

Abstract
Cellular quiescence is a reversible cell cycle arrest that is poised to re-enter the cell cycle in response to a combination of cell-intrinsic factors and environmental cues. In hematopoietic stem cells, a coordinated balance between quiescence and differentiating proliferation ensures longevity and prevents both genetic damage and stem cell exhaustion. However, little is known about how all these processes are integrated at the molecular level. We will briefly review the environmental and intrinsic control of stem cell quiescence and discuss a new model that involves a protein-to-protein interaction between G0S2 and the phospho-nucleoprotein nucleolin in the cytosol.
Keywords: G0S2, hematopoietic stem cells, quiescence
Introduction
Blood cells are continuously generated by the differentiation of hematopoietic stem cells (HSCs) in the bone marrow. Multipotency restricted to the hematopoietic system has been exploited for cell and gene therapy and the regeneration of the hematopoietic system in cancer patients. HSCs can enter the proliferation program or a non-proliferative state that can be reversible (quiescence) or irreversible (senescence); therefore, in the balance between active cell division and dormancy HSCs should avoid entering a terminal senescence. More than 80% of HSCs remain quiescent in specific regions of bone marrow (stem cell niches), ensuring stemness and longevity over the lifetime of an individual. Cell division imposes diverse fates in HSCs: differentiation into lymphoid or myeloid lineages, the generation of daughter cells with the same stem cell properties of the parental cell (self-renewal), the mobilization to peripheral tissues, or death. Multiple cell divisions are likely required for lineage differentiation, and thus the classical stepwise model of hematopoiesis needs to be viewed as a continuous process of differentiation, with the loss of stem cell-associated properties, such as quiescence, self-renewal, and multipotency (Fig. 1). Some blood cells (e.g., lymphocytes) retain, at some level, the properties of quiescence and self-renewal.
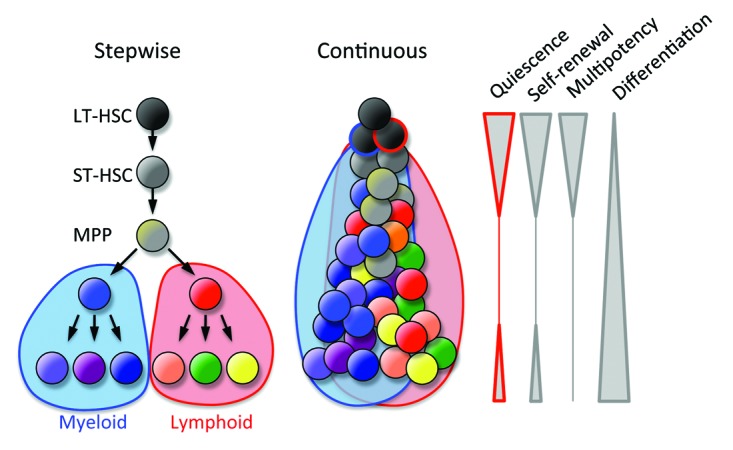
Figure 1. Models of bone marrow hematopoiesis. The current model of blood formation is a continuous process of the differentiation and progressive loss of stem cell features (quiescence, self-renewal, and multipotency).
The advent of genetically engineered mouse models and the identification of HSCs by multi-parameter flow cytometry have defined the ontogeny of bone marrow hematopoiesis and jump-started the identification of the genes involved in stem cell maintenance. Although the prospective identification of HSCs by the expression of cell surface markers is a powerful approach, the gold standard to define HSCs is their capacity to rescue an ablated host and reconstitute lymphoid and myeloid cells over a long period of time. Most immunophenotypic HSCs (Lin− Sca-1+ c-kit+ CD150+ CD48− or Lin− Sca-1+ c-kit+ CD34− Flt3−) are in the G0 phase of the cell cycle (quiescence), which is defined as cells with a 2n DNA content devoid of proliferation markers (e.g., Pyronin Y and Ki67). The bone marrow transplantation of purified cell populations has revealed that quiescent HSCs exhibit increased homing in competitive transplants.
In this review, we will summarize the environmental and cell-intrinsic regulators of quiescence and discuss in more detail a novel model in the control of HSC quiescence by G0S2-mediated sequestration of nucleolin in the cytosol.
Stem Cell-Extrinsic (SCE) Factors
In contrast to senescence, which is an irreversible cell cycle arrest, quiescent cells can re-enter the cell cycle, because they conserve their capacity to proliferate. As nutrients increase cellular growth and proliferation, quiescence was considered for many years to be a default state in the absence of growth factors. However, the current paradigm indicates that quiescence is an active process regulated by stem cell-intrinsic (SCI) and -extrinsic (SCE) factors (Fig. 2A). Some groups have used cellular approaches to identify HSC populations expressing the receptor of interest and study their role on HSC proliferation, and other groups have employed genetic approaches to delete the gene of interest (Table 1). Stem cell niches are specialized microenvironments in the bone marrow that provide signals to HSCs.1-3 The osteoblastic niche, localized in the endosteal region of the bone, provides a myriad of signals aimed at preserving quiescence and stemness of HSCs in contact with osteoblasts.4-6 Most relevant pro-quiescence signals from the stem cell niche are summarized in Table 1 and the references therein: angiopoietin (Ang-1), thrombopoietin (THPO), Wnt, stem cell factor (SCF), tumor growth factor β (TGFβ), stromal derived factor 1 (SDF-1), and osteopontin (OPN). HSCs read these signals via the corresponding receptors: Tie2, Mpl, frizzled, c-kit, TGFR, CXCR4, and integrin receptors. As a result of these cell-to-cell communications, HSCs become more resistant to proliferating signals, deepen their dormancy state, and undergo self-renewing divisions to maintain the HSC pool. In addition to the osteoblastic niche, the sinusoidal vascular niche is another stem cell microenvironment where endothelial cells secrete SCF to support the HSCs in this niche.7,8
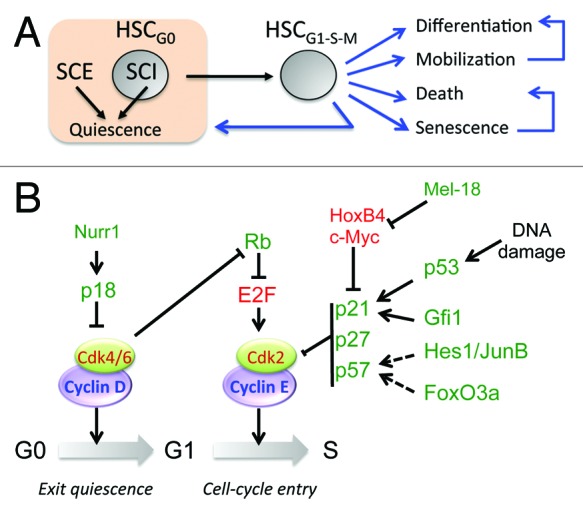
Figure 2. Control of HSC quiescence. (A) HSC quiescence is controlled by stem cell extrinsic (SCE) and intrinsic (SCI) factors. Proliferating HSCs face multiple fates: differentiation, mobilization, death, senescence, and re-entry to quiescence. (B) Regulation of the exit from quiescence and entry into cell division by cell cycle regulators (activating in green and inhibitory in red).
