μM AZO probe. After 30 min at 37°C, the fractions were separated by mild centrifugation and dissolved in 1% SDS. Labeled protein was retrieved with streptavidin agarose and separated by SDS-PAGE.
B) Experiment as in A), but now the fractions were dissolved in NP40 lysis buffer.
C) PDI was retrieved through IP from the lysates produced in A) to serve as a control for separation.
Detection of chlamydial DUBs in infected cells
Having established proof-of-principle, we focused our attention on labeling of DUBs in cells infected by Chlamydia trachomatis. HeLa cells were infected with Chlamydia 24hrs prior to incubation with the AZO UbVME probe. In order to exclusively visualize the Chlamydial proteins, the cells were cultured in the presence of [35S]cysteine/methionine, as well as cycloheximide, to block host protein synthesis while leaving bacterial protein synthesis intact.[29] The cells were permeabilized using PFO and incubated with the AZO probe, after which the supernatant was separated from the pellet fraction by centrifugation. After analysis by SDS-PAGE, we observed one specific UbVME-reactive protein in the pellet fraction, and three proteins in the supernatant fraction (arrowheads, Figure 5a), one of which has a molecular weight that corresponds to ChlaDUB2 modified with UbVME. None of these proteins were detected after inclusion of chloramphenicol, a potent inhibitor of bacterial (but not host) protein synthesis, further validating them as Chlamydial proteins. The Chlamydial inclusion remains intact during this procedure, as the outer membrane protein OMP1 is found exclusively in the pellet fraction (Figure 5b).
Figure 5.

The labeling of Chlamydial de-ubiquitylating enzymes.
A) HeLa cells infected with Chlamydia trachomatis were labeled with [35S]cysteine/methionine. Host protein synthesis was blocked by the addition of cycloheximide. Premeabilization of the cells using PFO and DUBs were labeled with 10 μM AZO probe and subsequently treated as described above for uninfected cells.
B) The supernatant and pellet fraction of the Chlamydia-infected cells were immunoblotted for GAPDH and the Chlamydial outer membrane protein (OMP1).
DUB-centered proteomics in semi-intact cells
To establish the identity of the UbVME reactive proteins in the Chlamydia-infected semi-intact cells, we subjected the DUBs and their interacting proteins to LC/MS/MS. We treated uninfected HeLa cells as in Figure 4b, separated the chemical eluate by SDS-PAGE and excised the bands after silver staining for analysis by LC/MS/MS. In total, 34 DUBs were identified, of which USP36 had not earlier been found in studies using ubiquitin activity-based probes (Table 1).
Table 1.
Deubiquitinating enzymes retrieved with Biotin-Azo-UbVME and identified by MS.
| Protein | Accession Number (NCBI) |
M W (k Da ) |
Uninfected HeLa cells (unique peptides/sequence converage) |
Chlamydia infected cells (unique peptides/sequence converage) |
||
|---|---|---|---|---|---|---|
|
|
||||||
| Pel. | Sup. | Pel. | Sup. | |||
| USPs | ||||||
| USP1 (UBP) | 31543910 | 88 | (16/23%) | (12/14%) | (1/1.3 %) |
(14/1 9%) |
| USP3 (SIH003, UBP) | 55770886 | 59 | (15/32%) | (3/6.5%) | (12/2 6%) |
