

Summary
Familial transthyretin amyloidosis (ATTR) is an autosomal-dominant protein-folding disorder caused by over 100 distinct mutations in the transthyretin (TTR) gene. In ATTR, protein secreted from the liver aggregates and forms fibrils in target organs, chiefly the heart and peripheral nervous system, highlighting the need for a model capable of recapitulating the multisystem complexity of this clinically variable disease. Here, we describe the directed differentiation of ATTR patient-specific iPSCs into hepatocytes that produce mutant TTR, and the cardiomyocytes and neurons normally targeted in the disease. We demonstrate that iPSC-derived neuronal and cardiac cells display oxidative stress and an increased level of cell death when exposed to mutant TTR produced by the patient-matched iPSC-derived hepatocytes, recapitulating essential aspects of the disease in vitro. Furthermore, small molecule stabilizers of TTR show efficacy in this model, validating this iPSC-based, patient-specific in vitro system as a platform for testing therapeutic strategies.
Highlights
-
•
Successful modeling of familial amyloidosis in vitro using iPSC technology
-
•
Proto-fibril formation leads to cellular damage in two target tissues of amyloidosis
-
•
iPSCs can be used in the testing of novel therapeutics for protein folding disorders
This work involves the modeling of hereditary transthyretin amyloidosis in vitro using iPSC technology. Murphy and colleagues demonstrate that it is possible to model a long-term, complex, multisystem disease using hepatic, cardiac, and neuronal lineages derived from patient-specific stem cells and validate this approach for the testing of therapeutic strategies.
Introduction
Amyloidosis refers to a group of diseases caused by the extracellular deposition of misfolded fibrillar proteins, leading to multiorgan failure and death (Falk et al., 1997). Familial amyloidosis (AF) occurs when inherited point mutations in the genes coding for abundant serum proteins such as transthyretin (TTR), fibrinogen, lysozyme, or apolipoproteins lead to clinical disease. The most common form of AF arises from aggregation of mutated TTR, a 55 kDa transport protein predominantly synthesized by the liver (Connors et al., 2003; Falk et al., 1997; Planté-Bordeneuve and Said, 2011). Familial transthyretin amyloidosis (ATTR) is a lethal, autosomal-dominant disease caused by single amino acid substitutions arising from 1 of more than 100 described mutations in the TTR gene (Connors et al., 2003; online amyloidosis mutation database in Rowczenio and Wechalekar [2010]). These single amino acid substitutions destabilize the native homotetrameric structure of circulating TTR, promoting release of amyloidogenic TTR monomers, fibril formation, and deposition as amyloid in target end organs (Ando et al., 2005; Falk et al., 1997). Most amyloidogenic TTR variants target the nervous system and heart, inducing neuropathy and cardiomyopathy (Jacobson et al., 1992, 1997; Planté-Bordeneuve and Said, 2011). The organ distribution and age of onset can vary across families and endemic areas, even with identical TTR mutations. Although transgenic mouse models and immortalized human cell lines have provided some insights into disease pathogenesis (Araki et al., 1994; Kohno et al., 1997; Reixach et al., 2004; Sousa et al., 2002; Tagoe et al., 2007), these systems are independent of the genetic context of the patient.
ATTR represents an important unmet medical need with significant morbidity and early mortality in affected families. Survival after ATTR disease onset is usually only 5–15 years (Benson et al., 2011; Planté-Bordeneuve and Said, 2011). Orthotopic liver transplantation is currently the only US-approved treatment for ATTR. However, only about one-third of patients are candidates for surgery (Monteiro et al., 2004), and cardiomyopathy and neuropathy may continue to worsen after transplantation. Recently, small molecule stabilizers of variant TTR (Tafamidis and diflunisal) shown to inhibit amyloid fibril formation in vitro are being studied in clinical trials (Berk et al., 2012; Coelho et al., 2012). Patient-specific, cell-based models are needed to facilitate the study of the genetic and epigenetic factors in this disease, permitting pharmacogenomic assessments of novel therapeutics.
The generation of induced pluripotent stem cells (iPSCs) through the reprogramming of somatic cells from patients with inherited diseases provides an unprecedented opportunity to study the effects of genetic abnormalities and disease progression. The derivation of unlimited quantities of the relevant cell types targeted and affected in patients with hereditary disease allows for the investigation of the cellular, molecular, and epigenetic events involved in a multisystem, genetic disease such as ATTR. Although most cases of ATTR are due to a single base pair mutation predominantly expressed in the liver, end organ damage occurs outside the liver, highlighting the need for a model capable of recapitulating the multisystem complexity of the disease.
Here, we describe the generation of ATTR patient-specific iPSCs and the directed differentiation of these cells into hepatic, neuronal, and cardiac lineages thereby modeling the three major tissue types involved in this disease (Figure 1). Our data are consistent with the clinical trial findings that known small molecule TTR stabilizers can inhibit TTR fibril formation in vivo, validating patient-specific iPSCs as a platform for testing therapeutics.
Figure 1.

Schematic Summary of the In Vitro iPSC Approach to Study ATTR
The cytokines that were used in each differentiation protocol are indicated. FAP, familial amyloid polyneuropathy; FAC, familial amyloid cardiomyopathy.
Results
Generation of ATTR Disease-Specific Human iPSCs
Using our highly efficient and reproducible STEMCAA reprogramming system (Somers et al., 2010; Sommer et al., 2009, 2010), we generated iPSC lines from tissue samples of a patient heterozygous for the leucine-55-proline (L55P) TTR mutation (see Figure S1A available online). This mutation, one of the most clinically aggressive variants of ATTR disease (Jacobson et al., 1992), produces a highly unstable and amyloidogenic TTR, clinically targeting the heart and the peripheral nervous system (Lashuel et al., 1999). The sequenced TTR transcript from the reprogrammed ATTR-iPSC derivatives was compared to that from control iPSCs to confirm that the ATTR iPSCs carried the L55P mutation (Figure S1B). Characterization of each iPSC clone demonstrated normal 46 XX karyotype, expression of stem cell marker genes, and pluripotency in teratoma assays (Figures S1C–S1F). Characterized clones were then differentiated into both the effector cells (hepatocytes) that produce mutant TTR and peripheral target cells (neurons and cardiomyocytes) that are damaged by exposure to aberrant protein in order to create an iPSC-based, in vitro model of ATTR (Figure 1).
ATTRL55P Disease-Specific Human iPSCs Can Be Directed into Hepatic-Lineage Cells that Secrete TTR
In ATTR, the deleterious effects of liver-produced TTR variants are seen in other organs, with the liver remaining phenotypically normal. The majority of patients with ATTR are heterozygous for one autosomal-dominant mutation in the TTR gene, although compound heterozygotes have been documented (Connors et al., 2003, 2009; Hammarström et al., 2001). Therefore, in most patients, TTR tetramers synthesized by the liver are comprised of a mix of normal and variant TTR monomers that assemble into stochastically formed homo- and heterotetramers (Schneider et al., 2001). To derive the full complement of TTR tetramer variants, containing posttranslational modifications that are normally present in patient serum TTR, we differentiated ATTRL55P iPSCs into hepatic-lineage cells with the aim of producing variant TTR protein from patient-derived cells.
