

Abstract
Objective
Brilliant blue G (BBG), a selective P2X7 receptor (P2X7R) antagonist, exhibits neuroprotective properties. This study examined whether BBG treatment ameliorates early brain injury (EBI) after experimental subarachnoid hemorrhage (SAH), specifically via inhibiting p38 mitogen-activated protein kinase (MAPK)-related proapoptotic pathways.
Design
Controlled in vivo laboratory study.
Setting
Animal research laboratory.
Subjects
One hundred-fifty four adult male Sprague-Dawley rats weighing 280–320g.
Interventions
SAH was induced in rats by endovascular perforation. Experiment 1 implemented sham-operated rats (sham) and SAH animals, which received vehicle (SAH+vehicle), BBG (SAH+BBG) or BBG plus BzATP (SAH+BBG+BzATP). The animals were intraperitoneally treated with BBG (30mg/kg) at 30 minutes after SAH. BzATP (50μg/rat), a P2X7R agonist, was intracerebroventricularly administered. Experiment 2 implemented sham-operated rats (sham) and SAH animals, which received vehicle (SAH+vehicle), scramble small interfering RNA (siRNA) (SAH+scramble siRNA) or P2X7R siRNA (SAH+P2X7R siRNA). SAH grading, neurobehavioral score and brain edema were evaluated at 24 and 72 hours after surgery. The expression of phosphorylated p38 MAPK, phosphorylated extracellular signal-regulated kinases (ERKs), phosphorylated c-Jun N-terminal kinases (JNKs), P2X7R, Bcl-2 and cleaved caspase-3 in the left cerebral hemisphere were determined by Western blot. Neuronal apoptosis was examined by double immunofluorescence staining using P2X7R, terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end-labeling (TUNEL) and NeuN.
Measurements and main results
BBG significantly improved neurobehavioral function and ameliorated brain water content at 24 and 72 hours after SAH. BzATP reversed these treatment effects. BBG attenuated neuronal apoptosis in the subcortex, which was associated with decreased expression of phosphorylated p38 MAPK and cleaved caspase-3, and an increased expression of Bcl-2 in the left cerebral hemisphere. The beneficial effects of P2X7R siRNA were also mediated by a p38 MAPK pathway.
Conclusions
Inhibition of P2X7R by BBG or P2X7R siRNA can prevent EBI via p38 MAPK after SAH.
Keywords: apoptosis, subarachnoid hemorrhage, early brain injury, P2X7 receptor, p38 mitogen-activated protein kinase, brilliant blue G
INTRODUCTION
Subarachnoid hemorrhage (SAH) is a fatal subtype of stroke. Although accounting for only 5% of all strokes, it presents a relevant burden on society and economy, as it affects mostly middle-aged patients, resulting in high mortality and disability rates (1, 2). Recently, early brain injury (EBI), starting immediately after aneurysm rupture to 72 hours, has been considered a primary target for research to combat SAH. Apoptosis of neuronal cells is believed to contribute to EBI after SAH (3).
The P2X7 receptor (P2X7R) is an adenosine triphosphate (ATP)-gated ion channel, which is known for its cytotoxic activity (4). P2X7R inhibition provides neuroprotection in various central nervous system (CNS) disease models, such as ischemia, traumatic brain injury, and neurodegenerative diseases (5–7). Stimulation of P2X7R activates mitogen-activated protein kinases (MAPKs) (8, 9), including extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinases (JNKs), and p38 MAPKs. ERKs are essential for cell survival, whereas JNKs and p38 MAPKs are stress activated and thus involved in apoptosis (10). However, it is not known whether P2X7R affects neuronal apoptosis via activation of MAPKs following SAH.
In the present study, we aim to investigate two hypotheses: (A) P2X7R inhibition via BBG, or via P2X7R gene silencing, ameliorates functional deficits and brain edema after experimental SAH in rats. (B) P2X7R inhibition reduces MAPK activation, thus decreasing neuronal apoptosis.
MATERIALS AND METHODS
Animals
All procedures were conducted following an institutionally approved protocol by the Institutional Animal Care and Use Committee (IACUC) at Loma Linda University, and in accordance with the NIH Guide for the Care and Use of Laboratory Animals. One hundred-fifty four male adult Sprague-Dawley rats (280–320g, Harlan, Indianapolis, IN) were housed in a light and temperature controlled environment with unlimited access to food and water.
SAH model and experimental design
The endovascular perforation model of SAH was conducted as previously described (11, 12). Briefly, anesthesia was maintained with 3% isoflurane in 70/30% medical air/oxygen. The external carotid (ECA) was ligated, cut, and shaped into a 3-mm stump. A sharpened 4-0 monofilament nylon suture was inserted into the ECA stump and then gently advanced into the internal carotid artery (ICA) until resistance was felt. The bifurcation of the anterior and middle cerebral artery was then punctured by inserting the suture an additional 3mm. The suture was immediately withdrawn from the ECA stump, to allow reperfusion of the ICA, resulting in SAH. Sham rats underwent the same procedures except for vessel puncture. After closing the skin incision, rats were kept at approximately 37°C on an electric heating blanket and were housed separately following complete recovery from anesthesia.