(4/11 %) |
| USP4 (UNP, Unph) | 40795665 | 10 9 |
(8/13%) | (1/1 %) |
(15/1 8%) |
|
| USP5 (ISOT1) | 148727247 | 93 | (40/54%) | (41/4 7%) |
||
| USP7 (HAUSP, TEF1) | 150378533 | 12 8 |
(47/37%) | (55/41%) | (23/2 0%) |
(63/5 2%) |
| USP8 (HumORF8; UBPY) | 190684690 | 12 8 |
(26/19%) | (24/21%) | (9/11 %) |
(11/1 3%) |
| USP10 (UBPO) | 119220605 | 87 | (18/32%) | (28/38%) | (10/1 5%) |
(22/2 9%) |
| USP11 (UHX1) | 24234683 | 11 0 |
(15/19%) | (4/7.4 %) |
||
| USP12 (UBH1, USP12L1) | 301500675 | 43 | (4/13%) | (7/13%) | (4/13 %) |
(5/16 %) |
| USP13 (IsoT-3, ISOT3) | 215598688 | 97 | (5/6.8%) | |||
| USP14 (TGT) | 4827050 | 56 | (29/56%) | (33/67%) | (22/4 4%) |
(25/4 5%) |
| USP15 (UNPH4, UNPH-2) | 355330276 | 11 2 |
(16/17%) | (57/57%) | (1/0.9 2%) |
(34/3 9%) |
| USP16 (UBP-M) | 5454156 | 94 | (9/9.7%) | (23/33%) | (14/8. 6%) |
(14/1 0%) |
| USP19 (ZMYND9) | 312596875 | 15 1 |
(5/4.3%) | (25/20%) | (8/5.2 %) |
(14/9. 9%) |
| USP24 | 260064009 | 29 4 |
(7/3.4%) | (1/0.4 6%) |
||
| USP25 (USP21) | 50312666 | 12 2 |
(4/4.5%) | (4/2.7 %) |
||
| USP28 | 16507200 | 12 2 |
(2/2.2%) | (2/2.1%) | (4/4.3 %) |
(15/2 1%) |
| USP33 (VDU1) | 42516561 | 94 | (4/7.6 %) |
|||
| USP36 (DUB1) | 122114651 | 12 3 |
(1/1.4%) | (9/8.5 %) |
(7/6.9 %) |
|
| USP47 (TRFP) | 1774197 | 14 7 |
(1/1.8%) | (23/22%) | (1/1.5 %) |
(17/9. 7%) |
| USP48 (RAP1GA1, USP31) | 52630449 | 11 9 |
(3/3.6%) | (7/5.3 %) |
||
| USP9X (DFFRX, FAF, FAM) | 145309309 | 29 2 |
(30/13%) | (17/6 %) |
||
| CYLD1 (BRSS, CDMT, EAC, MFT1, SBS, TEM, USPL2) |
109637774 | 10 7 |
(4/4.1 %) |
|||
| UCHs | ||||||
| BAP1 (HUCEP-13, hucep-6, TPDS, UCHL2) |
4757836 | 80 | (6/7.5%) | (3/3.6 %) |
(13/9. 7%) |
|
| UCHL3 (UCH-L3) | 5174741 | 26 | (5/29%) | (19/62%) | (4/24 %) |
(21/6 2%) |
| UCHL5 (CGI-70, INO80R, UCH37) |
7706753 | 38 | (18/58%) | (22/72%) | (11/3 3%) |
(23/6 6%) |
| Josephins | ||||||
| JOS (ATX3, JOS, MJD, MJD1, SCA3) |
13518019 | 41 | (2/6.4%) | (10/35%) | (1/3.3 %) |
(9/31 %) |
| JOS2 | 19923879 | 21 | (1/6.4%) | (2/7.4 %) |
||
| OTU domain | ||||||
| OTU1 (YOD1, DUBA8, OTUD2, PRO0907) |
62751964 | 38 | (10/20%) | (4/12 %) |
||
| OTUB1 (HSPC263) | 109148508 | 31 | (3/13%) | (4/14 %) |
||
| OTUB2 | 12962939 | 27 | (2/11%) | |||
| OTUD3 (UBA4) | 149192871 | 45 | (3/8.5%) | (4/8.8%) | (3/9.5 %) |
|
| OTUD5B (DUBA) | 209977019 | 60 | (5/11%) | (2/3.2 %) |
||
| OTUD6B (GI-77, DUBA5) | 157364937 | 37 | (4/19%) | (14/47%) | (1/4.3 %) |
(8/23 %) |
| OTUD7B (CEZANNE, ZA20D1) | 118026942 | 93 | (2/3.7%) | (19/32%) | (12/9. 3%) |
|
| ZRANB1 (TRABID) | 110815809 | 81 | (2/4.1 %) |
|||
| VCIP1 (DUBA3, VCIP135) | 36029914 | 13 4 |
(2/2.3%) | |||
| Chlamydial DUBs | ||||||
| ChlaDUB1 | 166154214 | 45 | (24/5 9%) |
(19/4 6%) |
||
| ChlaDUB2 | 166154213 | 38 | (4/14 %) |
|||
Where most DUBs retrieved belong to the USP class of ubiquitin hydrolases, we also observe 8 DUBs from the otubain family, 2 from the josephin family, and 3 from the family of ubiquitin C-terminal hydrolases. These results once again confirm the remarkable complexity of DUBs expressed by the typical mammalian cell, underscoring the need to carefully regulate both ubiquitylation and deubiquitylation reactions.