Table 1. Stem cell-extrinsic (SCE) regulation of HSC quiescence.
| Ligand | Receptor | Mouse model | Effect on quiescence | References |
|---|---|---|---|---|
| Ang-1 |
Tie2 |
Immunophenotypic identification and functional characterization of Tie2-positive HSCs |
Positive |
61 |
| THPO |
Mpl |
Immunophenotypic identification of Mpl-positive HSCs. Blocking signaling with anti-Mpl antibodies |
Positive |
62 |
| Wnt |
Frizzled |
Transgenic mice expressing the Wnt inhibitor Dkk1 driven by an osteoblast-specific promoter |
Positive |
63 |
| Cross of mice with the insertion of the Myc-tagged stable form of human β-catenin into the R26 locus with Mx1-Cre mice |
Negative |
64 |
||
| TGFβ |
TGFBR |
cKO TGFBRI (Mx1-Cre) |
No effect at steady-state |
65 |
| TGFβ levels during myelosuppression and blockade of TGFβ during recovery |
Reset quiescence after cytoablation |
11 |
||
| IFNγ |
IFNGR |
IFNγ −/−, Mycobacterium avium infection |
Negative |
9 |
| SDF1(CXCL12) |
CXCR4 |
Cxcr4 −/− |
Positive |
66 |
| OPN |
|
Opn −/− |
Positive |
67 and 68 |
| Hh |
Patched |
Gli1 −/− (downstream transcription factor) Ptc +/− |
Negative |
69 and 12 |
| SCF | c-kit | SCF isoform −/− as a recipient | Positive | 70 |
There is emerging evidence suggesting that pro-inflammatory cytokines (e.g., IFNα, IFNγ, and TGFβ) regulate HSC proliferation and thus modulate bone marrow hematopoiesis, supporting cross talk between the immune system and bone marrow hematopoiesis.9-11 In addition to inflammatory cytokines, the cytotoxicity of bone marrow can induce HSC proliferation while maintaining an appropriate balance of self-renewing and differentiating cell divisions aimed at maintaining the stem cell numbers, but yet producing adequate numbers of mature cells. For example, hedgehog (Hh) induces the cycling and expansion of HSCs under homeostatic conditions and during stressed hematopoiesis.12
Stem Cell-Intrinsic (SCI) Factors
Many stem cell-intrinsic factors are downstream effectors of the signaling pathways described above; however, they can also lower the threshold of HSC responsiveness to external proliferative signals independently as part of complex transcriptional circuits. Table 2 summarizes the main stem cell intrinsic (SCI) factors categorized based on their role in the cell cycle, transcription, and miscellaneous mechanisms. Most studies have used mice transgenic for the expression of Cre recombinase driven by the promoter of the gene encoding the myxovirus-resistance 1 polypeptide (Mx1-Cre) to induce the deletion of floxed genes by the administration of polyinosinic:polycytidylic acid (polyI:C). However, IFNα released by the systemic administration of polyI:C also induces HSC proliferation and exhaustion, which makes the interpretation of gene-specific phenotypes more difficult.10 Thus, research groups are testing other inducible (ER-ROSA-Cre) or lineage-specific cre (e.g., Tie2-Cre and vav-iCre) systems to study HSC maintenance.
Table 2. Stem cell intrinsic (SCI) regulation of HSC quiescence.