In our system, the derivation of hepatic-lineage cells from iPSCs was achieved through a sequential, growth factor-driven differentiation process (Hay et al., 2008; Sullivan et al., 2010) (Figure 2A). Mimicking signaling events that occur during embryonic patterning, high levels of activin A and Wnt3a were used to drive iPSCs toward a definitive endoderm progenitor state. At this stage, the majority of the cells stained positively for CXCR4 and/or cKit (Figure 2B), cell surface markers for definitive endoderm (Yasunaga et al., 2005). Further maturation of the hepatic-lineage cells was achieved through culturing in maturation media containing hepatocyte growth factor (HGF) and oncostatin M (OncM). As determined by quantitative PCR, the resultant hepatic-lineage cells express robust levels of hepatic markers apolipotein A1 (APOA1), albumin (ALB), alpha-fetoprotein (AFP), and, also importantly, TTR (Figure 2C).
Figure 2.
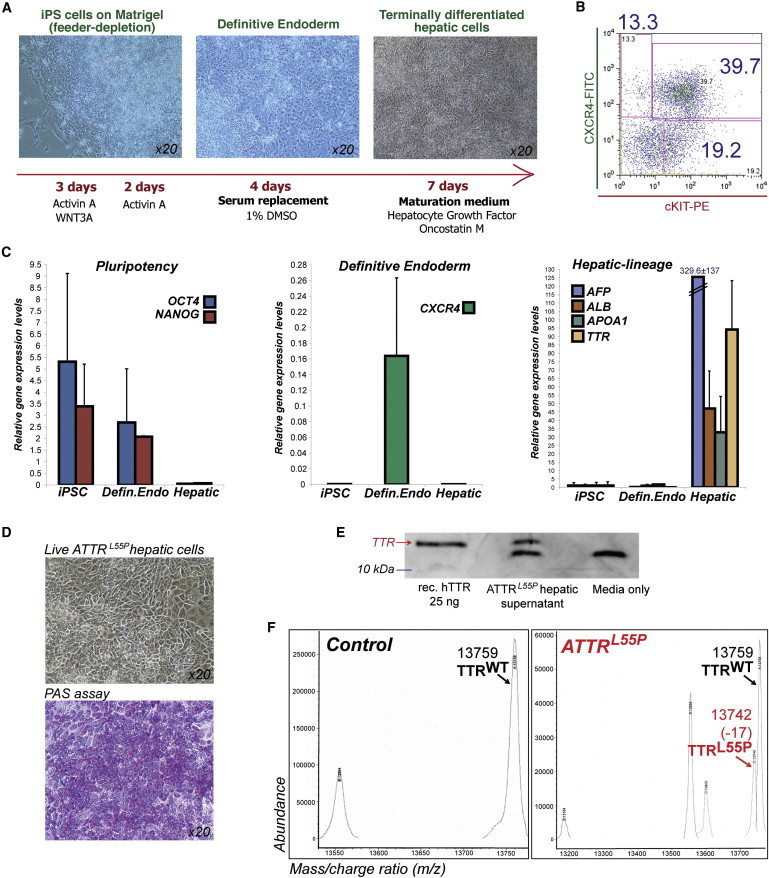
Generation and Characterization of ATTRL55P iPSC-Hepatic Lineage Cells
(A) Schematic diagram details the stepwise differentiation protocol used to obtain hepatic-lineage cells from iPSCs.
(B) Cells at day 5 of differentiation express the definitive endoderm cell surface markers CXCR4 and cKit, as determined by FACS analysis.
(C) Hepatic markers AFP, ALB, APOA1, and TTR are upregulated in hepatic-lineage cells. These markers are not expressed in iPSCs or (precursor cell type) definitive endoderm cells. All values are normalized to GAPDH. Error bars represent the SD (n = 3 up to six independent biological samples).
(D) Hepatic-lineage cells derived from ATTRL55P iPSCs morphologically resemble hepatocytes and are capable of glycogen storage as demonstrated by PAS staining.
(E) ATTRL55P iPSC hepatic-lineage cells produce and secrete TTR protein as demonstrated by immunoblot analysis with media supernatants. Recombinant human TTR (25 ng) and serum-free media alone serve as positive and negative controls, respectively. The lower band, seen in the ATTRL55P hs and media-only lanes, is a result of the nonspecific cross-reactivity of the TTR antibody to a product in the media formulation.
(F) Mass spectrometry analysis of control and ATTRL55P hs for TTR species is shown. hs generated from ATTRL55P hepatic-lineage cells contained both WT (13,759 kDa) and L55P (13,742 kDa) forms of TTR protein (labeled black and red arrows, respectively), whereas only the former was detectable in control samples.
Similarly, efficient derivation of hepatic-lineage cells and their precursor cell types was achieved from both ATTRL55P iPSCs and a control iPSC line as judged by comparable CXCR4 and cKit stainings at day 5 of differentiation (Figures 2B and S2A), TTR gene expression (Figure S2B), and functionally, the iPSC-derived, hepatic-lineage cells were capable of glycogen storage as determined by periodic acid Schiff (PAS) staining (Figures 2D and S2C). For the purposes of our in vitro disease model, we investigated whether the ATTRL55P iPSC-derived hepatic cells were capable of synthesizing and secreting TTR protein. Western blot analysis of serum-free conditioned media (hepatic supernatant [hs]) produced from ATTRL55P hepatic-lineage cells revealed the presence of TTR protein, indicating that iPSC-derived hepatic-lineage cells were indeed capable of TTR protein production and secretion, with the concentration of TTR in hs estimated to be in the range of 25–50 nM (Figure 2E). Moreover, mass spectrometric analysis was utilized to precisely characterize mutant and wild-type (WT) forms of TTR in hs, revealing the presence of the TTRL55P species only in supernatant derived from ATTRL55P hepatic-lineage cells (Figure 2F). As expected, the presence of TTRWT was detected in both control and ATTRL55P supernatants (Figure 2F). The ratio of ATTRL55P-to-TTRWT monomers in ATTR hs was calculated to be 1:2 rather than 1:1 as expected, suggesting that a proportion of the TTRL55P produced in hepatic-lineage cells is not secreted into the media, for reasons that are unknown.
Although it is the site of aberrant protein production in ATTR, the livers of patients with ATTR are thought to be relatively normal, escaping the cellular damage seen in other target tissues. However, there is some evidence to suggest that there could be physiological and molecular differences in ATTR livers. Examination of the liver in a transgenic murine model for a different form of amyloidosis, SSA (which involves the deposition of WT TTR), revealed a link between the levels of protein folding/chaperoning genes in the liver, and the degree of observable TTR deposition in the “target” cardiac tissue in aged transgenic mice. The livers of the young transgenic mice also exhibited increased expression of genes linked to protein trafficking and inflammation/immunity (Buxbaum et al., 2012).
Although control and ATTRL55P iPSC-derived hepatic cells express similar levels of hepatic markers, we were interested in the possible existence of gene expression signature differences between the two cell populations. To examine if this was the case, we performed microarray analysis to compare the transcriptomes of hepatic-stage cells derived from normal versus ATTRL55P iPSCs (three independent replicates per sample type). Hierarchical clustering analysis indicated segregation of the ATTRL55P iPSC-derived biological triplicates from the control samples (Figure S3A). KEGG and Biocarta analysis of the data set revealed that extracellular matrix and connective tissue genes including collagen, laminin, and integrin transcripts were overrepresented in control hepatic cells (Figures S3B and S3C). Interestingly, genes relating to protein folding and stress response, especially the heat shock protein 70 (hsp70) family genes, were among the main genes upregulated in ATTRL55P hepatic cells (Figures S3B–S3D), suggesting that the in-vitro-derived, disease-specific hepatic cells upregulate these genes in response to the aberrant TTR protein.