As our method was devised to allow identification of proteins with which DUBs engage in complex formation, we were encouraged by the presence of a number of well-studied DUB-protein complexes. An example is the recovery of most of the regulatory cap proteins of the 26S proteasome (Table 2). Other examples of protein complexes retrieved include host cell factor-1, galectin 7, p97, ras GTPase binding proteins 1 and 2, WD-repeat containing protein 48 and WD-repeat containing protein 20, and thioredoxin like protein, which interact with BAP, USP1, ataxin-3, USP10, USP12, and USP14 or UCH-L5 [4,30], respectively.
Table 2.
Interacting proteins retrieved with Biotin-Azo-UbVME and identified by MS
| MW (kDa) |
Uninfected HeLa cells (unique peptides/sequence converage) |
|||
|---|---|---|---|---|
| Protein | Accession Number (NCBI) | pellet | supernatant | |
| Proteasome subunits | ||||
| alpha subunits | ||||
| PSMA1 | 23110935 | 30 | 4/15% | 34/44% |
| beta subunits | ||||
| PSMB5 | 4506201 | 28 | 2/8.7% | 2/7.6% |
| 26S ATPase | ||||
| PSMC1; p56; S4 | 24430151 | 49 | 13/31% | 20/45% |
| PSMC2; MSS1; S7 | 4506209 | 49 | 16/42% | 25/48% |
| PSMC3; TBP1 | 21361144 | 49 | 13/36% | 28/62% |
| PSMC4; MIP224; S6 | 5729991 | 47 | 8/23% | 22/39% |
| PSMC5; p45; S8 | 312596881 | 45 | 15/48% | 27/69% |
| PSMC6; p42 | 195539395 | 46 | 17/49% | 18/50% |
| 26S non-ATPase | ||||
| PSMD1; P112; S1 | 25777600 | 106 | 16/23% | 34/44% |
| PSMD2; P97; S2 | 25777602 | 100 | 23/30% | 28/39% |
| PSMD3; P58; S3 | 25777612 | 61 | 15/30% | 17/31% |
| PSMD4; S5A | 5292161 | 41 | 16/41% | 15/45% |
| PSMD5; S5B | 4826952 | 56 | 3/7.1% | 16/38% |
| PSMD6; S10 | 7661914 | 46 | 9/28% | 9/26% |
| PSMD7; P40; S12 | 25777615 | 37 | 12/43% | 10/40% |
| PSMD8; p31; S14 | 156631005 | 40 | 4/14% | 2/7.1% |
| PSMD9; p27 | 18543329 | 25 | 5/23% | 7/34% |
| PSMD10; p28 | 4506217 | 24 | 6/35% | |
| PSMD11; p44.5; S9 | 28872725 | 47 | 11/28% | 14/35% |
| PSMD12; p55 | 4506221 | 53 | 6/12% | 9/19% |
| PSMD13; p40.5; S11 | 157502193 | 43 | 7/21% | 5/14% |
| PSMD14 | 5031981 | 35 | 3/16% | 5/21% |
| ADRM1; hRpn13 | 28373192 (+1) | 42 | 12/19% | 14/25% |
| Proteasome associated | ||||
| PAAF1; PAAF; WDR71 | 13376751 | 42 | 10/28% | 8/18% |
| Other interactors | ||||
| HCF-C1 | 98986457 | 209 | 15/4.9% | 1/1.2% |
| galectin-7 | 109948279 (+1) | 15 | 5/45% | |
| VCP; p97 | 6005942 | 89 | 31/43% | 13/22% |
| G3BP-1 | 38327552 (+1) | 52 | 8/18% | 10/21% |
| G3BP-2 | 19923399 (+1) | 54 | 4/9.8% | 6/12% |
| WD repeat-containing protein 48 | 18874090 | 76 | 33/47% | 26/41% |
| WD repeat-containing protein 20 isoform 7 | 334848139 | 67 | 6/13% | 4/8.5% |
| thioredoxin-like protein 1 | 4759274 | 32 | 13/55% | |
In parallel we performed this type of experiment using Chlamydia-infected cells. As a benchmark, ChlaDUB1 was identified with 59% and 46% sequence coverage in the supernatant and pellet fraction, respectively (24 unique peptides in the pellet fraction and 19 unique peptides in the supernatant fraction, see Table 1). In addition to ChlaDUB1, we were able to identify 2 additional host DUBs previously not labeled with UbVME: USP33 and TRABID in the Chlamydia-infected cells. More importantly, we also identified ChlaDUB2. This is the first time ChlaDUB2 has been identified in the course of infection. ChlaDUB2 was identified by mass spectrometry with 14% sequence coverage (4 unique peptides).
