

Abstract
Facilitates chromatin transcription (FACT) is a chromatin remodeling complex with two subunits: SSRP1 and SPT16. Mechanisms controlling FACT levels are of interest, since the complex is not expressed in most differentiated cells, but is frequently upregulated in cancer, particularly in poorly differentiated, aggressive tumors. Moreover, inhibition of FACT expression or function in tumor cells interferes with their survival. Here we demonstrate that SSRP1 and SPT16 protein levels decline upon induction of cellular differentiation or senescence in vitro and that similar declines in protein levels for both SSRP1 and SPT16 occur upon RNAi-mediated knockdown of either SSRP1 or SPT16. The interdependence of SSRP1 and SPT16 protein levels was found to be due to their association with SSRP1 and SPT16 mRNAs, which stabilizes the proteins. In particular, presence of SSRP1 mRNA is critical for SPT16 protein stability. In addition, binding of SSRP1 and SPT16 mRNAs to the FACT complex increases the stability and efficiency of translation of the mRNAs. These data support a model in which the FACT complex is stable when SSRP1 mRNA is present, but quickly degrades when SSRP1 mRNA levels drop. In the absence of FACT complex, SSRP1 and SPT16 mRNAs are unstable and inefficiently translated, making reactivation of FACT function unlikely in normal cells. Thus, we have described a complex and unusual mode of regulation controlling cellular FACT levels that results in amplified and stringent control of FACT activity. The FACT dependence of tumor cells suggests that mechanisms controlling FACT levels could be targeted for anticancer therapy.
Keywords: FACT, SSRP1, SPT16, mRNA stability, protein stability
Introduction
Facilitates Chromatin Transcription (FACT) is a conserved heterodimeric complex of two protein subunits, Structure Specific Recognition Protein 1 (SSRP1) and Suppressor of Ty 16 (SPT16). FACT is involved in chromatin remodeling through nucleosome stabilization, destabilization and histone exchange (see ref. 1 for review) and has been implicated in numerous biological processes that encompass chromatin. FACT assists in transcription driven by all three RNA polymerases,1-3 replication,4-6 recombination,7 DNA damage responses,7-9 and DNA methylation.10 With respect to its role in regulation of gene expression, FACT is not a general enhancer of transcription, but appears to be specifically required for expression of particular genes that have highly ordered chromatin structure and are induced under certain conditions rather than constitutively expressed.11,12 Despite these intriguing functions (and the possibility of additional functions that have yet to be described), FACT has been primarily studied in single cell models, such as yeast and cultured human tumor cells, and the physiological roles of FACT in multicellular organisms remain poorly understood.
A significant aspect of FACT’s activity in higher organisms was revealed by our recent identification of FACT as the molecular target of a novel class of small molecules (Curaxins) with potent anti-tumor activity.13 The importance of FACT for tumor cells was confirmed by experiments showing that reduced expression of either SSRP1 or SPT16 (via RNAi) led to tumor cell death13 and by our finding that both SSRP1 and SPT16 are expressed at higher levels in tumor cells than in corresponding normal cells and that FACT is expressed more frequently in aggressive poorly differentiated tumors with low overall survival.14
Importantly, while FACT expression is critical in tumor cells, it is not ubiquitously expressed in normal adult tissues of higher organisms. Rather, we and others have shown that FACT expression is generally limited to stem-like or undifferentiated cells such as germ cells, stem cells and progenitor cells in mammals and undifferentiated cells in growing parts of plants.15-17 Moreover, Hertel et al. demonstrated that protein levels of both FACT subunits decrease during senescence or differentiation of mouse fibroblasts18,19 and we found that levels of SSRP1 and, even more dramatically, SPT16, decline upon induction of in vitro differentiation of mouse myoblasts into myotubes.16
Given the very low or undetectable levels of expression of both FACT subunits in normal adult tissues, FACT has strong potential as a diagnostic/prognostic marker and therapeutic anticancer target, particularly for poorly differentiated, aggressive tumors.16,14 To facilitate development of strategies for therapeutic modulation of FACT, this study was aimed at characterizing the endogenous mechanisms that control cellular levels of FACT. Our previous work revealed a strong positive correlation between levels of SSRP1 and SPT16 on both the mRNA and protein levels in different cells and tissues under different conditions,16 suggesting that either their expression or stability is co-regulated. Here, we demonstrate that in undifferentiated cells both SSRP1 and SPT16 proteins are present and are extremely stable; however, upon induction of in vitro differentiation or upon RNAi-mediated knock-down of either subunit, protein levels of both subunits decline. The interdependence of SSRP1 and SPT16 protein levels was found to be due to their association with SSRP1 and SPT16 mRNAs, which stabilizes the proteins. In particular, presence of SSRP1 mRNA is critical for SPT16 protein stability. In addition, binding of SSRP1 and SPT16 mRNAs to the FACT complex increases the stability and efficiency of translation of the mRNAs. These data support a model in which the FACT complex is stable while SSRP1 mRNA is available, but quickly degrades when SSRP1 mRNA levels drop. In the absence of FACT complex, SSRP1 and SPT16 mRNAs are unstable and inefficiently translated, making reactivation of FACT function unlikely in normal cells. The observed inter-regulation of SSRP1 and SPT16 mRNA and protein levels creates an effective means for prompt disruption of an otherwise very stable complex in conditions when it is no longer required and, moreover, provides a barrier against reformation of the complex, which based on its association with cancer can be considered ‘unwanted’.