ATTRL55P iPSCs Can Be Directed into Target Cells of the Disease: Cardiomyocytes and Neurons
The liver is not a clinically important site of amyloid deposition in vivo. Recapitulation of the ATTR disease phenotype requires a multilineage system to model complex interactions between the liver and target organ systems, including epigenetic events required for the full clinical phenotype to develop. The flexibility of the iPSC-based system allows for the directed differentiation of other tissue types, such as cardiomyocytes and neurons, that are affected by the variant protein produced by the liver. In initial studies, ATTRL55P iPSC-derived cardiac and neuronal cells were characterized with regards to gene expression profiling and functional assays.
The derivation of cardiomyocytes from ATTRL55P iPSCs was achieved using two methods. First, a modified growth factor-driven approach (Kattman et al., 2011; Yang et al., 2008) was used to efficiently obtain beating cardiomyocytes after 21 days of differentiation. A traditional embryoid body (EB) differentiation approach also successfully yielded beating cardiomyocyte colonies after only 10 days of differentiation (Figure 3A; Movie S1). A side-by-side comparison of the two methods through quantitative PCR analysis indicated that the EB approach was more efficient at yielding cardiomyocyte cells (as judged by the end-stage cardiomyocyte marker cTNT) (Figure 3B; data not shown). In addition to this, a high proportion of cells in cardiac EB (cEB) cultures express the cardiac marker sarcomeric anti-actinin as determined by intracellular FACS analysis (Figure 3C), and this method was therefore used to generate cardiomyocytes for subsequent experiments.
Figure 3.

Generation of Beating Cardiomyocytes from ATTRL55P iPSCs
(A) Day 14 contracting EBs are presented.
(B) Quantitative PCR analysis of ATTRL55P iPSC and ATTRL55P day 14 cEB cultures is shown. The pluripotency marker Oct4 is downregulated, and cardiac markers (Nkx2.5, Tbx5, Islet1, cardiac actin, and the end-stage marker cardiac troponin) are upregulated in the latter sample type. All values are normalized to GAPDH. Independent sample sizes are iPSC n = 2 and cEB n = 3. Error bars, SD.
(C) Intracellular FACS was used to quantitate the proportion of cells that express the cardiac marker sarcomeric anti-actinin (blue peak) in day 13 cEB cultures. The isotype control peak is shown in red.
For the derivation of neuronal-lineage cells, a modified stepwise-directed differentiation approach was used to obtain neuronal-lineage cells from iPSCs based on a previously described protocol by Chambers et al. (2009) and Hu et al. (2009). Morphologically, the cells produced resemble neuronal cells with long dendritic processes and axon-like structures (Figure 4A). Immunofluorescence characterization revealed that early-stage cells were positive for neuronal class III β-tubulin (TUJ1) (Figure 4B), whereas late-stage differentiation cells expressed microtubule-associated protein 2 (MAP2) and choline acetyltransferase (ChAT), proteins that localize to the cytoplasm of neuronal cells and the latter an enzyme that is critical for the production of the neurotransmitter acetylcholine (Figure 4C). Real-time PCR analysis demonstrated the expression of transcripts for TUJ1 and homeobox protein 9 (HB9), a motor neuron-specific marker (Figure 4D).
Figure 4.
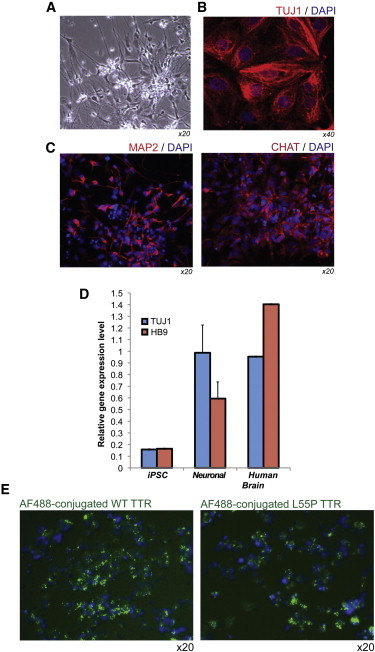
Derivation of Neuronal-Lineage Cells from ATTRL55P iPSCs that Are Capable of TTR Internalization
(A) Phase-contrast image shows neuronal cells derived from ATTRL55P iPSCs.
(B) Immunofluorescence staining presents ATTRL55P iPSC-derived neuronal cells for the neuronal marker TUJ1 (red). Nuclei were stained with DAPI (blue).
(C) Immunofluorescence staining shows ATTRL55P iPSC-derived neuronal cells for the MAP2 (red). Nuclei were stained with DAPI (blue).
(D) Quantitative PCR analysis of derived neuronal-lineage cells compared to undifferentiated iPSCs and human brain cDNA is shown. Neuronal cells exhibit upregulated levels of TUJ1 and motor neuron homeobox transcription factor HB9. All independent biological samples (n = 3) were normalized to GAPDH. Error bars, SD.
(E) ATTRL55P iPSC neuronal-lineage cells are capable of TTR (both WT and L55P) internalization as demonstrated by exposure to AF488-labeled human recombinant proteins. Foci of labeled proteins (green fluorescence) are visible in the cells. Nuclei were stained with DAPI (blue).
Studies have suggested that target cells affected by amyloidogenic TTR internalize and modify the variant protein in a process that may contribute to fibrillogenesis (Fleming et al., 2007, 2009). We examined whether ATTRL55P iPSC neuronal-lineage cells were capable of taking up fluorescently labeled WT or L55P recombinant human TTR protein. As demonstrated in Figure 4E, cells incubated with either labeled WT or L55P TTR contained foci of fluorescence in the cytoplasm, demonstrating that the cells were indeed capable of TTR internalization, similar to their in vivo counterparts. Interestingly, iPSC-derived cardiac cells did not demonstrate this property (data not shown).
Patient-Specific Recapitulation of Target Tissue Damage In Vitro
Having successfully derived the tissue types involved in ATTRL55P from patient-specific iPSCs, we then assessed the cellular and molecular effects of exposure to aberrant TTR in the directly differentiated target cells. To do this, serum-free conditioned media (hs) were harvested from control and ATTRL55P iPSC-derived hepatic cells for use in subsequent TTR exposure experiments in the neuronal- and cardiac-lineage target cells.
ATTRL55P iPSC-derived neuronal and cardiac-lineage cells were cultured with media containing either control or ATTRL55P hs, which was added to normal culture media in a 1:1 ratio. After an extended period of exposure (≥12 days), there was no noticeable difference in the morphology of the neuronal cells, and no visible protofilaments or aggregates were present in the cultures (Figure 5A). Similar observations were made for the cardiac cells after 6–10 days of exposure (Figure 5B). These results were in contrast to those seen when iPSC-derived neuronal cells and a neuroblastoma cell line (SY5Y) were exposed to human recombinant L55P TTR protein; clear aggregate formation was noted in these cultures, but not in cultures dosed with WT TTR protein (Figures S4A and S4B). The L55P protein aggregates stained positively for the ATTR amyloid binding chemical, thioflavin T (Figure S4C).
Figure 5.