| Category | Gene | Control of quiescence | Mouse model | References |
|---|---|---|---|---|
| Cell cycle |
p21 |
Positive |
−/− |
13 |
| p27 |
−/− |
16 |
||
| p18 |
−/− |
14 |
||
| p57 |
cKO (Mx1-cre) |
15 and 16 |
||
| Rb/p107/p130 |
cKO (Mx1-cre, Rosa26-cre ERt2) |
71 |
||
| CyclinD1/D2/D3 |
Negative |
−/− |
72 |
|
| Cdk4/6 |
−/− |
73 |
||
| Transcription factor |
JunB |
Positive |
cKO (MORE-cre) |
18 |
| Pbx1 |
−/−, cKO (Mx1-cre, Tie2-cre) |
19 |
||
| FoxO1/3/4 |
cKO (Mx1-cre) |
20 |
||
| FoxO3a |
−/− |
21 |
||
| Nurr1 |
Retroviral-transduction |
22 |
||
| Scl |
+/− |
74 |
||
| p53 |
−/− |
24 and 75 |
||
| Necdin |
−/− |
76 |
||
| Egr1 |
−/− |
28 |
||
| ELF4 |
Negative |
−/− |
23 |
|
| AML1/Runx1 |
cKO (Mx1-cre) |
27 |
||
| Stat5 |
cKO (Mx1-cre) |
29 |
||
| Other | Fbw7 |
Positive |
cKO (Mx1-cre), Retroviral expression |
30 and 77 |
| Lkb1 |
cKO (Mx1-cre, Rosa26-cre ERt2) |
9, 33, and 34 |
||
| Tsc1-mTOR |
cKO (Mx1-cre) |
36 |
||
| Cited2 |
cKO (Mx1-cre) |
38 and 39 |
||
| Txnip |
−/− |
40 |
||
| Atm |
−/− |
78 |
||
| Mi-2β |
cKO (Mx1-cre) |
79 |
||
| Paf |
−/− |
80 |
||
| Ott1 |
cKO (Mx1-cre) |
81 |
||
| G0S2 |
Retroviral expression and silencing |
41 |
||
| Akt1/2 |
Negative | −/− |
35 |
|
| Lnk | −/− | 82 and 83 |
Cell cycle
The transitions G0→G1 (exit from quiescence) and G1→S (cell cycle entry) are controlled by the Cyclin D/cdk-4/6 and Cyclin E/cdk-2 complexes, respectively. In turn, cyclin-dependent kinase inhibitors (CKI) of the INK (p15, p16, p18, p19) and CIP/KIP (p21, p27 and p57) families control the activity of cyclin/cdk complexes (Fig. 2B). Thus, the deletion of individual CKIs was examined to study HSC maintenance, and unsurprisingly, many of these deletions resulted in alterations of the cell cycle. The first cell cycle regulator studied in HSCs was the cyclin-dependent kinase inhibitor, Cdkn1a (p21). Immunophenotypic and functional assays showed increased homeostatic HSC proliferation in p21−/− mice, which led to stem cell exhaustion in serial bone marrow transplants.13 Conversely, the loss of p18 increased self-renewing cell divisions and long-term engraftment, indicating that quiescence and self-renewal are both regulated in the early G1 phase.14 In addition to alterations in the self-renewal capacity, p57−/− HSC displayed lower percentages in the G0 phase of the cell cycle.15 This effect on the maintenance of HSC quiescence was synergistic to p21; thus, the deletion of both p21 and p57 resulted in more severe defects in HSC function.15 More recently, a group linked TGFβ signaling with the p57-dependent resetting of HSC quiescence following myelosuppression.11 Although the loss of p27 showed no significant alterations at the stem cell level, the combined deficiency of p27 and p57 resulted in the nuclear localization of the heat shock cognate protein HSC70, the phosphorylation of retinoblastoma, and loss of HSC quiescence.16
Transcription factors
The expression of genes involved in quiescence, self-renewal, and differentiation are temporally expressed to modulate HSC function.17 For example, HSCs activate genes involved in quiescence and self-renewal while repressing genes associated with the differentiation to mature lineages to preserve stemness and dormancy. This expression signature is regulated by specific transcription factors for which their roles have been studied using gain-of-function (transgenic or retroviral expression) and loss-of-function (embryonic or somatic gene deletion) mouse models (Table 2). The analysis of loss-of-gene expression allows the identification of non-redundant functions that cannot be compensated by other transcriptional regulators present in the cell of interest. Elevated levels of specific transcription factors in HSCs usually correlate with a functional role in stem cell maintenance, although gene regulation requires the formation of functional transcriptional complexes. In addition to normal physiology, the deregulation of stem cell-associated genes is often associated with leukemogenesis or myeloproliferative disorders. For example, the loss of JunB leads to a myeloproliferative disease because of its role in the proliferation of HSCs downstream of the Notch1 and TGFβ pathways.18
The conditional deletion of the pre-B cell leukemia transcription factor 1 (PBX1) in HSCs leads to a reduction of quiescence and stem cell numbers in bone marrow in addition to an aberrant response to TGFβ stimulation; however, PBX1 plays an opposite role in the proliferation of progenitor cells.19 The homozygous deletion of the forkhead transcription factors FoxO1, FoxO2, and FoxO3 resulted in the reduction of the lineage-negative Sca-1+ c-kit+ (LSK) cells, a subset enriched in HSCs and progenitor cells, and defective long-term repopulating capacity that correlated with the increased proliferation and apoptosis of HSCs.20 Mice with a homozygous embryonic deletion of FoxO3a showed a decreased frequency of HSCs and a defective long-term reconstitution of cytoablated recipient mice in competitive bone marrow transplants, which was associated with the increased activity of p38 MAPK and elevated levels of reactive oxygen species (ROS), suggesting that FoxO3a regulates self-renewal downstream of the Pten/PI3K/Akt pathway.21 Although the authors proposed a disruption of quiescence in FoxO3a-null HSCs, it was measured using an LSK population that was known to contain progenitor cells in addition to HSCs. An area of transcription factors that has not been fully explored in HSCs is the nuclear receptor-type transcription factors. The retroviral expression of the orphan receptor Nurr1 (Nr4a2) in wild-type bone marrow cells increased HSC quiescence by upregulating the cell cycle inhibitor p18Ink4c, whereas the deletion of one allele augmented HSC proliferation.22 Lowering the expression of Scl/Tal1 either by gene silencing or haplo-insufficiency reduced the percentage of HSCs in the G0 phase of the cell cycle and impaired hematological reconstitution upon transplantation. Scl promotes the expression of Id1 and p21, which are involved in the inhibition of the G0→G1 transition and HSC quiescence.