Discussion
Here we describe the development and application of a catch-and-release strategy to identify DUB-protein complexes at endogenous levels of expression. The catch-and-release probes were crafted through tandem site-specific modification of ubiquitin, which combined intein chemistry with sortase-mediated transacylation. This strategy allows for rapid synthesis of the activity-based probe (UbVME), which is then modified with a handle of choice at the N-terminus, as illustrated by the production of three different catch-and-release probes. Neither of these strategies is limited to modification of ubiquitin, and we could easily envision the generation of other proteins, similarly modified with two bio-orthogonal substituents.
Protein modification with ubiquitin is a complex reaction, which relies not only on the necessary levels of substrate specificity (targets are not modified randomly), but also on the imposition of linkage types that produce ubiquitin chains of unique topology. While the number of possible substrates and the types of ubiquitin modifications they carry are sheer endless, the number of available DUBs that can reverse ubiquitylation and recycle ubiquitin is not. Approximately 80 DUBs are encoded by the human genome, not all of which need to be expressed simultaneously or in the same cell type. Where analyzed, individual DUBs often harbor specificity for a particular type of ubiquitin chain,[31] effectively further limiting the number of available DUBs to hydrolyze ubiquitin and complete the modification cycle.
To fully understand how de-ubiquitylation is regulated, and with it the relevant cell biology, the protein complexes that harbor this DUB activity must be analyzed at their endogenous expression level. To this end, we combined our new probes with a delivery method that enables DUB labeling in the cytosol of PFO permeabilized cells with minimal dilution of cytosol. A high concentration of probe is delivered through the PFO-induced pores to enable labeling under conditions as close to physiological as possible. This method allowed us to retrieve 36 individual DUBs expressed in HeLa cells, and retrieve proteins in complex with these DUBs, amongst which, for example, the regulatory cap of the proteasome (see Table 2), confirming earlier observations with a first generation probe [6]. This value for the number of expressed and active DUBs is probably representative for different mammalian cell types, based on the complexity of banding patterns observed in immunoblots on HA-UbVME modified cell extracts.
The sensitivity of our method is further illustrated by the detection of ChlaDUB2 in HeLa cells infected with Chlamydia trachomatis, a DUB whose expression in the course of infection had not been demonstrated experimentally. It is clear that similar experiments can be undertaken for other intracellular pathogens. Our analysis here is limited to snap-shots of DUB activity in a population of cells. The nature of the experimental set-up, which does not rely on genetic expression of the ABP, allows for a more dynamic approach. How does the cellular ubiquitin machinery react to external changes? As an example we demonstrated the expression of active DUBs by the pathogen Chlamydia trachomatis, as assessed by ABP labeling. Our results suggest that the cellular environment in which Chlamydia seeks to survive and expand necessitates the delivery of different bacterial countermeasures: not only is ChlaDUB1 expressed, but also ChlaDUB2, raising the interesting question of how these Chlamydial enzymes differ in their substrate specificity from the host set of DUBs, and of course the identity of their preferred targets. In the absence of a genetic system with which to ablate either or both of the genes that encode the ChlaDUBs, their contribution to successful completion of the Chlamydial life cycle will have to await the development of such tools.
Material and Methods
Preparation of G3UbVME
Ubiquitin (1-75) N-terminally fused to thrombin cleavage site followed by GGG (MGSSHHHHHHSSGLVPRGGG) and C-terminally fused to intein was cloned into pTYB2. The vector was transformed into BL21(DE3)pLysS. The ubiquitin-intein constructed was expressed, purified and converted into the UbVME adduct as previously described for HA-tagged UbVME.[6] Cleaving the thrombin sequence using a Thrombin CleanCleave kit (sigma Aldrich) exposed the N-terminal Gly-Gly-Gly sequence.