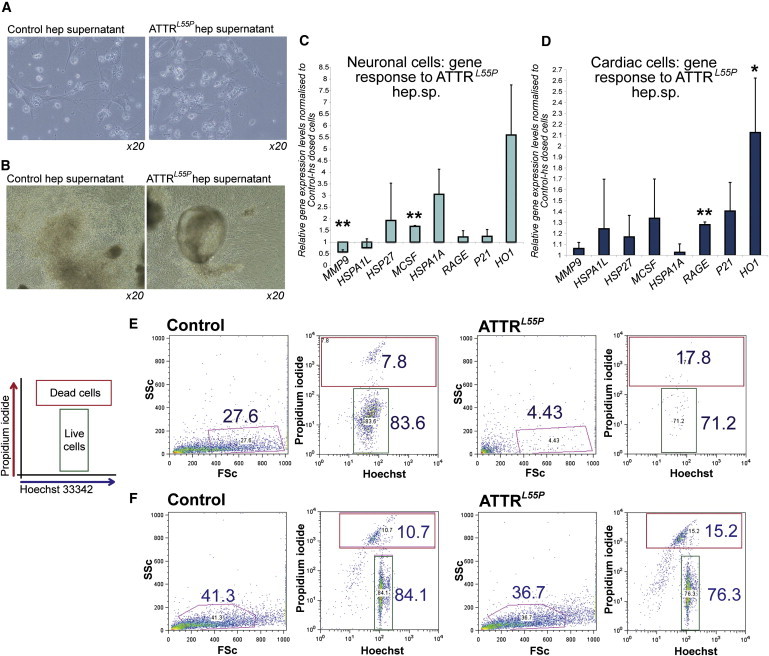
iPSC-Based Modeling of ATTR In Vitro; ATTRL55P hs Deleteriously Affects iPSC-Derived Target Cells
(A) Mature ATTRL55P iPSC neuronal-lineage cells were exposed to hs from either control iPSCs or ATTRL55P iPSC hepatic-lineage cells. No discernable differences in cell morphology and no visible protein aggregation were seen after 10 days. Images, 20× magnification.
(B) Cardiomyocytes derived from ATTRL55P iPSCs were exposed to hs for 6 days. There were no visible differences in the cultures containing control hs and ATTRL55P hs (20× magnification).
(C and D) Gene expression analysis of day 12-dosed ATTRL55P neuronal cells (C) and day 10-dosed ATTRL55P cardiac cells (D) for a panel of markers associated with stress response and protein folding. Error bars, SD. Asterisks (∗, ∗∗) denote t test significance at the 5% and 1% level, respectively. Sample size is n = 2 from independent experiments. ATTRL55P samples were normalized to controls for each experiment.
(E) ATTR neuronal cells cultured in media containing hs for 19 days were analyzed for the uptake of PI and Hoechst 33342. Cultures dosed with ATTRL55P hs contained fewer live cells (forward versus side scatter gate; Hoechst-positive gate, annotated) compared to control hs cultures.
(F) ATTRL55P cardiac cells dosed for 10 days with media containing ATTRL55P hs exhibited a decrease in cell viability compared to control cultures.
Although there was no observable difference in the morphology of cells exposed to either control or ATTRL55P hs, a number of genes previously reported to be associated with ATTR disease progression were upregulated in both iPSC-derived neuronal and cardiac cells exposed to ATTRL55P hs. In particular, the stress-response markers, macrophage colony stimulating factor (M-CSF) and heme oxygenase 1 (HO1), were upregulated (Figures 5C and 5D; asterisks denote statistically significant differences as compared to control samples, as determined by Student’s t test analysis). These genes were previously reported to be associated with ATTR in murine model systems and in neuronal biopsy samples of patients with ATTR (Sousa et al., 2001a, 2001b, 2005). The expression of protein folding genes was also examined in control and ATTRL55P hs-exposed cells based on the observation that higher levels of these genes were seen in ATTRL55P iPSC-derived hepatic-lineage cells (compared to disease-free controls) by microarray analysis (Figure S3). Of the interrogated genes, the hsp70 gene, hspA1A, was upregulated in neuronal-lineage cells exposed to ATTRL55P hs, but not to a significant level (Figures 5C and 5D).
In addition to these marked gene expression changes, Hoechst 33342 and propidium iodide (H-PI) staining revealed that both neuronal and cardiac cells exposed to ATTRL55P hs displayed statistically significant decreased cell survival, although no overt morphological changes were noted. Cell survival was ascertained by a decrease in the proportion of cells in the FSC versus SSC gate and an increase in PI-positive cells (representative flow cytometry results in Figures 5E and 5F with compiled results in Figures 6B and 6D). Similar results had previously been observed in preliminary experiments ascertaining the effect of recombinant human WT or L55P TTR on cell survival in iPSC-derived neuronal (Figure S4A) and cardiac (Figure S4B) cultures.
Figure 6.
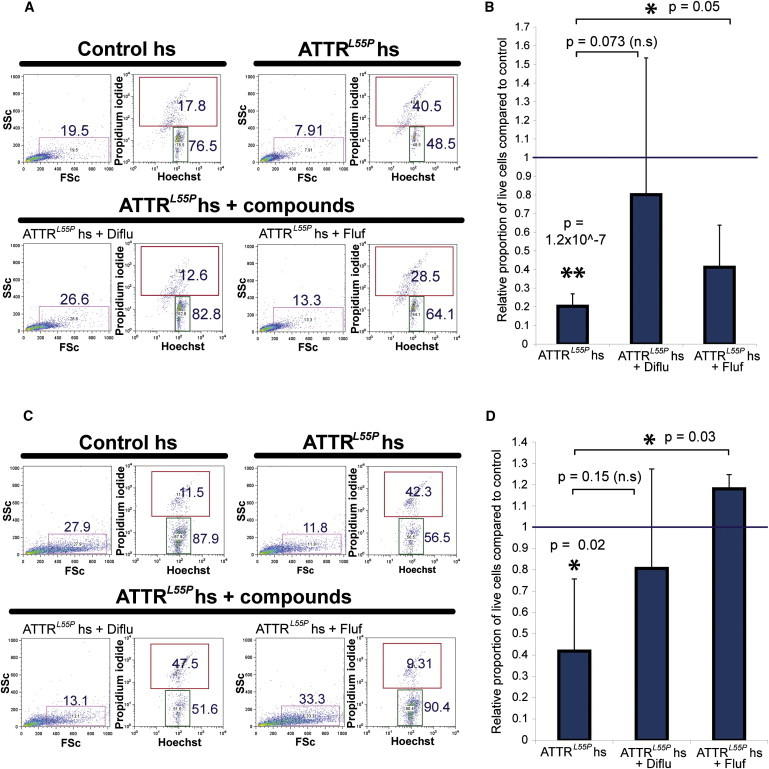
The Deleterious Effects of ATTRL55P hs Can Be Abrogated by Small Molecule TTR-Stabilizing Agents
(A) ATTRL55P iPSC-derived neuronal-lineage cells were exposed to control and ATTRL55P hs in the presence or absence of diflunisal (20 μM) and flufenamic acid (10 μM). Cells were harvested and examined for their H-PI positivity by flow cytometry. Increased apoptosis and cell death seen in ATTRL55P hs cultures were rescued in cultures also containing the small molecule compounds.
(B) Neuronal live-cell proportions in cultures dosed with ATTRL55P hs for 12 days, ± compounds, compared to control hs-dosed cells. Error bars, SD. Asterisks (∗, ∗∗) denote t test significance at the 5% and 1% level, respectively. p Values are denoted in the graph. Sample sizes (from independent experiments) are ATTRL55P n = 4, ATTRL55P + Diflu n = 3, and ATTRL55P + Fluf n = 3. n.s., not significant.
(C) ATTRL55P iPSC-derived cardiac cells were dosed for 6 days and analyzed for Hoechst-PI positivity in the same manner.
(D) Cardiac live-cell proportions in cultures dosed with ATTRL55P hs, ± compounds, compared to control hs-dosed cells. Error bars, SD. Asterisks (∗, ∗∗) denote t test significance at the 5% and 1% level, respectively. p Values are denoted in the graph. Sample sizes (from independent experiments) are ATTRL55P n = 3, ATTRL55P + Diflu n = 3, and ATTRL55P + Fluf n = 3.