We reported that the transcription factor ELF4 (also known as MEF), a member of the ETS family of winged helix-turn-helix transcription factors, negatively regulates HSC quiescence by promoting cell cycle entry in the steady state but not during regenerative hematopoiesis.23 ELF4 is one of the few known negative regulators of HSC quiescence (Table 2). The loss of ELF4 resulted in the accumulation of mitotically quiescent HSCs in vivo, and, consequently, Elf4-null mice exhibited increased resistance to cell cycle-dependent cytotoxicity induced by chemodrugs (e.g., 5-fluorouracil and busulfan) or radiation.23 This regulation was later shown to be dependent on the tumor suppressor p53, which regulates the quiescence and self-renewal of HSCs during homeostasis by targeting Gfi1 and Necdin.24 Necdin restricts the proliferation of HSCs in the steady state; therefore, a lower percentage of necdin-null HSCs were found in the G0 phase of the cell cycle. In the absence of Mdm2, the E3 ubiquitin ligase, which targets p53 for degradation, and ROS-induced postnatal p53 activity depletes the HSCs.25 Of note, p53 has also been shown to promote cellular quiescence by suppressing senescence, although this mechanism has not been tested in the hematopoietic system.26 Finally, AML1 (also known as Runx1), a binding partner of ELF4, was originally identified at the breakpoint of the chromosomal translocation t(8;21). Interestingly, AML1-deficiency increased the number of side population fractions in bone marrow, suggesting the enrichment of quiescent HSCs.27
Early growth response 1 (Egr1) is a zinc-finger transcription factor involved in cell growth, development, and stress responses in a variety of tissues. The Egr1 gene is expressed in quiescent HSCs and downregulated by the induction of cell division and migration. Egr1-null mice showed increased numbers of dividing HSCs and the increased expression of Bmi-1 and Cdk4 in the steady state, suggesting that Egr1 promotes HSC quiescence.28 Stat5 (signal transducer and activator of transcription 5) maintains quiescence and the number of HSCs during steady-state hematopoiesis; therefore, the conditional deletion of Stat5 in HSCs causes the loss of quiescence.29 Stat5-induced HSC self-renewal is mediated at least in part via the upregulation of hypoxia-induced factor 2α (HIF2α).
Other mechanisms
In addition to the cell cycle and transcriptional regulation of HSC proliferation, there are molecules that indirectly control quiescence by modulating metabolism, transcript and protein stability. For example, the E3 ubiquitin ligase Fbw7 controls HSC proliferation and its capacity to reconstitute hosts upon transplantation by regulating the half-life of the c-Myc protein.30 Low levels of c-Myc rescued the functional deficiencies observed in Fbw7−/− HSC; conversely, the enforced expression of Fbw7 increased HSC quiescence by reducing the protein levels of both c-Myc and Notch1. Interestingly, two groups recently showed that Fbw7 and its target, c-Myc, also control the initiation and progression of chronic myeloid leukemia by activating the p53-dependent apoptosis of leukemia-initiating cells.31,32
There is emerging evidence that metabolic fitness influences the proliferative capacity of stem cells by controlling energy availability. Loss of the tumor suppressor Lkb1 increased the proliferation of HSCs, exhaustion of the stem cell pool, pancytopenia, and impaired the repopulating capacity of HSCs.33 Although one group showed dependency on AMPK in the control of the mitochondrial potential and pool of ATP, another group postulated that these effects were independent of the Lkb1 regulation of AMPK and mTOR.34 AKT inhibits the activity of the Tsc1-Tsc2 complex by the phosphorylation of the inhibitor of mTOR signaling, thereby activating mTOR activity. The deletion of AKT1 and AKT2 resulted in increased HSC quiescence and lowered the levels of ROS.35 Interestingly, Tsc1 maintains HSC quiescence by limiting mitochondrial biogenesis and ROS levels.36 Similar to HSCs, the inhibition of mTOR can drive cells to quiescence over senescence in other cell lineages during aging.37
Gene expression is regulated at the transcriptional level by transcription factors and co-activators/repressors and epigenetic regulation. The conditional deletion of the transcriptional co-regulator Cited2 causes the loss of HSC function and defective hematopoiesis, which can be rescued by the deletion of p16, p19, or p53.38 The authors concluded that this effect was due in part to elevated HIF-1 activity and the associated decreased expression of p57, Egr1, and Hes1.39 The transcriptional repressor thioredoxin-interacting protein (Txnip) induces cell cycle arrest and quiescence in HSCs; therefore, Txnip-null HSCs exhibit increased proliferation and exhaustion associated with a lower retention in the HSC niche.40 Other genes involved in diverse functions, such as kinase (Atm), nucleosome deacetylase (Mi-2β), proliferating cell nuclear antigen (PCNA), and RNA binding protein (Ott1), have shown a reduction of HSC quiescence by somatic deletion (Table 2).