Introduction of the Biotin-cleavable linker
G3UbVME (58 μM final concentration) was incubated with Sortase A of S. aureus (150 μM final concentration, 4.5× stock in 50 mM Tris, pH 7.4, 150 mM NaCl) and biotin-cleavable linker-LPETGG peptides 1-3 (0.5 mM final concentration, 10× stock, for synthesis see SI) in sortase reaction buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 10 mM CaCl2). The resulting mixture was incubated at 37°C for 3h, acidified by the addition of 1% TFA in H2O and purified by reverse phase HPLC (30→45% B in 20 min, 3 mL/min). The resulting purified protein was neutralized with sat. aq. NaHCO3 concentrated in vacuo, redissolved in H2O and quantified by gel-electrophoresis. The protein was analyzed by LC/MS. Biotin-Azo-UbVME: Rt 7.76 min; linear gradient 5→45% B in 20 min; ESI/MS: m/z = 10140 (M+H)+. Biotin-Lev-UbVME: Rt 7.57 min; linear gradient 5→45% B in 20 min; ESI/MS: m/z = 10244 (M+H)+. Biotin-CLB-UbVME: Rt 7.30 min; linear gradient 5→45% B in 20 min; ESI/MS: m/z = 9928 (M+H)+.
Antibodies, Cell lines, Constructs and Reagents
Antibodies against YOD1 and PDI were raised in rabbits against purified whole protein and have been described[26]. GAPDH antibody was obtained from Abcam. HEK293T and HeLa cells were purchased from American Type Culture Collection. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM).
The YOD1 construct in a pcDNA3.1(+) plasmid used for transfection experiments has been described[26]. HEK293T cells were transiently transfected using Trans-IT (Takara Mirus Bio) according to the manufacturer’s instructions. Cycloheximide, anisomycin and chloramphenicol were obtained from Sigma Aldrich.
Metabolic labeling, immunoprecipitations and SDS-PAGE
To achieve steady-state protein labeling, cells were incubated overnight with 200μCi of [35S]methionine/cysteine (Perkin Elmer) per ml of methionine/cysteine-free DMEM supplemented with 10% dialyzed IFS at 37°C.
Cells were either lysed in NP40 lysis buffer (0.5% NP40, 10 mM Tris-HCl, 150 mM NaCl, 5 mM MgCl2, pH 7.4) -if indicated supplemented with a complete protease inhibitor cocktail (Roche)-, or in 1%SDS. Prior to immunoprecipitation, the SDS lysate was diluted to 0.1% SDS in NP40 lysis buffer.
Immunoprecipitation was performed using either 30 μL streptavidin agarose (Sigma) or 30 μL of immobilized Protein A (IPA 300, Repligen) with the relevant antibodies.
Protein samples were separated by SDS-PAGE and visualized by autoradiography.
Immunoblotting
Protein samples were separated by SDS-PAGE, blotted onto PVDF membrane and probed with the relevant antibodies.
Probe delivery to semi-intact cells
Cells were collected on ice, and resuspended in 100 μL 0.5 HBSS, containing 0.1 μM purified PFO (for preparation of PFO, see [28]). After addition of the relevant probe, cells were transferred to 37°C for 30 min. After 30 min, the cells were returned to 4°C and fractions were separated by centrifugation at 3000 rpm for 5 min. The supernatant fraction was withdrawn, after which the pellet fraction was washed in HBSS followed by another round of centrifugation.
Labeling of HeLa cells infected with Chlamydia Trachomatis L2/434/Bu
HeLa cells and Chlamydia Trachomatis L2/434/Bu were propagated and stored as described before.[11] [35S]methionine/cysteine labeling and PFO permeabilization were performed analogous to the protocol described by Kleba et. al. [29]. Briefly, HeLa cells were infected with Chlamydia (MOI of 3-10), and cultured at 37°C. After 20 hrs, the DMEM was replaced with methionine/cysteine-free DMEM supplemented with 10% dialyzed IFS, cycloheximide (225 μg/mL), anisomycin (30 μg/mL) and 200μCi of [35S]methionine/cysteine. As a control bacterial proteins synthesis was blocked with chloramphenicol (100 μg/mL). The cells were incubated at 37°C for 4 hrs. Azo-UbVME was introduced by permeabilization with PFO as described in the general method above.