Thus, iPSC-derived hs containing amyloidogenic L55P TTR had a deleterious effect on cell survival compared to supernatant from control-iPSC hepatocytes. Through the examination of previously identified ATTR-associated genes and genes identified through our iPSC-derived hepatic-lineage microarray screen, we were able to determine that ATTRL55P hs exposure upregulated several ATTR disease-associated genes in target cells, in a manner that is similar to some reported observations in both in vivo and in vitro systems.
Small Molecules Can Ameliorate the Damaging Effects of ATTRL55P Exposure in an iPSC-Based, Multisystem Model of ATTR
In vitro and clinical data demonstrate that small molecules complexing with TTR at the thyroxine binding site can stabilize the native tetramer, preventing amyloid fibril formation, and inhibiting disease progression (Coelho et al., 2012; Miller et al., 2004; Peterson et al., 1998; Sekijima et al., 2006; Tojo et al., 2006). To determine the effect of TTR stabilizers in our iPSC-derived multiorgan model of ATTR, we examined the effect of identified TTR-responsive small molecules on ATTRL55P hs-induced apoptosis of iPSC-derived cardiac and neuronal-lineage cells.
To ascertain whether diflunisal and flufenamic acid, two thyroxine-mimetic small molecule TTR stabilizers, negate the deleterious effects of ATTRL55P hs, these compounds were added to ATTRL55P hs for dosing experiments with neuronal and cardiac cells at final concentrations of 20 and 10 μM, respectively. As demonstrated in Figure 6, the addition of the small molecules to ATTRL55P hs had a protective effect on both neuronal and cardiac cell survival compared to cells exposed to ATTRL55P hs alone, as judged by H-PI stainings of dosed cells. Although exposure of both neuronal and cardiac cells to ATTRL55P hs led to a statistically significant decrease in cell survival compared to control hs samples (Figures 6B and 6D), addition of small molecule compounds resulted in increased cell survival (close to, but not significantly different compared to ATTRL55P hs for ATTRL55P+diflunisal; significant for ATTRL55P+flufenamic acid). These drugs were also capable of preventing recombinant human L55P TTR protein from forming microscopically visible protofilaments in cultures of patient iPSC-derived neuronal cells and SY5Y neuroblastoma cells, as assessed by thioflavin T binding (Figures S4C–S4E). These experiments demonstrate that an iPSC-based, multisystem, in vitro model can serve as a valuable platform for the testing of novel therapeutics in the genetic context of the patient.
Discussion
Human iPSC-based disease modeling provides an opportunity to recapitulate a disease phenotype in the context of a patient’s exact genetic background. In the case of ATTR, patients with the same genetic mutations have been shown to exhibit variable patterns of disease progression (Planté-Bordeneuve and Said, 2011), highlighting the involvement of genetic modifiers of disease.
ATTR is an unusual hereditary disease that manifests clinically in adulthood with no reproductive pressure against transmission of the disease. Until recently, diagnostics were not available, and carriers could not be identified before clinical disease onset, which typically occurs in the fourth decade or later. In patients affected with the TTRL55P mutation, the age of disease onset is earlier and the clinical outcome more severe (Jacobson et al., 1992), characteristics that reflect greater instability of the mutant TTR tetramer (Lashuel et al., 1998, 1999). Our work documents the successful modeling of ATTR in vitro with the use of iPSC technology, demonstrating that it is possible to model a long-term, complex, multisystem disease in a relatively short space of time, using lineage-specified cells derived from patient-specific stem cells.
iPSCs have been utilized to model genetic diseases in a single lineage in which the variant protein functions, for example, Fanconi anemia (Raya et al., 2009), alpha 1 antitrypsin deficiency (Rashid et al., 2010), and amyotrophic lateral sclerosis (Dimos et al., 2008). Our work demonstrates that iPSC technology can be used to model a multisystem disease in which the effects of variant protein produced by one organ manifests disease in other target tissues.
The mechanism by which variant TTR adversely affects cells is still being elucidated. It is postulated that abundant extracellular amyloid fibril deposits seen in patient biopsies have a disruptive effect on tissue architecture and organ function, but recent evidence suggests that TTR monomers and/or oligomeric prefibrillar protein structures are toxic to cells (Reixach et al., 2004; Sousa et al., 2001a, 2002). We have recapitulated a part of this process in a patient-specific fashion in vitro. In our iPSC-based disease model, the lack of visible protofilament/aggregate material in ATTRL55P hs cultures suggests that the negative effect of ATTRL55P hs is likely to be due to monomer or oligomer-induced cytotoxicity, reported by others to be a mechanism of cellular damage (Reixach et al., 2004; Sousa et al., 2002).
Although extended exposure to ATTRL55P hs increased apoptosis in both neuronal and cardiac cells, we were interested in examining some of the molecular changes in the exposed target-lineage cells. Based on the previously discussed microarray data obtained through the comparison of hepatic cells derived from normal or ATTRL55P-specific iPSCs (Figure S2), we focused on expression of hsp70 family genes that are involved in protein folding and upregulated in response to cellular stressors. These genes have been studied in a range of protein-misfolding disorders, including Alzheimer’s disease (Hoshino et al., 2011). Intriguingly, an ATTR mouse model with impaired heat shock response exhibits a comparatively accelerated amyloid deposition pattern, implicating the importance of these genes for the regulation of the process of ATTR amyloidogenesis (Santos et al., 2010). We also examined the expression of markers previously reported to be associated with ATTR in other model systems and patient tissues, such as M-CSF, p21, and antioxidant enzyme HO1 (Sousa et al., 2001a, 2001b, 2005), and the receptor for advanced glycation end products (RAGE), a marker that has been postulated as a mechanism through which variant TTR targets certain tissues for amyloid deposition and cytotoxic effects (Sousa et al., 2001b). Through the examination of these markers in the exposed target-lineage cells, we were able to determine a pattern of gene expression upregulation in ATTRL55P hs-exposed cells that was accordant with previous observations. This iPSC-based, in vitro system may therefore prove to be a valuable model system in which to further dissect the molecular mechanisms through which aberrant TTR affects ATTR disease target cells and tissues. Moreover, it is interesting that in our system, iPSC-derived ATTRL55P hepatic cells exhibit gene expression differences compared to control cells because the livers of patients with ATTR are thought to be comparatively normal despite being the site of aberrant TTR production. Recent findings from a murine SSA model also suggest that the liver has a role to play in the degree of amyloidogenesis in target tissues, through a novel long-range chaperoning effect (Buxbaum et al., 2012). The iPSC-based system may prove to be an ideal platform in which to further investigate the role of the liver in ATTR amyloidogenesis.