Twenty-seven SAH rats were excluded from this study because of mild bleeding. Experiment 1 implemented sham-operated rats (sham group, n=27) and SAH animals, which received vehicle (SAH+vehicle group, n=36), BBG (SAH+BBG group, n=31) or BBG plus receptor agonist BzATP (SAH+BBG+BzATP group, n=6). BzATP is a P2X7R agonist (13). 30 minutes after SAH-induction, animals were intraperitoneally treated with the vehicle (normal saline, 2ml) or BBG (30mg/kg, 2ml). BzATP (50μg/rat) was intracerebroventricularly administered at 1 hour before SAH surgery, in order to reverse the noncompetitive inhibition of BBG. For 72 hours study, BBG was administered at 0.5, 24 and 48 hours after SAH-induction by intraperitoneal injection. Experiment 2 implemented sham-operated rats (sham group, n=6) and SAH animals, which received vehicle (SAH+vehicle group, n=7), scramble small interfering RNA (siRNA) (SAH+scramble siRNA group, n=7) or P2X7R siRNA (SAH+P2X7R siRNA group, n=7).
All drugs and P2X7R siRNA were purchased from Sigma-Aldrich (St Louis, MO). Scramble siRNA was purchased from Dharmacon/Thermo Fisher Scientific (Lafayette, CO).
Intracerebroventricular infusion
Anesthetized rats were fixed onto a stereotaxic head apparatus under continuous isoflurane administration (2–3%). The 26 gauge needle of a 10μL Hamilton syringe (Microliter #701; Hamilton Company, Reno, NV) was inserted into the left lateral ventricle through a cranial burr hole, at the following coordinates relative to bregma: 1.5mm posterior; 1.0mm lateral; 3.2mm below the horizontal plane of bregma. In order to enhance the gene silence efficiency, two different P2X7R siRNA were mixed: (a) sense,5′-CAGUGAAUGAGUACUACUA-3′; antisense, 5′-UAGUAGUACUCAUUCACUG-3′ (b) sense,5′-CUCUUGAGGAGCGCCGAAA-3′; antisense, 5′-UUUCGGCGCUCCUCAAGAG-3′. The nonsilencing RNA was used as the control siRNA. 500pmol SiRNA in 2μL sterile saline was injected intracerebroventricularly by a microinfusion pump (Harvard Apparatus, Holliston, MA) at a rate of 0.5μL/min at 24 hours before SAH production (14). The needle was left in place for an additional 15 minutes after the infusion finishing. Finally, the incision was closed with sutures. Sham animals received a cranial burr hole, but no needle was inserted.
Assessment of Behavioral Outcome
Neurological scores, consisting of the modified Garcia score and the beam balance test, were blindly evaluated at 24 and 72 hours after SAH (15). There are six tests in modified Garcia score: spontaneous activity; spontaneous movement of all limbs; forepaw outstretching; climbing; body proprioception; and response to whisker stimulation. The evaluation consisted of six tests that can be scored from either 0–3 (spontaneous activity, movement of all limbs, and forepaw outstretching) or 1–3 (climbing, body proprioception, and response to whisker stimulation). For the beam balance test, rats were allowed to walk on a 15mm-wide wooden beam for 1 minute. The score ranged from 0 to 4 was given according to the walking distance. The average score of three consecutive trials was calculated. Higher scores indicated better test performance.
Measurement of Brain Water Content
Brain water content was measured at 24 and 72 hours after SAH. Briefly, rats were decapitated under lethal isoflurane anesthesia. Brains were quickly removed and separated into four parts: left hemisphere, right hemisphere, cerebellum and brain stem. Brain specimens were weighed to obtain the wet weight using an analytic microbalance (APX-60, Denver Instrument, Bohemia, NY). Brain specimens were dried at 100°C for 72 hours before determining the dry weight. The brain water content (%) was calculated as: (wet weight-dry weight)/wet weight×100% (16, 17).
Western Blotting
The left brain hemispheres (perforation side) were harvested at 24 hours after SAH and processed as previously described (18). Equal amounts of protein (50 μg) were separated by SDA-PAGE gel, electrophoresed, then transferred onto nitrocellulose membranes and incubated with the respective primary antibodies: Rabbit polyclonal anti-cleaved caspase-3 (1:1000) (Cell Signaling Technology, Danvers, MA), rabbit polyclonal anti-P2X7R (1:1000), mouse monoclonal anti-phosphorylated p38 MAPK(1:1000), goat polyclonal anti-phosphorylated JNK (1:1000), mouse monoclonal anti-phosphorylated ERK1/2 (1:1000), goat polyclonal β-actin (1:5000) (Santa Cruz Biotechnology, Santa Cruz, CA). Immunoblots were processed with appropriate secondary antibodies (1:2000, Santa Cruz Biotechnology, Santa Cruz, CA) for 2 hours at room temperature. Bands were visualized with the ECL Plus chemiluminescence reagent kit (Amersham Bioscience, Arlington Heights, IL) and density quantified, using the software Image J (National Institutes of Health). Results are expressed as relative density ratio, normalized to the average value of the sham group.
Immunofluorescence Staining
Rats were euthanized at 24 hours after SAH. Double immunofluorescence staining was performed as previously described (19, 20) using the neuronal marker of neuronal nuclei (NeuN) (1:100, Millipore, Temecula, CA) and P2X7R (1:100, Santa Cruz Biotechnology, Santa Cruz, CA). Brain sections were incubated with a mixture of the above mentioned primary antibodies over night at 4°C, followed by a mixture of Texas Red- and AMCA-conjugated secondary antibodies (Jackson Immunoresearch, West Grove, PA) for 2 hours at room temperature. Microphotographs were analyzed with a fluorescent microscope (Olympus OX51).