Results
Protein and mRNA levels of SSRP1 and SPT16 subunits of FACT are reduced upon induction of differentiation or senescence, but with different kinetics
Analysis of multiple publicly available high-content gene expression data sets suggested that mRNA levels of SSRP1 and SPT16 decrease upon differentiation and senescence of mammalian cells.16 Here, we used three in vitro systems to model these processes in vitro and directly measure accompanying changes in SSRP1 and SPT16 mRNA and protein levels: 1) differentiation of mouse C2C12 myoblast cells into myotubes, (2) senescence of BJ human diploid fibroblasts via tamoxifen-regulated expression of the H-ras12V oncogene, and 3) etoposide-induced senescence of WI38 human diploid fibroblasts. In all three experimental systems, we observed that protein levels of SPT16 and SSRP1 both decline upon induction of differentiation or senescence, but with different kinetics (Fig. 1A–C). SPT16 protein levels undergo rapid and dramatic reduction, while the reduction in SSRP1 protein levels is more modest and gradual (Fig. 1A–C). Interestingly, however, levels of the two mRNAs respond in an opposite manner, with SPT16 mRNA levels declining more slowly and less dramatically than SSRP1 mRNA levels (Fig. 1D and data not shown). We proposed that this unusual pattern might be due to differences in the SSRP1 and SPT16 protein and mRNA half-lives. However, measurement of these parameters showed that both mRNA and protein half-lives were similar for SSRP1 and SPT16. The intermediate half-life of both mRNAs was between 4 and 8 h (Fig. 1E). In contrast, the half-life of both proteins was very long, more than 48 h (Fig. 1F), suggesting that SSRP1 and SPT16 are very stable proteins.

Figure 1. Levels of the FACT subunits SSRP1 and SPT16 decline upon induction of differentiation or senescence in vitro.(A)Western blot analysis of SSRP1 and SPT16 protein levels in lysates of C2C12 cells collected before and after differentiation from myoblasts into myotubes. C2C12 myoblasts were grown to high density and maintained for 2 d (day −2 to day 0). At day 0, differentiation was induced (see Materials and Methods). Myosin heavy chain (MHC) was analyzed as a marker of myotube differentiation.(B)Western blot analysis of SSRP1, SPT16, and β-actin protein levels in lysates of BJ human diploid fibroblasts collected at different times after induction of senescence via tamoxifen (TMX)-regulated expression of the H-rasV12 oncogene (see Materials and Methods).(C)Western blot analysis of SSRP1, SPT16, and β-actin protein levels in lysates of WI38 cells collected at different times after induction of senescence by addition of 1mg/ml etoposide.(D)Quantitative RT-PCR (qRT-PCR) analysis of SSRP1 and SPT16 mRNA levels in WI38 treated as in (C). mRNA levels are shown relative to the level at “time 0” (before addition of etoposide), which was set at 1.0. E-F. Stability of SSRP1 and SPT16 mRNAs (E) and proteins (F) assessed using qRT-PCR and western blotting in cells with blocked transcription (E, actinomycin D, 100 ng/ml) or translation (F, cycloheximide, 50 μg/ml) for the indicated periods of time. SE and LE panels are correspondingly short and long expositions of the same membrane.
These data indicate that the abrupt reduction in SPT16 protein level observed upon induction of differentiation or senescence cannot be simply due to cessation of SPT16 gene expression. The half-lives of the mRNA and protein suggest that SPT16 protein levels should persist for a substantial amount of time even after transcription of the gene is turned off and all existing mRNA has decayed. Therefore, we hypothesized that a mechanism affecting the stability of the SPT16 protein itself is involved in the loss of FACT upon cellular differentiation/senescence.
The protein level of each FACT subunit depends on the presence of the other subunit
In our previous work, RNA interference (RNAi) technology was used to specifically knockdown (KD) each individual subunit of FACT and demonstrate that FACT is essential for survival of tumor cells.13 During the course of those studies, we noticed that RNAi-mediated KD of SSRP1 or SPT16 led to reduction of not only the subunit targeted by the si/shRNA but also the other FACT complex subunit (Fig. 2). Thus, HeLa cells transduced with either shSSRP1 constructs or shSPT16 constructs showed reduced levels of both SSRP1 and SPT16 proteins, but not an unrelated control protein (β-actin) (Fig. 2A). The responses of SPT16 and SSRP1 protein levels to anti-SSRP1 shRNA (Fig. 2A, B, and D) were similar to those observed upon induction of differentiation and senescence (Fig. 1A–C): a sharp drop for SPT16 and a more gradual decline for SSRP1. Coordinated reduction of SSRP1 and SPT16 protein levels was observed in several different cell lines (Fig. 2A–C) with multiple different anti-SSRP1 and anti-SPT16 shRNA constructs, thus indicating that the observed effect was not due to ‘off-target’ effects of shRNAs. In contrast to the effect of FACT subunits KD on protein levels, levels of SSRP1 and SPT16 mRNAs were not reduced in cells transduced with siRNA constructs targeting the opposite subunit (Fig. 2D, E).