The ability of diflunisal and flufenamic acid, known TTR tetramer stabilizers, to prevent iPSC-derived target cell death in the presence of ATTRL55P hs validates this system as a potential platform in which to test novel therapeutic agents. Different approaches to therapeutically target the cause and symptoms of ATTR have been tested in in vitro and in vivo systems, with several progressing onto human clinical trials. Examples include the reduction of serum level TTR through the use of antisense technology (Ackermann et al., 2012), and the design of small molecule compounds with the aim of either kinetically stabilizing aberrant TTR heterotetramers (Alhamadsheh et al., 2011; Connelly et al., 2010) (thus slowing the rate-limiting step in the process of amyloidogenesis) or disruption of existing amyloid fibrils and prefibrillar aggregates (Cardoso et al., 2010; Ferreira et al., 2012). Patients with ATTR form amyloidogenic protein based on 1 of over 100 point mutations in the TTR gene (Connors et al., 2003). The binding affinity of small molecule inhibitors to different variant TTRs varies significantly, predicting mutation-unique responses to specific TTR stabilizers (Tojo et al., 2006), an indicator that a one-size-fits-all approach to ATTR treatment may not be feasible. Generation of differentiated, iPSC-derived, ATTR disease-specific cells permits variant TTR-specific screening of potential therapeutic molecules, overcoming issues of incomplete mutation representation in clinical trials of this rare orphan disease. These cells also provide a model system in which to study genetic modifiers of TTR secretion, and cytoprotective agents that inhibit toxic signaling pathways in the target cells. Based on the fact that these disease-specific cells are pluripotent and can be stored and expanded indefinitely, they will serve as a permanent repository for researchers around the world as a valuable tool for investigating potential new targets for drugs modulating protein misfolding and aggregation.
Experimental Procedures
STEMCCA Lentivirus Production, iPSC Generation and Maintenance, and Teratoma Formation Assay
hSTEMCCA virus production and concentration (Sommer et al., 2009, 2010) and iPSC generation, maintenance, and validation were carried out as previously described by Somers et al. (2010). For this study, fibroblasts from a 27-year-old female patient heterozygous for the L55P TTR mutation were used to create three clonal disease-specific lines. The three control iPSC lines used in this study were obtained from the Boston University and Boston Medical Center’s Center for Regenerative Medicine (CReM) comprehensive iPSC bank (http://www.bumc.bu.edu/stemcells/crem-ips-cell-bank/) and were matched with the disease-specific lines for age, gender, and ethnicity.
Generation of Hepatic-Lineage Cells from iPSCs
Differentiation of iPSCs toward the hepatic lineage was carried out as described by Hay et al. (2008) and Sullivan et al. (2010). For the collection of media supernatants from hepatic-lineage cells, the end-stage differentiated cells were washed with PBS and incubated in serum-free media (L-15 [Leibovitz] medium, 10% KnockOut Serum Replacement [KSR], and Penicillin-Streptomycin-Glutamine, which were all from Invitrogen). The media were harvested and replaced every 24 hr up to 3 days. See the Supplemental Experimental Procedures for details regarding microarray analysis of iPSC-derived hepatic-lineage cells. For the FACS analysis of definitive endoderm markers, trypsin-dissociated cells were stained with human CXCR4-biotin/strepavidin-FITC and cKit-PE (eBiosciences) antibodies and analyzed on an LSRII (BD Biosciences) using FACsDiva software.
Generation of Cardiomyocytes from iPSCs
Adherent cardiac cell cultures were specified based upon a previously described protocol by Kattman et al. (2011) and Yang et al. (2008). For the EB differentiation protocol, confluent iPSC colonies were treated with 1 mg/ml collagenase IV (Invitrogen), then washed from the wells using a p1000 pipette tip. Cell clusters were transferred to methylcellulose dishes in DMEM, 20% FBS (Hyclone), 1% NEAA (Invitrogen), 100 mM 2-mercaptethanol for 7 days. The resultant EBs were transferred to gelatin-coated tissue culture plates. Media were changed every 3 days.
For intracellular staining of cEB cells for sarcomeric anti-actinin, trypsin-dissociated 4% paraformaldehyde (PFA):PBS-fixed cells were washed and stained in PBS/5% FBS/0.5% saponin. The primary antibody was sarcomeric anti-actinin (Sigma-Aldrich), the isotype control was mouse IgG1 κ, and the secondary antibody was FITC anti-mouse IgG (both from BD PharMingen). Analysis was carried out on an LSRII flow cytometer.
Generation of Neuronal-Lineage Cells from ATTRL55P iPSCs
Neuronal-lineage cells were generated as previously described by Chambers et al. (2009) and Hu et al. (2009). For immunofluorescence visualization of TUJ1, MAP2, and ChAT, cells were grown on 18 mm glass coverslips coated with 5 μg/ml fibronectin and 10 μg/ml laminin (for TUJ1, MAP2 stainings) or Matrigel (for ChAT stainings). Cells were fixed with 4% PFA in PBS and permeabilized and blocked with 5% BSA in PBS with 0.1% Triton X-100. Primary and secondary antibody stainings were carried out in PBS/0.005% Triton X-100. Primary antibodies were mouse anti-human Tuj1, mouse anti-human MAP2 (Sigma-Aldrich), and rabbit anti-human ChAT (Millipore). Secondary antibodies were donkey anti-mouse 594 nm and donkey anti-rabbit 488 nm (Jackson ImmunoResearch). Cell nuclei were stained with DAPI (Molecular Probes) after antibody incubations and final washes.
PAS Assays
PAS assays were carried out on adherent cell cultures in 6-well plates, based on manufacturer recommendations (Invitrogen). Cells were visualized and photographed using a Nikon Eclipse TS100 microscope and SPOT software (SPOT Imaging Solutions).
Immunoblot Analysis
A total of 25 ng of purified human serum TTR (Sigma-Aldrich), hepatic-iPSC supernatant, and KSR-based media control samples were analyzed by SDS/PAGE using a 12% homogeneous gel under reducing conditions. Rabbit anti-human TTR (DAKO) and HRP-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology) were used for TTR detection. The immunoblot was developed using the ECL Plus Western Blotting Detection Reagent (GE Healthcare).
Mass Spectrometry
TTR protein was purified from culture supernatants by immunoprecipitation and characterized by electrospray ionization (ESI) mass spectrophotometry (Lim et al., 2002).
Thioflavin T Assay
Cell and media cytospins were fixed in 4% PFA for 10 min, washed with distilled water, and stained with 1% thioflavin T solution for 5 min. The cells were then differentiated in 70% ethanol and rinsed twice with distilled water before being mounted for microscopy under polarized light.
RNA Extraction, and Semiquantitative and Quantitative PCR
The RNeasy Micro Kit (QIAGEN), DNA-free Kit (Ambion), and High Capacity cDNA Reverse Transcription Kit (Applied Biosciences) were used for the extraction and purification of RNA and cDNA synthesis, all according to manufacturer instructions. Quantitative PCR was conducted using SYBR Green Real-Time PCR Master Mix (Applied Biosystems) and StepOne Real-Time PCR machines (Applied Biosystems). See Table S1 for a list of primers. Where appropriate, Student’s t test statistical analyses were carried out to determine if there were significant differences between sample groups.
Cell Viability Assays
For dosing experiments involving hs, filtered supernatants collected from iPSC-hepatic cultures were diluted 1:1 with neuronal/cardiac growth media as appropriate before addition to cell cultures. For experiments involving small molecules, diflunisal and flufenamic acid were added to supernatants and incubated at 4°C overnight before addition to cell cultures (final concentrations, 20 and 10 μM, respectively). Dosed cell cultures were trypsin dissociated and washed with PBS-5% FBS before incubation with 8.8 ng/ml Hoechst 33342 (Sigma-Aldrich) at 37°C for 15 min. After washes, the cells were incubated with PI (Invitrogen) and immediately analyzed by flow cytometry (LSRII). For statistical analyses, a value for cell survival for each sample was calculated from the proportion of cells in the FSc versus SSc gate and the proportion of Hoechst-positive, PI-negative cells within this population. Samples were normalized to the control sample for each experiment. Student’s t tests were conducted to compare ATTRL55P to control, ATTRL55P plus diflunisal, and ATTRL55P plus flufenamic acid groups. See the Supplemental Experimental Procedures for details of experiments involving recombinant TTR protein.