Double immunofluorescence staining was processed with anti-NeuN (1:100, Millipore, Temecula, CA) and terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling (TUNEL) (In situ Cell Death Detection Kit, Fluorescein, Roche Inc, Mannheim, Germany). TUNEL-positive neurons were counted in a blinded manner. The extent of neuronal damage was evaluated by an apoptotic index, which was calculated as the average number of TUNEL-positive neurons in six sections per brain at ×400 magnification. The data were expressed as cells/mm2.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 5 and SPSS 16.0 software. Data were expressed as mean±SEM and statistically analyzed with one-way ANOVA followed by Tukey post hoc test. Fisher’s exact test was used in two group comparisons for mortality analysis. All behavior data were expressed as median±25th–75th percentiles and analyzed with Kruskal-Wallis one-way ANOVA on ranks, followed by the Student-Newman-Keuls method. Statistical significant level was considered at p<0.05.
RESULTS
Mortality and SAH grading
No significant changes of the physiological parameters (body temperature, blood gases and body weight) were observed between different experimental groups (data not shown). No sham-operated animal died after surgery. In experiment 1, the mortality in the SAH+vehicle group reached 22.22% (8 of 36 rats), 19.35% (6 of 31 rats) in the SAH+BBG group and 16.67% (1 of 6 rats) in the SAH+BBG+BzATP group (p>0.05). In experiment 2, the mortality in the SAH+vehicle group reached 14.29% (1 of 7 rats), 14.29% (1 of 7 rats) in the SAH+scramble siRNA group and 14.29% (1 of 7 rats) in the SAH+P2X7R siRNA group (p>0.05).
Brain injury in the SAH model is directly related to the severity of bleeding (21). There was no significant difference in SAH grading score, at each time point, among the groups (Fig.1A, Fig.2A and Fig.5A).
Figure 1.
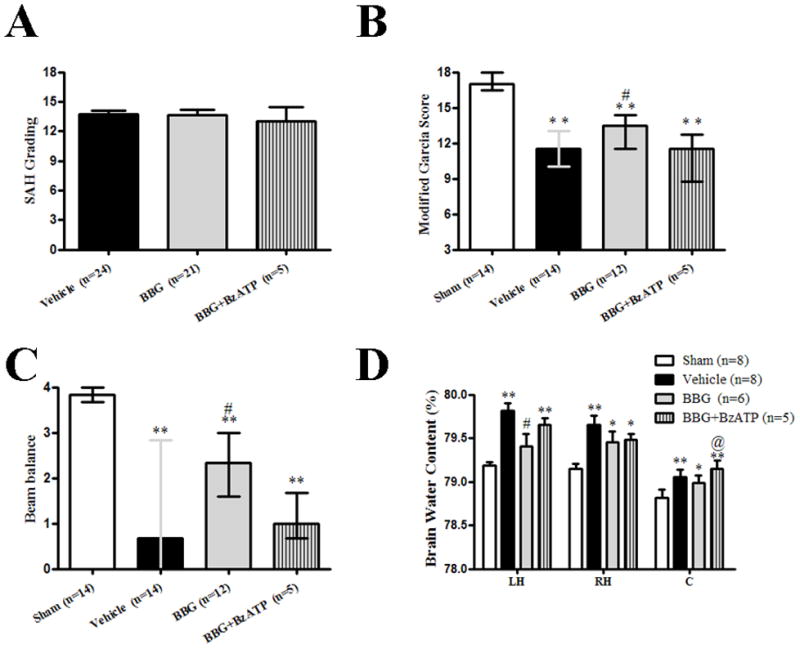
Effects of Brilliant blue G (BBG) treatment on subarachnoid hemorrhage (SAH) grading, Modified Garcia and Beam balance score, and Brain water content at 24 hours after SAH. A: Similar SAH grading was observed in the SAH+vehicle group (n=24), SAH+BBG group (n=21) and SAH+BBG+ BzATP (n=5). B and C: BBG treatment increased modified Garcia and beam balance score at 24 hours after SAH (*p<0.05 and **p<0.01 vs. sham; #p<0.05 vs. vehicle) respectively, BzATP tended to reversed the effect of BBG (p>0.05). D: BBG treatment decreased brain water content significantly in the left hemisphere at 24 hours after SAH, BzATP aggravated post-SAH brain edema in cerebellum (*p<0.05 and **p<0.01 vs. sham; #p<0.05 vs. vehicle; @p<0.05 vs. BBG, sham: n=8, vehicle: n=8, BBG: n=6, BBG+BzATP: n=5). A and D: error bar=SEM, B and C: error bar=25th–75th interquarile percentiles
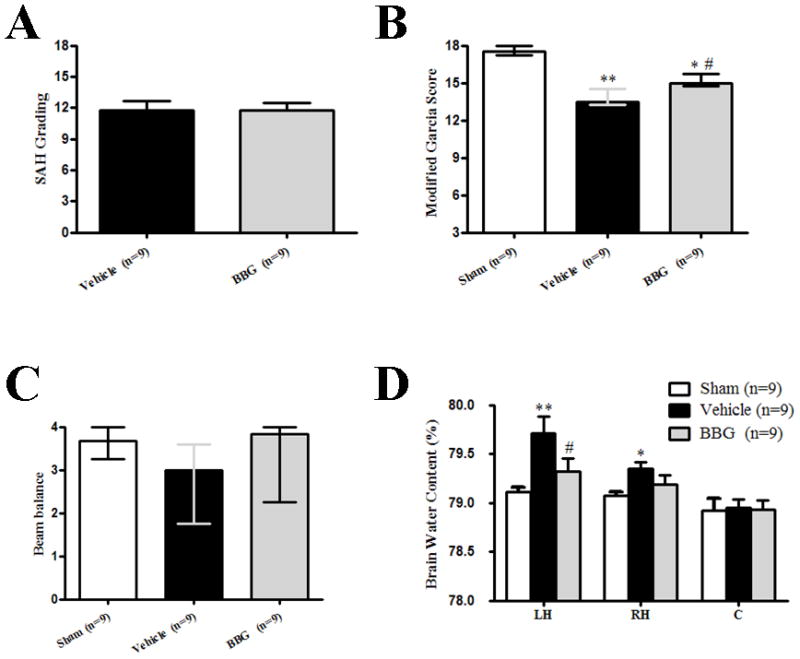
Figure 2.