Control of HSC Quiescence by G0S2 and Nucleolin Interaction
As discussed above, HSCs are able to balance proliferation and differentiation by integrating complex mechanisms at the transcriptional and post-translational levels, which are regulated by intrinsic and extrinsic cell factors. We recently described a novel mechanism that controls HSC quiescence mediated by the cytosolic protein G0S2.41 G0S2 is a basic protein with an ill-defined function and was discovered more than 20 years ago from lectin-activated lymphocytes.42 A putative role in the control of the G0/G1 phase of the cell cycle was initially ascribed to G0S2 by either releasing lymphocytes from quiescence (G0→G1 transition) or by promoting proliferation (G1→S phase transition).42,43 However, disparate functions other than proliferation have emerged for G0S2 in recent years, suggesting that G0S2 is at the crossroads of multiple cellular pathways, including lipid metabolism,44-46 carcinogenesis and chemoresistance,47,48 inflammation,49-53 and senescence.54
The elevated levels of G0S2 in HSCs, defined immunophenotypically as Lin– Sca-1+ c-kit+ CD150+ CD48− CD41− cells, are downregulated with the differentiation to progenitor and mature blood cells.41 The expression of G0S2 is also downregulated in HSCs induced to proliferate in vivo by the administration of 5-fluorouracil, a feature of genes involved in the regulation of stem cell quiescence, further supporting an inhibitory function in HSC proliferation.17 Consistent with this observation, the retroviral expression of G0S2 pushed HSCs deeper into quiescence by increasing the percentage in the G0 phase; conversely, silencing endogenous G0S2 expression increased HSC proliferation.41
The subcellular localization of G0S2 showed a perinuclear distribution in hematopoietic cells, co-localizing with markers of the mitochondria, endoplasmic reticulum, and early endosomes.41 This finding raised the question of how a cytosolic protein can regulate HSC proliferation. One hypothesis proposed that G0S2 controls the choice to proliferate or remain quiescent by interacting with cell cycle regulators. Co-immunoprecipitation combined with mass spectrometry led to the identification of nucleolin and ribonucleoproteins (L3, L6, L13) as the main G0S2 protein partners in EL4 cells, which was later confirmed in primary bone marrow cells.41 Nucleolin is a multifunctional protein localized predominantly in the nucleolus, although it can also localize to the nucleoplasm, cytosol, and cell membrane.55 Although the expression of nucleolin has been associated with highly proliferative cells such as cancer cells, how nucleolin controls cell growth and cell division is not clear. Nucleolin is believed to promote proliferation by regulating the transcription of ribosomal DNA in nucleoli, the maturation of pre-rRNA in nucleus, and the transport of ribonucleoproteins and ribosomal particles to the cytosol for final assembly.56 In addition, nucleolin also stabilizes mRNA, enhances translation, and shuttles proteins into the nucleus.57-59 Because the complete loss of nucleolin is not compatible with cell viability, the nucleolin-to-G0S2 interaction can modulate the proliferative functions of nucleolin without causing cell death.
The G0S2 protein contains a conserved hydrophobic domain (HD) that interacts with bcl-2 and adipose triglyceride lipase.46,60 On the other side, nucleolin contains several RNA-binding domains (RBD) in the N-terminus and arginine-glycine-glycine (RGG) domains in the C-terminal, which are known to bind transcripts and ribonucleoproteins, respectively. The HD domain of G0S2 and the RGG domain of nucleolin are required for protein-to-protein interactions, suggesting a supramolecular assembly of nucleolin, ribonucleoproteins, and G0S2.41 How could this interaction inhibit proliferation? The immunofluorescence analysis showed that G0S2 overexpression caused a re-distribution of nucleolin from nucleoli to the cytosol. In purified Lin– Sca-1+ c-kit+ CD150+ CD48− cells (> 80% in the G0 phase), the elevated levels of G0S2 were associated with a ‘ring-like’ perinuclear distribution of nucleolin caused by its retention in the cytosol (Fig. 3).41 Interestingly, nucleolin returns to the nucleoli as G0S2 expression wanes in ‘proliferating’ Lin– Sca-1+ c-kit+ CD150+ CD48− cells. These findings suggest that the interaction of nucleolin and G0S2 is a dynamic and reversible process triggered by increased cellular levels of the G0S2 protein in quiescent HSCs. Collectively, we proposed a novel model that controls HSC quiescence in which G0S2 sequesters nucleolin in the cytosol of quiescent HSCs, precluding its pro-proliferation functions in the nucleolus, whereas low levels of G0S2 in proliferating HSCs restores the function of nucleolin (Fig. 3).

Figure 3. G0S2 promotes quiescence by interacting with nucleolin. (A) The arginine-glycine-glycine (RGG) domain of nucleolin interacts with a highly conserved hydrophobic domain (HD) of G0S2. (B) The ectopic expression of G0S2 retains nucleolin in the cytosol of quiescent HSCs (Lin− Sca-1+ c-kit+ CD48− CD150+ cells). (C) Diagram depicting a novel model that controls the quiescence of HSCs mediated by the interaction between G0S2 and nucleolin.
Acknowledgments
This work was supported by the Gabrielle’s Angel Foundation for Cancer Research (to HDL) and the National Institutes of Health Grants R01-AI077536 (to HDL) and R01-AI077536-02S1 (to HDL).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25416
References
- 1.Zhang J, Li L. Stem cell niche: microenvironment and beyond. J Biol Chem. 2008;283:9499–503. doi: 10.1074/jbc.R700043200. [DOI] [PubMed] [Google Scholar]
- 2.Kiel MJ, Morrison SJ. Uncertainty in the niches that maintain haematopoietic stem cells. Nat Rev Immunol. 2008;8:290–301. doi: 10.1038/nri2279. [DOI] [PubMed] [Google Scholar]