Mass spectrometry based analysis of the labeled proteins
Proteins were labeled, immunoprecipitated and released from the affinity matrix as described above. The retrieved proteins were separated on a gel and visualized by silver staining. Gel lanes were excised; the proteins were reduced, alkylated and digested with trypsin overnight at 37°C. The resulting tryptic fragments were extracted, concentrated and separated on a Waters NanoAcquity HPLC equipped with a self-packed Jupiter C18 column (3 μm, 0.075×10 mm) using standard reverse-phase gradients. The eluted peptides were analyzed using a Thermo LQT linear ion trap mass spectrometer (nanospray configuration) operated in a data dependent manner. The SEQUEST database was used to correlate the fragmentation spectra. Scaffold 3 was used to analyze and report the resulting data.
Supplementary Material
Acknowledgments
The authors would like to thank the members of the Ploegh lab for insightful discussions. We are grateful to Sumana Sanyal for providing recombinant PFO and to Herman Overkleeft and Paul Geurink for providing the levulinoyl ester based cleavable linker. David Rosmarin is acknowledged for setting up Chlamydial cultures. MDW is supported by a Rubicon grant from The Netherlands Organisation for Scientific Research and JHLC by the Boehringer Ingelheim Fonds. HLP receives grants from the NIH.
7. References
- [1].Komander D, Rape M. Annu. Rev. Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- [2].Nijman SMB, Luna-Vargas MPA, Velds A, Brummelkamp TR, Dirac AMG, Sixma TK, Bernards R. Cell. 2005;123:773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- [3].Kessler BM, Edelmann MJ. Cell Biochem. Biophys. 2011;60:21–38. doi: 10.1007/s12013-011-9176-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sowa ME, Bennett EJ, Gygi SP, Harper JW. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ernst R, Claessen JHL, Mueller B, Sanyal S, Spooner E, van der Veen AG, Kirak O, Schlieker CD, Weihofen WA, Ploegh HL. PLoS Biol. 2011;8:e1000605. doi: 10.1371/journal.pbio.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Borodovsky A, Ovaa H, Kolli N, Gan-Erdene T, Wilkinson KD, Ploegh HL, Kessler BM. Chem. Biol. 2002;9:1149–1159. doi: 10.1016/s1074-5521(02)00248-x. [DOI] [PubMed] [Google Scholar]
- [7].Hemelaar J, Galardy PJ, Borodovsky A, Kessler BM, Ploegh HL, Ovaa H. J. Proteome Res. 2004;3:268–276. doi: 10.1021/pr0341080. [DOI] [PubMed] [Google Scholar]
- [8].McGouran JF, Kramer HB, Mackeen MM, di Gleria K, Altun M, Kessler BM. Org. Biomol. Chem. 2012;10:3379–3383. doi: 10.1039/c2ob25258a. [DOI] [PubMed] [Google Scholar]
- [9].de Jong A, Merkx R, Berlin I, Rodenko B, Wijdeven RHM, Atmioui D. El, Yalçin Z, Robson CN, Neefjes JJ, Ovaa H. ChemBioChem. 2012;13:2251–2258. doi: 10.1002/cbic.201200497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Love KR, Pandya RK, Spooner E, Ploegh HL. ACS Chem Biol. 2009;4:275–287. doi: 10.1021/cb9000348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Misaghi S, Balsara ZR, Catic A, Spooner E, Ploegh HL, Starnbach MN. Mol. Microbiol. 2006;61:142–150. doi: 10.1111/j.1365-2958.2006.05199.x. [DOI] [PubMed] [Google Scholar]
- [12].Frickel E-M, Quesada V, Muething L, Gubbels M-J, Spooner E, Ploegh H, Artavanis-Tsakonas K. Cell. Microbiol. 2007;9:1601–1610. doi: 10.1111/j.1462-5822.2007.00896.x. [DOI] [PubMed] [Google Scholar]