Effects of Brilliant blue G (BBG) treatment on subarachnoid hemorrhage (SAH) grading, Modified Garcia and Beam balance score, and Brain water content at 72 hours after SAH. A: Similar SAH grading was observed in the SAH+vehicle group and SAH+BBG group. B and C: modified Garcia score decreased at 72 hours after surgery. BBG treatment significantly increased modified Garcia score, but not beam balance test (*p<0.05 and **p<0.01 vs. sham; #p<0.05 vs. vehicle). D: BBG treatment significantly decreased brain water content in the left hemisphere. (*p<0.05 and **p<0.01 vs. sham; #p<0.05 vs. vehicle, n=9). A and D: error bar=SEM, B and C: error bar=25th–75th interquarile percentiles.
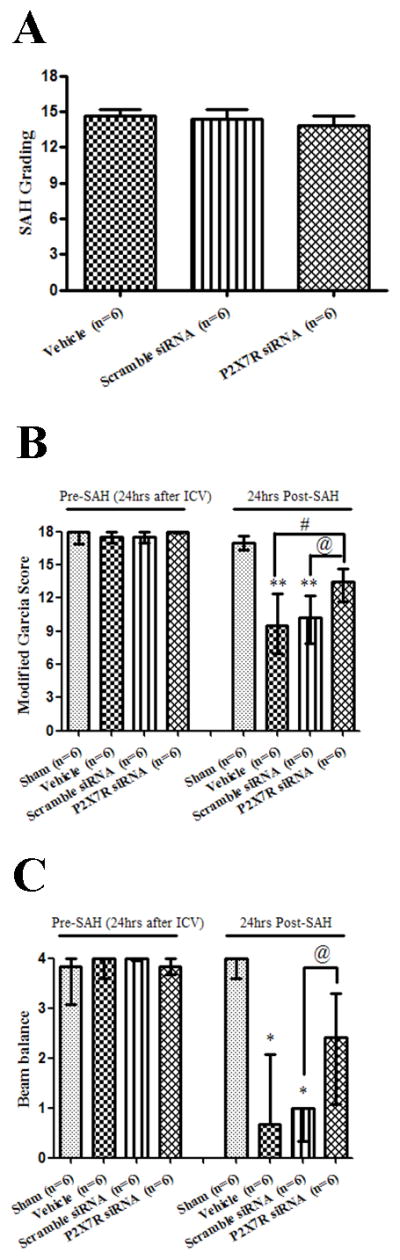
Figure 5.
Effects of P2X7 recptor (P2X7R) siRNA on subarachnoid hemorrhage (SAH) grading, Modified Garcia and Beam balance score at pre-SAH and 24 hours after SAH. A: Similar subarachnoid hemorrhage (SAH) grading was observed in the SAH+vehicle group, SAH+scramble siRNA group and SAH+P2X7R siRNA (p>0.05). B and C: P2X7R siRNA treatment increased modified Garcia score and beam balance at 24 hours after SAH (*p<0.05 and **p<0.01 vs. sham; #p<0.05 vs. vehicle; @ p<0.05 vs. scramble siRNA, n=6) respectively. A: error bar=SEM, B and C: error bar=25th–75th interquarile percentiles.
BBG improved neurobehavioral functions and attenuated brain edema after SAH
As compared with sham group, neurobehavioral function impairment was evident in SAH subjects. Post-SAH administration of BBG significantly improved neurobehavioral deficits at 24 hours after SAH (p<0.05 vs. SAH+vehicle; Fig.1B and Fig.1C). Pre-SAH administration of BzATP, an agonist of P2X7R, tended to reduce the neuroprotective effect of BBG without significant difference (p>0.05 vs. SAH+BBG; Fig.1B and Fig.1C). At 72 hours post SAH, BBG treatment resulted in significantly improved neurobehavioral functions, evaluated via the modified Garcia score (p<0.05 vs. SAH+vehicle; Fig.2B).