Acknowledgments
Funding was provided by the Amyloid Foundation (to G.J.M. and A.L.), grant NIH R01AG031804 (to L.H.C. and C.M.K.), grants R01 NS051306 and FDA FD-R-002532 (to J.L.B.), the Young Family Amyloid Research Fund (to L.H.C., C.M.K., and J.L.B.), and the Boston University Clinical and Translational Science Institute (CTSI) (to G.J.M.).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Supplemental Information
References
- Ackermann E.J., Guo S., Booten S., Alvarado L., Benson M., Hughes S., Monia B.P. Clinical development of an antisense therapy for the treatment of transthyretin-associated polyneuropathy. Amyloid. 2012;19(Suppl 1):43–44. doi: 10.3109/13506129.2012.673140. [DOI] [PubMed] [Google Scholar]
- Alhamadsheh M.M., Connelly S., Cho A., Reixach N., Powers E.T., Pan D.W., Wilson I.A., Kelly J.W., Graef I.A. Potent kinetic stabilizers that prevent transthyretin-mediated cardiomyocyte proteotoxicity. Sci. Transl. Med. 2011;3:97ra81. doi: 10.1126/scitranslmed.3002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando Y., Nakamura M., Araki S. Transthyretin-related familial amyloidotic polyneuropathy. Arch. Neurol. 2005;62:1057–1062. doi: 10.1001/archneur.62.7.1057. [DOI] [PubMed] [Google Scholar]
- Araki S., Yi S., Murakami T., Watanabe S., Ikegawa S., Takahashi K., Yamarnura K. Systemic amyloidosis in transgenic mice carrying the human mutant transthyretin (Met 30) gene. Pathological and immunohistochemical similarity to human familial amyloidotic polyneuropathy, type I. Mol. Neurobiol. 1994;8:15–23. doi: 10.1007/BF02778004. [DOI] [PubMed] [Google Scholar]
- Benson M.D., Teague S.D., Kovacs R., Feigenbaum H., Jung J., Kincaid J.C. Rate of progression of transthyretin amyloidosis. Am. J. Cardiol. 2011;108:285–289. doi: 10.1016/j.amjcard.2011.03.040. [DOI] [PubMed] [Google Scholar]
- Berk J.L., Suhr O.B., Sekijima Y., Yamashita T., Heneghan M., Zeldenrust S.R., Ando Y., Ikeda S., Gorevic P., Merlini G., Familial Amyloidosis Consortium The Diflunisal Trial: study accrual and drug tolerance. Amyloid. 2012;19(Suppl 1):37–38. doi: 10.3109/13506129.2012.678509. [DOI] [PubMed] [Google Scholar]
- Buxbaum J.N., Tagoe C., Gallo G., Walker J.R., Kurian S., Salomon D.R. Why are some amyloidoses systemic? Does hepatic “chaperoning at a distance” prevent cardiac deposition in a transgenic model of human senile systemic (transthyretin) amyloidosis? FASEB J. 2012;26:2283–2293. doi: 10.1096/fj.11-189571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso I., Martins D., Ribeiro T., Merlini G., Saraiva M.J. Synergy of combined doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: studies in FAP mouse models. J. Transl. Med. 2010;8:74. doi: 10.1186/1479-5876-8-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers S.M., Fasano C.A., Papapetrou E.P., Tomishima M., Sadelain M., Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho T., Maia L.F., Martins da Silva A., Waddington Cruz M., Planté-Bordeneuve V., Lozeron P., Suhr O.B., Campistol J.M., Conceição I.M., Schmidt H.H. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79:785–792. doi: 10.1212/WNL.0b013e3182661eb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly S., Choi S., Johnson S.M., Kelly J.W., Wilson I.A. Structure-based design of kinetic stabilizers that ameliorate the transthyretin amyloidoses. Curr. Opin. Struct. Biol. 2010;20:54–62. doi: 10.1016/j.sbi.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors L.H., Lim A., Prokaeva T., Roskens V.A., Costello C.E. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid. 2003;10:160–184. doi: 10.3109/13506120308998998. [DOI] [PubMed] [Google Scholar]
- Connors L.H., Prokaeva T., Lim A., Théberge R., Falk R.H., Doros G., Berg A., Costello C.E., O’Hara C., Seldin D.C., Skinner M. Cardiac amyloidosis in African Americans: comparison of clinical and laboratory features of transthyretin V122I amyloidosis and immunoglobulin light chain amyloidosis. Am. Heart J. 2009;158:607–614. doi: 10.1016/j.ahj.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Dimos J.T., Rodolfa K.T., Niakan K.K., Weisenthal L.M., Mitsumoto H., Chung W., Croft G.F., Saphier G., Leibel R., Goland R. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- Falk R.H., Comenzo R.L., Skinner M. The systemic amyloidoses. N. Engl. J. Med. 1997;337:898–909. doi: 10.1056/NEJM199709253371306. [DOI] [PubMed] [Google Scholar]
- Ferreira N., Saraiva M.J., Almeida M.R. Epigallocatechin-3-gallate as a potential therapeutic drug for TTR-related amyloidosis: “in vivo” evidence from FAP mice models. PLoS One. 2012;7:e29933. doi: 10.1371/journal.pone.0029933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming C.E., Saraiva M.J., Sousa M.M. Transthyretin enhances nerve regeneration. J. Neurochem. 2007;103:831–839. doi: 10.1111/j.1471-4159.2007.04828.x. [DOI] [PubMed] [Google Scholar]
- Fleming C.E., Mar F.M., Franquinho F., Saraiva M.J., Sousa M.M. Transthyretin internalization by sensory neurons is megalin mediated and necessary for its neuritogenic activity. J. Neurosci. 2009;29:3220–3232. doi: 10.1523/JNEUROSCI.6012-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarström P., Schneider F., Kelly J.W. Trans-suppression of misfolding in an amyloid disease. Science. 2001;293:2459–2462. doi: 10.1126/science.1062245. [DOI] [PubMed] [Google Scholar]
- Hay D.C., Fletcher J., Payne C., Terrace J.D., Gallagher R.C.J., Snoeys J., Black J.R., Wojtacha D., Samuel K., Hannoun Z. Highly efficient differentiation of hESCs to functional hepatic endoderm requires ActivinA and Wnt3a signaling. Proc. Natl. Acad. Sci. USA. 2008;105:12301–12306. doi: 10.1073/pnas.0806522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino T., Murao N., Namba T., Takehara M., Adachi H., Katsuno M., Sobue G., Matsushima T., Suzuki T., Mizushima T. Suppression of Alzheimer’s disease-related phenotypes by expression of heat shock protein 70 in mice. J. Neurosci. 2011;31:5225–5234. doi: 10.1523/JNEUROSCI.5478-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B.Y., Du Z.W., Li X.J., Ayala M., Zhang S.C. Human oligodendrocytes from embryonic stem cells: conserved SHH signaling networks and divergent FGF effects. Development. 2009;136:1443–1452. doi: 10.1242/dev.029447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson D.R., McFarlin D.E., Kane I., Buxbaum J.N. Transthyretin Pro55, a variant associated with early-onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement. Hum. Genet. 1992;89:353–356. doi: 10.1007/BF00220559. [DOI] [PubMed] [Google Scholar]
- Jacobson D.R., Pastore R.D., Yaghoubian R., Kane I., Gallo G., Buck F.S., Buxbaum J.N. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N. Engl. J. Med. 1997;336:466–473. doi: 10.1056/NEJM199702133360703. [DOI] [PubMed] [Google Scholar]
- Kattman S.J., Witty A.D., Gagliardi M., Dubois N.C., Niapour M., Hotta A., Ellis J., Keller G. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell. 2011;8:228–240. doi: 10.1016/j.stem.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Kohno K., Palha J.A., Miyakawa K., Saraiva M.J., Ito S., Mabuchi T., Blaner W.S., Iijima H., Tsukahara S., Episkopou V. Analysis of amyloid deposition in a transgenic mouse model of homozygous familial amyloidotic polyneuropathy. Am. J. Pathol. 1997;150:1497–1508. [PMC free article] [PubMed] [Google Scholar]