- [13].Artavanis-Tsakonas K, Weihofen WA, Antos JM, Coleman BI, Comeaux CA, Duraisingh MT, Gaudet R, Ploegh HL. J. Biol. Chem. 2010;285:6857–6866. doi: 10.1074/jbc.M109.072405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Le Negrate G, Krieg A, Faustin B, Loeffler M, Godzik A, Krajewski S, Reed JC. Cell. Microbiol. 2008;10:1879–1892. doi: 10.1111/j.1462-5822.2008.01178.x. [DOI] [PubMed] [Google Scholar]
- [15].Domon B, Aebersold R. Nat. Biotechnol. 2010;28:710–721. doi: 10.1038/nbt.1661. [DOI] [PubMed] [Google Scholar]
- [16].Dirksen A, Yegneswaran S, Dawson PE. Angew. Chem. Int. Ed. 2010;49:2023–2027. doi: 10.1002/anie.200906756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Verhelst SHL, Fonovic M, Bogyo M. Angew. Chem. Int. Ed. 2007;46:1284–1286. doi: 10.1002/anie.200603811. [DOI] [PubMed] [Google Scholar]
- [18].Geurink PP, Florea BI, Li N, Witte MD, Verasdonck J, Kuo C-L, van der Marel GA, Overkleeft HS. Angew. Chem. Int. Ed. 2010;49:6802–6805. doi: 10.1002/anie.201001767. [DOI] [PubMed] [Google Scholar]
- [19].Iphöfer A, Kummer A, Nimtz M, Ritter A, Arnold T, Frank R, van den Heuvel J, Kessler BM, Jänsch L, Franke R. ChemBioChem. 2012;13:1416–1420. doi: 10.1002/cbic.201200261. [DOI] [PubMed] [Google Scholar]
- [20].Antos JM, Chew G-L, Guimaraes CP, Yoder NC, Grotenbreg GM, Popp MW, Ploegh HL. J. Am. Chem. Soc. 2009;131:10800–10801. doi: 10.1021/ja902681k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yamamoto T, Nagamune T. Chem. Commun. 2009:1022–1024. doi: 10.1039/b818792d. [DOI] [PubMed] [Google Scholar]
- [22].Witte MD, Cragnolini JJ, Dougan SK, Yoder NC, Popp MW, Ploegh HL. Proc. Natl. Acad. Sci. Usa. 2012;109:11993–11998. doi: 10.1073/pnas.1205427109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].van Kasteren SI, Kramer HB, Jensen HH, Campbell SJ, Kirkpatrick J, Oldham NJ, Anthony DC, Davis BG. Nature. 2007;446:1105–1109. doi: 10.1038/nature05757. [DOI] [PubMed] [Google Scholar]
- [24].Neumann H, Wang K, Davis L, Garcia-Alai M, Chin JW. Nature. 2010;464:441–444. doi: 10.1038/nature08817. [DOI] [PubMed] [Google Scholar]
- [25].Misaghi S, Galardy P, Meester WJN, Ovaa H, Ploegh HL, Rachelle G. J. Biol. Chem. 2005;280:1512–1520. doi: 10.1074/jbc.M410770200. [DOI] [PubMed] [Google Scholar]
- [26].Ernst R, Mueller B, Ploegh HL, Schlieker C. Mol. Cell. 2009;36:28–38. doi: 10.1016/j.molcel.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Budin G, Moune-Dimala M, Leriche G, Saliou J-M, Papillon J, Sanglier-Cianférani S, Van Dorsselaer A, Lamour V, Brino L, Wagner A. ChemBioChem. 2010;11:2359–2361. doi: 10.1002/cbic.201000574. [DOI] [PubMed] [Google Scholar]
- [28].Sanyal S, Claessen JHL, Ploegh HL. J. Biol. Chem. 2012;287:23594–23603. doi: 10.1074/jbc.M112.365312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kleba B, Stephens RS. Infect. Immun. 2008;76:4842–4850. doi: 10.1128/IAI.00715-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Boeddrich A, Gaumer S, Haacke A, Tzvetkov N, Albrecht M, Evert BO, Müller EC, Lurz R, Breuer P, Schugardt N. EMBO J. 2006;25:1547–1558. doi: 10.1038/sj.emboj.7601043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Virdee S, Ye Y, Nguyen DP, Komander D, Chin JW. Nat. Chem. Biol. 2010;6:750–757. doi: 10.1038/nchembio.426. [DOI] [PubMed] [Google Scholar]
- [32].Yang Y-Y, Ascano JM, Hang HC. J. Am. Chem. Soc. 2010;132:3640–3641. doi: 10.1021/ja908871t. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.