- 3.Kiel MJ, Morrison SJ. Maintaining hematopoietic stem cells in the vascular niche. Immunity. 2006;25:862–4. doi: 10.1016/j.immuni.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Yin T, Li L. The stem cell niches in bone. J Clin Invest. 2006;116:1195–201. doi: 10.1172/JCI28568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–41. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- 6.Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–6. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 7.Kopp HG, Avecilla ST, Hooper AT, Rafii S. The bone marrow vascular niche: home of HSC differentiation and mobilization. Physiology (Bethesda) 2005;20:349–56. doi: 10.1152/physiol.00025.2005. [DOI] [PubMed] [Google Scholar]
- 8.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–62. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465:793–7. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–8. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 11.Brenet F, Kermani P, Spektor R, Rafii S, Scandura JM. TGFβ restores hematopoietic homeostasis after myelosuppressive chemotherapy. J Exp Med. 2013;210:623–39. doi: 10.1084/jem.20121610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trowbridge JJ, Scott MP, Bhatia M. Hedgehog modulates cell cycle regulators in stem cells to control hematopoietic regeneration. Proc Natl Acad Sci U S A. 2006;103:14134–9. doi: 10.1073/pnas.0604568103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–8. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 14.Yuan Y, Shen H, Franklin DS, Scadden DT, Cheng T. In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4C. Nat Cell Biol. 2004;6:436–42. doi: 10.1038/ncb1126. [DOI] [PubMed] [Google Scholar]
- 15.Matsumoto A, Takeishi S, Kanie T, Susaki E, Onoyama I, Tateishi Y, et al. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell. 2011;9:262–71. doi: 10.1016/j.stem.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 16.Zou P, Yoshihara H, Hosokawa K, Tai I, Shinmyozu K, Tsukahara F, et al. p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell. 2011;9:247–61. doi: 10.1016/j.stem.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Venezia TA, Merchant AA, Ramos CA, Whitehouse NL, Young AS, Shaw CA, et al. Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol. 2004;2:e301. doi: 10.1371/journal.pbio.0020301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santaguida M, Schepers K, King B, Sabnis AJ, Forsberg EC, Attema JL, et al. JunB protects against myeloid malignancies by limiting hematopoietic stem cell proliferation and differentiation without affecting self-renewal. Cancer Cell. 2009;15:341–52. doi: 10.1016/j.ccr.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ficara F, Murphy MJ, Lin M, Cleary ML. Pbx1 regulates self-renewal of long-term hematopoietic stem cells by maintaining their quiescence. Cell Stem Cell. 2008;2:484–96. doi: 10.1016/j.stem.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–39. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 21.Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1:101–12. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 22.Sirin O, Lukov GL, Mao R, Conneely OM, Goodell MA. The orphan nuclear receptor Nurr1 restricts the proliferation of haematopoietic stem cells. Nat Cell Biol. 2010;12:1213–9. doi: 10.1038/ncb2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lacorazza HD, Yamada T, Liu Y, Miyata Y, Sivina M, Nunes J, et al. The transcription factor MEF/ELF4 regulates the quiescence of primitive hematopoietic cells. Cancer Cell. 2006;9:175–87. doi: 10.1016/j.ccr.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4:37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abbas HA, Maccio DR, Coskun S, Jackson JG, Hazen AL, Sills TM, et al. Mdm2 is required for survival of hematopoietic stem cells/progenitors via dampening of ROS-induced p53 activity. Cell Stem Cell. 2010;7:606–17. doi: 10.1016/j.stem.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010;107:9660–4. doi: 10.1073/pnas.1002298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ichikawa M, Goyama S, Asai T, Kawazu M, Nakagawa M, Takeshita M, et al. AML1/Runx1 negatively regulates quiescent hematopoietic stem cells in adult hematopoiesis. J Immunol. 2008;180:4402–8. doi: 10.4049/jimmunol.180.7.4402. [DOI] [PubMed] [Google Scholar]
- 28.Min IM, Pietramaggiori G, Kim FS, Passegué E, Stevenson KE, Wagers AJ. The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell. 2008;2:380–91. doi: 10.1016/j.stem.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 29.Wang Z, Li G, Tse W, Bunting KD. Conditional deletion of STAT5 in adult mouse hematopoietic stem cells causes loss of quiescence and permits efficient nonablative stem cell replacement. Blood. 2009;113:4856–65. doi: 10.1182/blood-2008-09-181107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reavie L, Della Gatta G, Crusio K, Aranda-Orgilles B, Buckley SM, Thompson B, et al. Regulation of hematopoietic stem cell differentiation by a single ubiquitin ligase-substrate complex. Nat Immunol. 2010;11:207–15. doi: 10.1038/ni.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reavie L, Buckley SM, Loizou E, Takeishi S, Aranda-Orgilles B, Ndiaye-Lobry D, et al. Regulation of c-Myc ubiquitination controls chronic myelogenous leukemia initiation and progression. Cancer Cell. 2013;23:362–75. doi: 10.1016/j.ccr.2013.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takeishi S, Matsumoto A, Onoyama I, Naka K, Hirao A, Nakayama KI. Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer Cell. 2013;23:347–61. doi: 10.1016/j.ccr.2013.01.026. [DOI] [PubMed] [Google Scholar]