At 24 hours post SAH, brain water content significantly increased by 0.63% in the left hemisphere (p<0.01), 0.51% in the right hemisphere (p<0.05), and 0.24% in the cerebellum (p<0.05) compared to sham animals; however, brain water content did not change significantly in the brain stem (Fig.1D). Brain water content in the left hemisphere significantly was reduced by BBG (SAH+BBG, 79.40 ± 0.14 vs. SAH+vehicle, 79.81 ± 0.09, p<0.05; Fig.1D). BzATP administration aggravated post-SAH brain water content in the cerebellum (SAH+BBG+BzATP, 79.15 ± 0.09 vs. SAH+BBG, 78.98 ± 0.08, p<0.05; Fig.1D), and tendentially in the left hemisphere (SAH+BBG+BzATP, 79.65 ± 0.08 vs. SAH+BBG, 79.40 ± 0.14; p=0.0782; Fig.1D). At 72 hours post SAH, the brain water content in the left brain hemisphere was significantly reduced by BBG (SAH+BBG, 79.32 ± 0.13 vs. SAH+vehicle, 79.71 ± 0.17, p<0.05; Fig.2D).
BBG reduced p38 MAPK activation and cleaved caspase-3 level, but increased Bcl-2 level in the left hemisphere at 24 hours following SAH
The expression of phosphorylated p38 MAPK increased by 274% 24 hours after SAH when compared with the sham group (p<0.05), and 151% elevation was detected in SAH+BBG group (p<0.05 vs. SAH+vehicle, Fig.3A). The other MAPKs (phosphorylated JNK and phosphorylated Erk1/2) also increased after SAH when compared with the sham group, but did not reach statistical significance (Fig.3B and Fig.3C). The protein expression of Bcl-2 was significantly decreased 24 hours after SAH (p<0.05 vs. sham), while administration of BBG increased its expression (p<0.05 vs. SAH+vehicle; Fig.3D). The protein expression of cleaved caspase-3 was significantly increased 24 hours after SAH (p<0.05 vs. sham), while administration of BBG reduced its expression (p<0.05 vs. SAH+vehicle; Fig.4B).
Figure 3.

Effects of Brilliant blue G (BBG) treatment on phosphorylated p38 MAPK (A), phosphorylated JNK (B), phosphorylated ERK (C) and Bcl-2 (D) expressions in the left cerebral hemisphere at 24 hours after subarachnoid hemorrhage (SAH). BBG reduced the level of p38 MAPK (A) and increased Bcl-2 (D) (*p<0.05 and **p<0.01 vs. sham; #p<0.05 vs. vehicle, n=6). error bar=SEM.
Figure 4.
Effects of Brilliant blue G (BBG) treatment on Cleaved caspase-3 expression and Neuronal Apoptosis at 24 hours after subarachnoid hemorrhage (SAH). A: Representative photographs of immunofluorescence staining for P2X7R (red) and neuron (NeuN, red) in the subcortical area at 24 hours following SAH. Scale bar: 30μm. B: western blotting showed BBG reduced the level of cleaved caspase-3 in the left hemisphere at 24 hours after SAH (*p<0.05 and **p<0.01 vs. sham; #p<0.05 vs. vehicle, n=6). C and D: Neuronal apoptosis quantification representative microphotographs showing colocalization of NeuN (Texas Red/red) and TUNEL (green). BBG treatment decreased TUNEL-positive neuron (n=4 or 5). Scale bar: 50μm. error bar=SEM.
BBG reduced neuronal apoptosis in the subcortex at 24 hours after SAH
Immunofluorescence staining of P2X7R and NeuN were generally colocalized in the vehicle group at 24 hours after SAH (Fig.4A). The total number of TUNEL and NeuN double-stained cells was significantly increased in vehicle group (p<0.05 vs. sham; Fig.4C and Fig.4D), but TUNEL-positive neurons were less apparent after BBG treatment (p<0.05 vs. SAH+vehicle; Fig.4C and Fig.4D).
Gene Silencing of P2X7R improved neurobehavioral functions
Neurobehavioral functions (Fig.5B and Fig.5C) were evaluated at 24 hours following SAH. No neurological impairment was caused by siRNA injection. The results revealed that vehicle and scramble siRNA treated animals developed neurological deficits compared to sham animals (p<0.01). No significant difference was seen between the vehicle group and scramble siRNA group (p>0.05). P2X7R siRNA treated rats showed better performances in during modified Garcia (p<0.05 vs. vehicle and scramble siRNA, respectively; Fig.5B) and beam balance tests (p<0.05 vs. scramble siRNA; Fig5C).
Gene Silencing of P2X7R decreased P2X7R expression and p38 MAPK activation, reducing cleaved caspase-3 in the left hemisphere at 24 hours following SAH
Western blot results revealed that P2X7R expression did not increase at 24 hours after SAH, but decreased in the P2X7R siRNA group, compared with the vehicle and the scramble siRNA group (p<0.05; Fig.6A). The level of phosphorylated p38 MAPK and cleaved caspase-3 were significantly increased in the SAH+vehicle group and as well as in the SAH+scramble siRNA (p<0.05, vs. sham; Fig.6B and Fig.6C). P2X7R siRNA significantly reduced the subsequent production of phosphorylated p38 MAPK and cleaved caspase-3 (p<0.05, vs. vehicle and scramble siRNA, respectively; Fig.6B and Fig.6C).
Figure 6.