- Lashuel H.A., Lai Z., Kelly J.W. Characterization of the transthyretin acid denaturation pathways by analytical ultracentrifugation: implications for wild-type, V30M, and L55P amyloid fibril formation. Biochemistry. 1998;37:17851–17864. doi: 10.1021/bi981876+. [DOI] [PubMed] [Google Scholar]
- Lashuel H.A., Wurth C., Woo L., Kelly J.W. The most pathogenic transthyretin variant, L55P, forms amyloid fibrils under acidic conditions and protofilaments under physiological conditions. Biochemistry. 1999;38:13560–13573. doi: 10.1021/bi991021c. [DOI] [PubMed] [Google Scholar]
- Lim A., Prokaeva T., McComb M.E., O’Connor P.B., Théberge R., Connors L.H., Skinner M., Costello C.E. Characterization of transthyretin variants in familial transthyretin amyloidosis by mass spectrometric peptide mapping and DNA sequence analysis. Anal. Chem. 2002;74:741–751. doi: 10.1021/ac010780+. [DOI] [PubMed] [Google Scholar]
- Miller S.R., Sekijima Y., Kelly J.W. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab. Invest. 2004;84:545–552. doi: 10.1038/labinvest.3700059. [DOI] [PubMed] [Google Scholar]
- Monteiro E., Freire A., Barroso E. Familial amyloid polyneuropathy and liver transplantation. J. Hepatol. 2004;41:188–194. doi: 10.1016/j.jhep.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Peterson S.A., Klabunde T., Lashuel H.A., Purkey H., Sacchettini J.C., Kelly J.W. Inhibiting transthyretin conformational changes that lead to amyloid fibril formation. Proc. Natl. Acad. Sci. USA. 1998;95:12956–12960. doi: 10.1073/pnas.95.22.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planté-Bordeneuve V., Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10:1086–1097. doi: 10.1016/S1474-4422(11)70246-0. [DOI] [PubMed] [Google Scholar]
- Rashid S.T., Corbineau S., Hannan N., Marciniak S.J., Miranda E., Alexander G., Huang-Doran I., Griffin J., Ahrlund-Richter L., Skepper J. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J. Clin. Invest. 2010;120:3127–3136. doi: 10.1172/JCI43122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raya A., Rodríguez-Pizà I., Guenechea G., Vassena R., Navarro S., Barrero M.J., Consiglio A., Castellà M., Río P., Sleep E. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature. 2009;460:53–59. doi: 10.1038/nature08129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reixach N., Deechongkit S., Jiang X., Kelly J.W., Buxbaum J.N. Tissue damage in the amyloidoses: transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc. Natl. Acad. Sci. USA. 2004;101:2817–2822. doi: 10.1073/pnas.0400062101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowczenio, D., and Wechalekar, A. (2010). Mutations in hereditary amyloidosis. http://amyloidosismutations.com/attr.html. [DOI] [PubMed]
- Santos S.D., Fernandes R., Saraiva M.J. The heat shock response modulates transthyretin deposition in the peripheral and autonomic nervous systems. Neurobiol. Aging. 2010;31:280–289. doi: 10.1016/j.neurobiolaging.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Schneider F., Hammarström P., Kelly J.W. Transthyretin slowly exchanges subunits under physiological conditions: a convenient chromatographic method to study subunit exchange in oligomeric proteins. Protein Sci. 2001;10:1606–1613. doi: 10.1110/ps.8901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekijima Y., Dendle M.A., Kelly J.W. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13:236–249. doi: 10.1080/13506120600960882. [DOI] [PubMed] [Google Scholar]
- Somers A., Jean J.-C., Sommer C.A., Omari A., Ford C.C., Mills J.A., Ying L., Sommer A.G., Jean J.M., Smith B.W. Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette. Stem Cells. 2010;28:1728–1740. doi: 10.1002/stem.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C.A., Stadtfeld M., Murphy G.J., Hochedlinger K., Kotton D.N., Mostoslavsky G. Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem Cells. 2009;27:543–549. doi: 10.1634/stemcells.2008-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C.A., Sommer A.G., Longmire T.A., Christodoulou C., Thomas D.D., Gostissa M., Alt F.W., Murphy G.J., Kotton D.N., Mostoslavsky G. Excision of reprogramming transgenes improves the differentiation potential of iPS cells generated with a single excisable vector. Stem Cells. 2010;28:64–74. doi: 10.1002/stem.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa M.M., Cardoso I., Fernandes R., Guimarães A., Saraiva M.J. Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: evidence for toxicity of nonfibrillar aggregates. Am. J. Pathol. 2001;159:1993–2000. doi: 10.1016/s0002-9440(10)63050-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa M.M., Du Yan S., Fernandes R., Guimaraes A., Stern D., Saraiva M.J. Familial amyloid polyneuropathy: receptor for advanced glycation end products-dependent triggering of neuronal inflammatory and apoptotic pathways. J. Neurosci. 2001;21:7576–7586. doi: 10.1523/JNEUROSCI.21-19-07576.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa M.M., Fernandes R., Palha J.A., Taboada A., Vieira P., Saraiva M.J. Evidence for early cytotoxic aggregates in transgenic mice for human transthyretin Leu55Pro. Am. J. Pathol. 2002;161:1935–1948. doi: 10.1016/S0002-9440(10)64469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa M.M., do Amaral J.B., Guimarães A., Saraiva M.J. Up-regulation of the extracellular matrix remodeling genes, biglycan, neutrophil gelatinase-associated lipocalin, and matrix metalloproteinase-9 in familial amyloid polyneuropathy. FASEB J. 2005;19:124–126. doi: 10.1096/fj.04-2022fje. [DOI] [PubMed] [Google Scholar]
- Sullivan G.J., Hay D.C., Park I.-H., Fletcher J., Hannoun Z., Payne C.M., Dalgetty D., Black J.R., Ross J.A., Samuel K. Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology. 2010;51:329–335. doi: 10.1002/hep.23335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagoe C.E., Reixach N., Friske L., Mustra D., French D., Gallo G., Buxbaum J.N. In vivo stabilization of mutant human transthyretin in transgenic mice. Amyloid. 2007;14:227–236. doi: 10.1080/13506120701464396. [DOI] [PubMed] [Google Scholar]
- Tojo K., Sekijima Y., Kelly J.W., Ikeda S. Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci. Res. 2006;56:441–449. doi: 10.1016/j.neures.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Yang L., Soonpaa M.H., Adler E.D., Roepke T.K., Kattman S.J., Kennedy M., Henckaerts E., Bonham K., Abbott G.W., Linden R.M. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. 2008;453:524–528. doi: 10.1038/nature06894. [DOI] [PubMed] [Google Scholar]
- Yasunaga M., Tada S., Torikai-Nishikawa S., Nakano Y., Okada M., Jakt L.M., Nishikawa S., Chiba T., Era T., Nishikawa S. Induction and monitoring of definitive and visceral endoderm differentiation of mouse ES cells. Nat. Biotechnol. 2005;23:1542–1550. doi: 10.1038/nbt1167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.