- 33.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–8. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gurumurthy S, Xie SZ, Alagesan B, Kim J, Yusuf RZ, Saez B, et al. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature. 2010;468:659–63. doi: 10.1038/nature09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juntilla MM, Patil VD, Calamito M, Joshi RP, Birnbaum MJ, Koretzky GA. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood. 2010;115:4030–8. doi: 10.1182/blood-2009-09-241000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Liu Y, et al. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008;205:2397–408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY) 2012;4:159–65. doi: 10.18632/aging.100443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kranc KR, Schepers H, Rodrigues NP, Bamforth S, Villadsen E, Ferry H, et al. Cited2 is an essential regulator of adult hematopoietic stem cells. Cell Stem Cell. 2009;5:659–65. doi: 10.1016/j.stem.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Du J, Chen Y, Li Q, Han X, Cheng C, Wang Z, et al. HIF-1α deletion partially rescues defects of hematopoietic stem cell quiescence caused by Cited2 deficiency. Blood. 2012;119:2789–98. doi: 10.1182/blood-2011-10-387902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeong M, Piao ZH, Kim MS, Lee SH, Yun S, Sun HN, et al. Thioredoxin-interacting protein regulates hematopoietic stem cell quiescence and mobilization under stress conditions. J Immunol. 2009;183:2495–505. doi: 10.4049/jimmunol.0804221. [DOI] [PubMed] [Google Scholar]
- 41.Yamada T, Park CS, Burns A, Nakada D, Lacorazza HD. The cytosolic protein G0S2 maintains quiescence in hematopoietic stem cells. PLoS One. 2012;7:e38280. doi: 10.1371/journal.pone.0038280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Russell L, Forsdyke DR. A human putative lymphocyte G0/G1 switch gene containing a CpG-rich island encodes a small basic protein with the potential to be phosphorylated. DNA Cell Biol. 1991;10:581–91. doi: 10.1089/dna.1991.10.581. [DOI] [PubMed] [Google Scholar]
- 43.Cristillo AD, Heximer SP, Russell L, Forsdyke DR. Cyclosporin A inhibits early mRNA expression of G0/G1 switch gene 2 (G0S2) in cultured human blood mononuclear cells. DNA Cell Biol. 1997;16:1449–58. doi: 10.1089/dna.1997.16.1449. [DOI] [PubMed] [Google Scholar]
- 44.Zandbergen F, Mandard S, Escher P, Tan NS, Patsouris D, Jatkoe T, et al. The G0/G1 switch gene 2 is a novel PPAR target gene. Biochem J. 2005;392:313–24. doi: 10.1042/BJ20050636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teunissen BE, Smeets PJ, Willemsen PH, De Windt LJ, Van der Vusse GJ, Van Bilsen M. Activation of PPARdelta inhibits cardiac fibroblast proliferation and the transdifferentiation into myofibroblasts. Cardiovasc Res. 2007;75:519–29. doi: 10.1016/j.cardiores.2007.04.026. [DOI] [PubMed] [Google Scholar]
- 46.Yang X, Lu X, Lombès M, Rha GB, Chi YI, Guerin TM, et al. The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 2010;11:194–205. doi: 10.1016/j.cmet.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tokumaru Y, Yamashita K, Osada M, Nomoto S, Sun DI, Xiao Y, et al. Inverse correlation between cyclin A1 hypermethylation and p53 mutation in head and neck cancer identified by reversal of epigenetic silencing. Cancer Res. 2004;64:5982–7. doi: 10.1158/0008-5472.CAN-04-0993. [DOI] [PubMed] [Google Scholar]
- 48.Kusakabe M, Watanabe K, Emoto N, Aki N, Kage H, Nagase T, et al. Impact of DNA demethylation of the G0S2 gene on the transcription of G0S2 in squamous lung cancer cell lines with or without nuclear receptor agonists. Biochem Biophys Res Commun. 2009;390:1283–7. doi: 10.1016/j.bbrc.2009.10.137. [DOI] [PubMed] [Google Scholar]
- 49.Kao LC, Germeyer A, Tulac S, Lobo S, Yang JP, Taylor RN, et al. Expression profiling of endometrium from women with endometriosis reveals candidate genes for disease-based implantation failure and infertility. Endocrinology. 2003;144:2870–81. doi: 10.1210/en.2003-0043. [DOI] [PubMed] [Google Scholar]
- 50.Ma Y, Koza-Taylor PH, DiMattia DA, Hames L, Fu H, Dragnev KH, et al. Microarray analysis uncovers retinoid targets in human bronchial epithelial cells. Oncogene. 2003;22:4924–32. doi: 10.1038/sj.onc.1206728. [DOI] [PubMed] [Google Scholar]
- 51.Nakamura N, Shimaoka Y, Tougan T, Onda H, Okuzaki D, Zhao H, et al. Isolation and expression profiling of genes upregulated in bone marrow-derived mononuclear cells of rheumatoid arthritis patients. DNA Res. 2006;13:169–83. doi: 10.1093/dnares/dsl006. [DOI] [PubMed] [Google Scholar]
- 52.Kobayashi S, Ito A, Okuzaki D, Onda H, Yabuta N, Nagamori I, et al. Expression profiling of PBMC-based diagnostic gene markers isolated from vasculitis patients. DNA Res. 2008;15:253–65. doi: 10.1093/dnares/dsn014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koczan D, Guthke R, Thiesen HJ, Ibrahim SM, Kundt G, Krentz H, et al. Gene expression profiling of peripheral blood mononuclear leukocytes from psoriasis patients identifies new immune regulatory molecules. Eur J Dermatol. 2005;15:251–7. [PubMed] [Google Scholar]
- 54.Yoon IK, Kim HK, Kim YK, Song IH, Kim W, Kim S, et al. Exploration of replicative senescence-associated genes in human dermal fibroblasts by cDNA microarray technology. Exp Gerontol. 2004;39:1369–78. doi: 10.1016/j.exger.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 55.Storck S, Shukla M, Dimitrov S, Bouvet P. Functions of the histone chaperone nucleolin in diseases. Subcell Biochem. 2007;41:125–44. doi: 10.1007/1-4020-5466-1_7. [DOI] [PubMed] [Google Scholar]