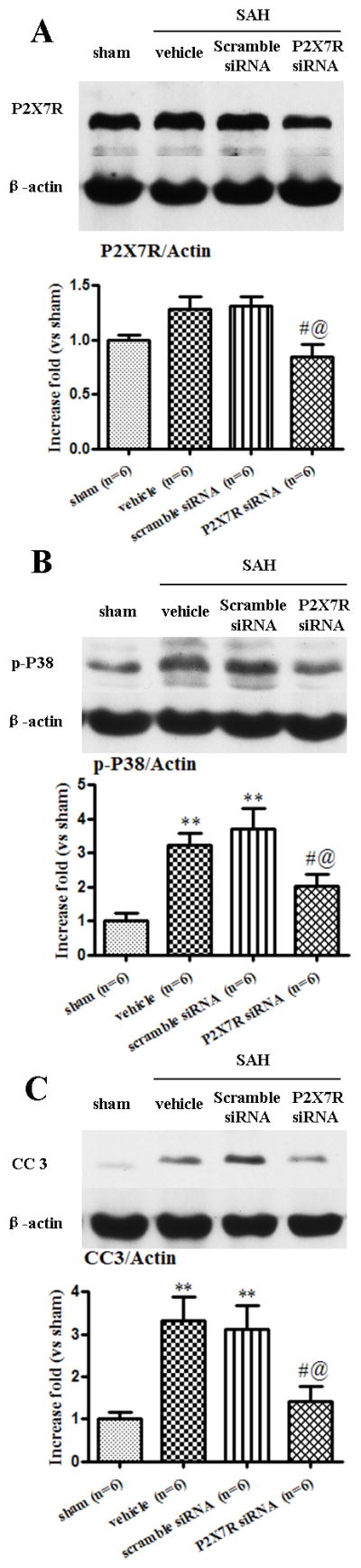
Effects of P2X7 recptor (P2X7R) siRNA on P2X7R, phosphorylated p38 MAPK, and Cleaved caspase-3 in the left cerebral hemisphere at 24 hours after subarachnoid hemorrhage (SAH). P2X7R siRNA reduced the level of P2X7R (A), phosphorylated p38 MAPK (B) and cleaved caspase-3 (C) (*p<0.05 and **p<0.01 vs. sham; #p<0.05 vs. vehicle; @ p<0.05 vs. scramble siRNA, n=6). error bar=SEM.
DISCUSSION
In this study, we found that SAH induced activation of p38 MAPK but not of JNK and ERK, which resulted in neuronal apoptosis. Additionally, treatment with either P2X7R siRNA or the selective P2X7R antagonist BBG effectively reversed p38 MAPK activation, thus reducing neuronal apoptosis after SAH. These data are consistent with our hypothesis suggesting that inhibition of P2X7R would attenuate neuronal apoptosis through reduction of phosphorylated and therefore activated p38 MAPK after SAH.
MAPKs are serine/threonine kinases, including: ERK, p38 MAPK, and JNK. Among them, p38 MAPK is a pro-apoptotic protein (22). The activation of p38 MAPK leads to the production of cleaved of caspase-3 and reduction of bcl-2, which result in apoptotic cell death (23). Previously, our study and others research found significant activation of p38 MAPK was obtained in brain after SAH (24, 25). Recently, inhibition of p38 MAPK suppressed apoptosis of the basilar artery in a rabbit SAH model (26). In the present study, we found SAH induced the increase of phosphorylated p38 MAPK. Hence, we speculated that p38 MAPK may be involved in SAH-induced neuronal apoptosis.
P2X7R is an ion channel, gated by extracellular ATP, which rapidly accumulates from the ruptured cells in stroke events (27). In the CNS, functional P2X7R have been localized in microglia and astrocytes, as well as in neuronal cells (28). In the present study, we focus on neuronal apoptosis, therefore, we performed immunofluorescence staining to localize our findings and suggested P2X7R may act as important regulators of neuronal cell death in response to SAH insult. P2X7R activation results in formation of the channel pore and the development of permeability to molecules as large as 900 Da, which may potentially exacerbate CNS disorders (5–7). Preventing its activation might ameliorate immune-mediated CNS disorders. Recently, various selective P2X7R antagonists are undergoing clinical trials (29). Moreover, P2X7R stimulates a diverse range of cellular responses, including ion fluxes along the cell membrane (Ca2+ and Na+ influx, K+ efflux), phospholipase A/D, MAPK, nuclear factor-κB and apoptosis (4). Intracellular Ca2+ mobilization induced by the P2X7R activation, could serve as a mechanistic link for P2X7R amplification of cellular responses (30). In primary astrocyte cultures, activation of the purinergic P2X7R lead to the activation of the ERK1/2, and p38 MAPK, which was measured by Western blotting (9). BBG, a P2X7 receptor blocker, could inhibit the ATP-provoked activation of JNK and p38 MAPK, but not ERK (31). Most importantly, in vivo, the P2X7R antagonist, oxidized ATP, inhibited LPS-induced activation of p38 MAPK and reduced numbers of caspase-3 positive neurons and increased neuronal survival in the inflamed brain (32). In the present study, BBG administered after SAH improved neurological function, reduced brain edema and decreased phosphorylated p38 MAPK. Furthermore, BBG treatment increased Bcl-2 and decreased cleaved caspase-3 expression, as well as neuronal cell death at 24 hours after SAH. Based on these data, we suggest that the improvement of neurological function by inhibition of P2X7R after SAH was ascribed to their effect on reducing p38 MAPK-related excessive neuron apoptosis. Moreover, siRNA is more specific than pharmacological inhibitor. Our data also demonstrated gene silencing of P2X7R reduced phosphorylated p38 MAPK and subsequently prevented caspase-3 activation after SAH.