- 56.Mongelard F, Bouvet P. Nucleolin: a multiFACeTed protein. Trends Cell Biol. 2007;17:80–6. doi: 10.1016/j.tcb.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 57.Fähling M, Steege A, Perlewitz A, Nafz B, Mrowka R, Persson PB, et al. Role of nucleolin in posttranscriptional control of MMP-9 expression. Biochim Biophys Acta. 2005;1731:32–40. doi: 10.1016/j.bbaexp.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 58.Otake Y, Soundararajan S, Sengupta TK, Kio EA, Smith JC, Pineda-Roman M, et al. Overexpression of nucleolin in chronic lymphocytic leukemia cells induces stabilization of bcl2 mRNA. Blood. 2007;109:3069–75. doi: 10.1182/blood-2006-08-043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abdelmohsen K, Tominaga K, Lee EK, Srikantan S, Kang MJ, Kim MM, et al. Enhanced translation by Nucleolin via G-rich elements in coding and non-coding regions of target mRNAs. Nucleic Acids Res. 2011;39:8513–30. doi: 10.1093/nar/gkr488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Welch C, Santra MK, El-Assaad W, Zhu X, Huber WE, Keys RA, et al. Identification of a protein, G0S2, that lacks Bcl-2 homology domains and interacts with and antagonizes Bcl-2. Cancer Res. 2009;69:6782–9. doi: 10.1158/0008-5472.CAN-09-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149–61. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 62.Yoshihara H, Arai F, Hosokawa K, Hagiwara T, Takubo K, Nakamura Y, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell. 2007;1:685–97. doi: 10.1016/j.stem.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 63.Fleming HE, Janzen V, Lo Celso C, Guo J, Leahy KM, Kronenberg HM, et al. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell. 2008;2:274–83. doi: 10.1016/j.stem.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kirstetter P, Anderson K, Porse BT, Jacobsen SE, Nerlov C. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat Immunol. 2006;7:1048–56. doi: 10.1038/ni1381. [DOI] [PubMed] [Google Scholar]
- 65.Larsson J, Blank U, Helgadottir H, Björnsson JM, Ehinger M, Goumans MJ, et al. TGF-beta signaling-deficient hematopoietic stem cells have normal self-renewal and regenerative ability in vivo despite increased proliferative capacity in vitro. Blood. 2003;102:3129–35. doi: 10.1182/blood-2003-04-1300. [DOI] [PubMed] [Google Scholar]
- 66.Nie Y, Han YC, Zou YR. CXCR4 is required for the quiescence of primitive hematopoietic cells. J Exp Med. 2008;205:777–83. doi: 10.1084/jem.20072513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stier S, Ko Y, Forkert R, Lutz C, Neuhaus T, Grünewald E, et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med. 2005;201:1781–91. doi: 10.1084/jem.20041992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nilsson SK, Johnston HM, Whitty GA, Williams B, Webb RJ, Denhardt DT, et al. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood. 2005;106:1232–9. doi: 10.1182/blood-2004-11-4422. [DOI] [PubMed] [Google Scholar]
- 69.Merchant A, Joseph G, Wang Q, Brennan S, Matsui W. Gli1 regulates the proliferation and differentiation of HSCs and myeloid progenitors. Blood. 2010;115:2391–6. doi: 10.1182/blood-2009-09-241703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Driessen RL, Johnston HM, Nilsson SK. Membrane-bound stem cell factor is a key regulator in the initial lodgment of stem cells within the endosteal marrow region. Exp Hematol. 2003;31:1284–91. doi: 10.1016/j.exphem.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 71.Viatour P, Somervaille TC, Venkatasubrahmanyam S, Kogan S, McLaughlin ME, Weissman IL, et al. Hematopoietic stem cell quiescence is maintained by compound contributions of the retinoblastoma gene family. Cell Stem Cell. 2008;3:416–28. doi: 10.1016/j.stem.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, et al. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004;118:477–91. doi: 10.1016/j.cell.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 73.Malumbres M, Sotillo R, Santamaría D, Galán J, Cerezo A, Ortega S, et al. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118:493–504. doi: 10.1016/j.cell.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 74.Lacombe J, Herblot S, Rojas-Sutterlin S, Haman A, Barakat S, Iscove NN, et al. Scl regulates the quiescence and the long-term competence of hematopoietic stem cells. Blood. 2010;115:792–803. doi: 10.1182/blood-2009-01-201384. [DOI] [PubMed] [Google Scholar]
- 75.Abbas HA, Pant V, Lozano G. The ups and downs of p53 regulation in hematopoietic stem cells. Cell Cycle. 2011;10:3257–62. doi: 10.4161/cc.10.19.17721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Asai T, Liu Y, Di Giandomenico S, Bae N, Ndiaye-Lobry D, Deblasio A, et al. Necdin, a p53 target gene, regulates the quiescence and response to genotoxic stress of hematopoietic stem/progenitor cells. Blood. 2012;120:1601–12. doi: 10.1182/blood-2011-11-393983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Iriuchishima H, Takubo K, Matsuoka S, Onoyama I, Nakayama KI, Nojima Y, et al. Ex vivo maintenance of hematopoietic stem cells by quiescence induction through Fbxw7α overexpression. Blood. 2011;117:2373–7. doi: 10.1182/blood-2010-07-294801. [DOI] [PubMed] [Google Scholar]
- 78.Maryanovich M, Oberkovitz G, Niv H, Vorobiyov L, Zaltsman Y, Brenner O, et al. The ATM-BID pathway regulates quiescence and survival of haematopoietic stem cells. Nat Cell Biol. 2012;14:535–41. doi: 10.1038/ncb2468. [DOI] [PubMed] [Google Scholar]
- 79.Yoshida T, Hazan I, Zhang J, Ng SY, Naito T, Snippert HJ, et al. The role of the chromatin remodeler Mi-2beta in hematopoietic stem cell self-renewal and multilineage differentiation. Genes Dev. 2008;22:1174–89. doi: 10.1101/gad.1642808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Amrani YM, Gill J, Matevossian A, Alonzo ES, Yang C, Shieh JH, et al. The Paf oncogene is essential for hematopoietic stem cell function and development. J Exp Med. 2011;208:1757–65. doi: 10.1084/jem.20102170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xiao N, Jani K, Morgan K, Okabe R, Cullen DE, Jesneck JL, et al. Hematopoietic stem cells lacking Ott1 display aspects associated with aging and are unable to maintain quiescence during proliferative stress. Blood. 2012;119:4898–907. doi: 10.1182/blood-2012-01-403089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bersenev A, Wu C, Balcerek J, Tong W. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J Clin Invest. 2008;118:2832–44. doi: 10.1172/JCI35808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Suzuki N, Yamazaki S, Ema H, Yamaguchi T, Nakauchi H, Takaki S. Homeostasis of hematopoietic stem cells regulated by the myeloproliferative disease associated-gene product Lnk/Sh2b3 via Bcl-xL. Exp Hematol. 2012;40:166–74, e3. doi: 10.1016/j.exphem.2011.11.003. [DOI] [PubMed] [Google Scholar]