There are several weaknesses in our study. Firstly, BBG may inhibit other P2X receptors, even it is at least 1000-fold more potent at P2X7R than other P2X receptors in rats (33). Therefore, we further used P2X7R siRNA to confirm our results, which is specific targeting this receptor. Secondly, blockade of P2X7R will also contribute anti-inflammatory effect (34). Hence, we cannot exclude possibility that the anti-inflammatory and even other effects also play a role in the neuroprotective effect of BBG. In addition, isoflurane has been shown to be neuroprotective and its neuroprotective effects should be considered in the current study. Furthermore, we did not investigate whether BzATP increases SAH-induced phosphorylation of p38 MAPK. At last, intracellular calcium mobilization induced by the P2X7R antagonists could serve as a mechanistic link for P2X7R amplification of cellular responses. Future studies measuring intracellular calcium mobilization will help us confirm the link between P2X7R and p38 MAPK.
In conclusion, we demonstrate for the first time that inhibition of P2X7R can prevent EBI via p38 MAPK-related apoptotic pathway after SAH. BBG treatment for SAH is promising, and our findings warrant further research.
Acknowledgments
Financial support: This study was supported by NIH NS053407 to JH Zhang and by Natural Science Foundation of China (No.81171096); Natural Science Foundation of Zhejiang province, China (No.Z2090200) to JM Zhang.
We thank Prof. Yi Shen (Department of Epidemiology & Health Statistics, Zhejiang University) for his statistical assistance.
Footnotes
Conflicts of Interest: None.
References
- 1.Venti M. Subarachnoid and intraventricular hemorrhage. Front Neurol Neurosci. 2012;30:149–153. doi: 10.1159/000333625. [DOI] [PubMed] [Google Scholar]
- 2.Chou SH, Feske SK, Simmons SL, et al. Elevated Peripheral Neutrophils and Matrix Metalloproteinase 9 as Biomarkers of Functional Outcome Following Subarachnoid Hemorrhage. Transl Stroke Res. 2011;2:600–607. doi: 10.1007/s12975-011-0117-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hasegawa Y, Suzuki H, Sozen T, et al. Apoptotic mechanisms for neuronal cells in early brain injury after subarachnoid hemorrhage. Acta Neurochir Suppl. 2011;110:43–48. doi: 10.1007/978-3-7091-0353-1_8. [DOI] [PubMed] [Google Scholar]
- 4.Skaper SD, Debetto P, Giusti P. The P2X7 purinergic receptor: from physiology to neurological disorders. FASEB J. 2010;24:337–345. doi: 10.1096/fj.09-138883. [DOI] [PubMed] [Google Scholar]
- 5.Takenouchi T, Sekiyama K, Sekigawa A, et al. P2X7 receptor signaling pathway as a therapeutic target for neurodegenerative diseases. Arch Immunol Ther Exp (Warsz) 2010;58:91–96. doi: 10.1007/s00005-010-0069-y. [DOI] [PubMed] [Google Scholar]
- 6.Chu K, Yin B, Wang J, et al. Inhibition of P2X7 receptor ameliorates transient global cerebral ischemia/reperfusion injury via modulating inflammatory responses in the rat hippocampus. J Neuroinflammation. 2012;9:69. doi: 10.1186/1742-2094-9-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kimbler DE, Shields J, Yanasak N, et al. Activation of P2X7 promotes cerebral edema and neurological injury after traumatic brain injury in mice. PLoS One. 2012;7:e41229. doi: 10.1371/journal.pone.0041229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Papp L, Vizi ES, Sperlagh B. P2X7 receptor mediated phosphorylation of p38MAP kinase in the hippocampus. Biochem Biophys Res Commun. 2007;355:568–574. doi: 10.1016/j.bbrc.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 9.Panenka W, Jijon H, Herx LM, et al. P2X7-like receptor activation in astrocytes increases chemokine monocyte chemoattractant protein-1 expression via mitogen-activated protein kinase. J Neurosci. 2001;21:7135–7142. doi: 10.1523/JNEUROSCI.21-18-07135.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 11.Zhan Y, Chen C, Suzuki H, et al. Hydrogen gas ameliorates oxidative stress in early brain injury after subarachnoid hemorrhage in rats. Crit Care Med. 2012;40:1291–1296. doi: 10.1097/CCM.0b013e31823da96d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duris K, Manaenko A, Suzuki H, et al. Sampling of CSF via the Cisterna Magna and Blood Collection via the Heart Affects Brain Water Content in a Rat SAH Model. Transl Stroke Res. 2011;2:232–237. doi: 10.1007/s12975-010-0063-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mishra A, Chintagari NR, Guo Y, et al. Purinergic P2X7 receptor regulates lung surfactant secretion in a paracrine manner. J Cell Sci. 2011;124:657–668. doi: 10.1242/jcs.066977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki H, Hasegawa Y, Kanamaru K, et al. Mechanisms of osteopontin-induced stabilization of blood-brain barrier disruption after subarachnoid hemorrhage in rats. Stroke. 2010;41:1783–1790. doi: 10.1161/STROKEAHA.110.586537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki H, Hasegawa Y, Chen W, et al. Recombinant osteopontin in cerebral vasospasm after subarachnoid hemorrhage. Ann Neurol. 2010;68:650–660. doi: 10.1002/ana.22102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yabluchanskiy A, Sawle P, Homer-Vanniasinkam S, et al. CORM-3, a carbon monoxide-releasing molecule, alters the inflammatory response and reduces brain damage in a rat model of hemorrhagic stroke. Crit Care Med. 2012;40:544–552. doi: 10.1097/CCM.0b013e31822f0d64. [DOI] [PubMed] [Google Scholar]
- 17.He Y, Karabiyikoglu M, Hua Y, et al. Ischemic Preconditioning Attenuates Brain Edema After Experimental Intracerebral Hemorrhage. Transl Stroke Res. 2012;3:180–187. doi: 10.1007/s12975-012-0171-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altay O, Hasegawa Y, Sherchan P, et al. Isoflurane delays the development of early brain injury after subarachnoid hemorrhage through sphingosine-related pathway activation in mice. Crit Care Med. 2012;40:1908–1913. doi: 10.1097/CCM.0b013e3182474bc1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou J, Huang WQ, Li C, et al. Intestinal ischemia/reperfusion enhances microglial activation and induces cerebral injury and memory dysfunction in rats. Crit Care Med. 2012;40:2438–2448. doi: 10.1097/CCM.0b013e3182546855. [DOI] [PubMed] [Google Scholar]
- 20.Xiong Y, Zhang Y, Mahmood A, et al. Erythropoietin Mediates Neurobehavioral Recovery and Neurovascular Remodeling Following Traumatic Brain Injury in Rats by Increasing Expression of Vascular Endothelial Growth Factor. Transl Stroke Res. 2011;2:619–632. doi: 10.1007/s12975-011-0120-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sugawara T, Ayer R, Jadhav V, et al. A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Methods. 2008;167:327–334. doi: 10.1016/j.jneumeth.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang X, Zhou G, Ren T, et al. beta-Arrestin prevents cell apoptosis through pro-apoptotic ERK1/2 and p38 MAPKs and anti-apoptotic Akt pathways. Apoptosis. 2012;17:1019–1026. doi: 10.1007/s10495-012-0741-2. [DOI] [PubMed] [Google Scholar]
- 23.Morisco C, Marrone C, Trimarco V, et al. Insulin resistance affects the cytoprotective effect of insulin in cardiomyocytes through an impairment of MAPK phosphatase-1 expression. Cardiovasc Res. 2007;76:453–464. doi: 10.1016/j.cardiores.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 24.Sasaki T, Kasuya H, Onda H, et al. Role of p38 mitogen-activated protein kinase on cerebral vasospasm after subarachnoid hemorrhage. Stroke. 2004;35:1466–1470. doi: 10.1161/01.STR.0000127425.47266.20. [DOI] [PubMed] [Google Scholar]
- 25.Kusaka G, Ishikawa M, Nanda A, et al. Signaling pathways for early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2004;24:916–925. doi: 10.1097/01.WCB.0000125886.48838.7E. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X, Zhao XD, Shi JX, et al. Inhibition of the p38 mitogen-activated protein kinase (MAPK) pathway attenuates cerebral vasospasm following experimental subarachnoid hemorrhage in rabbits. Ann Clin Lab Sci. 2011;41:244–250. [PubMed] [Google Scholar]
- 27.Melani A, Amadio S, Gianfriddo M, et al. P2X7 receptor modulation on microglial cells and reduction of brain infarct caused by middle cerebral artery occlusion in rat. J Cereb Blood Flow Metab. 2006;26:974–982. doi: 10.1038/sj.jcbfm.9600250. [DOI] [PubMed] [Google Scholar]
- 28.Surprenant A, North RA. Signaling at purinergic P2X receptors. Annu Rev Physiol. 2009;71:333–359. doi: 10.1146/annurev.physiol.70.113006.100630. [DOI] [PubMed] [Google Scholar]
- 29.Baroja-Mazo A, Pelegrin P. Modulating P2X7 Receptor Signaling during Rheumatoid Arthritis: New Therapeutic Approaches for Bisphosphonates. J Osteoporos. 2012;2012:408242. doi: 10.1155/2012/408242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lenertz LY, Gavala ML, Zhu Y, et al. Transcriptional control mechanisms associated with the nucleotide receptor P2X7, a critical regulator of immunologic, osteogenic, and neurologic functions. Immunol Res. 2011;50:22–38. doi: 10.1007/s12026-011-8203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzuki T, Hide I, Ido K, et al. Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J Neurosci. 2004;24:1–7. doi: 10.1523/JNEUROSCI.3792-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi HB, Ryu JK, Kim SU, et al. Modulation of the purinergic P2X7 receptor attenuates lipopolysaccharide-mediated microglial activation and neuronal damage in inflamed brain. J Neurosci. 2007;27:4957–4968. doi: 10.1523/JNEUROSCI.5417-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang LH, Mackenzie AB, North RA, et al. Brilliant blue G selectively blocks ATP-gated rat P2X(7) receptors. Mol Pharmacol. 2000;58:82–88. [PubMed] [Google Scholar]
- 34.Arulkumaran N, Unwin RJ, Tam FW. A potential therapeutic role for P2X7 receptor (P2X7R) antagonists in the treatment of inflammatory diseases. Expert Opin Investig Drugs. 2011;20:897–915. doi: 10.1517/13543784.2011.578068. [DOI] [PMC free article] [PubMed] [Google Scholar]