Figure 2. Interdependence of SSRP1 and SPT16 levels: RNAi-mediated knock-down (KD) of either SSRP1 or SPT16 leads to reduced levels of both proteins. (A–C) Western blot analysis of SSRP1, SPT16, and β-actin protein levels in lysates of cells with KD of SSRP1 or SPT16: (A) HeLa cells transduced with lentiviral shRNA constructs targeting SSRP1 (three different shRNAs), SPT16 (two different shRNAs), or GFP (control) followed by puromycin selection; (B) HeLa and HT1080 cells transduced with a lentiviral tet-on regulated shRNA construct targeting SSRP1 and treated with doxycycline (Dox) for the indicated periods of time; (C) MCF10A, MCF7 and MBA-MB-231 cells transduced with lentiviral shRNA constructs targeting SSRP1 (#3), SPT16 (#2), or GFP (control) followed by puromycin selection as in (A). (D and E) SSRP1 and SPT16 protein levels are reduced earlier than their corresponding mRNAs upon siRNA-mediated KD of the opposite subunit.(D)RT-PCR (upper three panels) and western blot analysis (lower 3 panels) of SPT16 and SSRP1 mRNA and protein levels, respectively, in HT1080 cells at the indicated times after transfection of control siRNA, siSSRP1, or siSPT16.(E)Levels of SSRP1 and SPT16 mRNA were measured in HeLa cells at different time points after transfection of the indicated siRNAs using qPCR. Mean of 3 replicates +/− standard deviation.
The finding that targeting of either SSRP1 or SPT16 on the mRNA level leads to loss of both proteins suggested that the stability of the SSRP1 and SPT16 proteins may be dependent upon the presence of the opposite subunit and FACT complex formation. To test this possibility, we measured the half-lives of SSRP1 and SPT16 proteins upon RNAi-induced KD of the opposite subunit. Under these conditions, the half-life for both SSRP1 and SPT16 was less than 10 h (as compared with >48 h without specific KD of either subunit) (Fig. 3A and B). The reduced stability of SSRP1 and SPT16 proteins following KD of another subunit appears to involve proteasome-mediated degradation, since bortezomib, an inhibitor of proteasome activity, was found to have a stabilizing effect on SPT16 and to a lesser extent SSRP1 proteins in cells treated with shSSRP1 or shSPT16 correspondingly, but not an irrelevant control shRNA (shGFP) (Fig. 3C).

Figure 3. Stability of the SSRP1 and SPT16 proteins is reduced when expression of mRNA is inhibited via RNAi.(A)Western blot analysis of SPT16, SSRP1, β-actin and p53 protein levels in HeLa cells transduced with shRNAs targeting SPT16, SSRP1 or GFP using cell lysates prepared at the indicated times after addition of 50 μg/ml cycloheximide (CHX). β-actin and p53 were evaluated as examples of proteins with long and short half-lives, respectively.(B)Quantitation of data shown in (A) corresponding to SPT16, and SSRP1 proteins normalized to the intensity of β-actin bands. Normalized intensities of bands at ‘time 0’ were set at 1.0.(C)Western blot analysis of SPT16, SSRP1, and β-actin protein levels in HeLa cells transfected with siRNA against GFP, SSRP1, or SPT16 and treated with 100 nM bortezomib (Btz) for 4 h.
Stability of FACT complex subunits depends on the presence of mRNA
The data described above suggested that loss of FACT expression in vivo (e.g., upon induction of differentiation or senescence), might be due to disruption of the FACT complex leading to reduced stability and enhanced degradation of both subunits, with a particularly strong effect on SPT16. However, a model in which SSRP1 and SPT16 protein levels depend solely upon the opposite subunit protein cannot fully explain the effects of SSRP1 KD: rapid degradation of SSRP1 mRNA accompanied by gradual/moderate loss of SSRP1 protein, and paradoxically, rapid/drastic loss of SPT16 protein (Fig. 2). In this experimental system, the stability of SPT16 protein appears more closely tied to the presence of SSRP1 mRNA than SSRP1 protein. Therefore, we hypothesized that the stability of the SSRP1/SPT16 protein complex depends on the presence of SSRP1 mRNA. To test this hypothesis, we compared the half-lives of SSRP1 and SPT16 proteins in untreated cells with normal endogenous mRNA content and in cells depleted of mRNAs by treatment with the general inhibitor of transcription, actinomycin D. We found that the half-lives of both SSRP1 and SPT16 proteins were reduced under conditions of mRNA depletion to less than 20 h (as compared with more than 48 h in untreated cells) (Fig. 4). Inhibition of RNA synthesis by actinomycin D had a stronger effect on FACT subunit protein levels than inhibition of translation by cyclohexamide and reduced the half-lives of the proteins to nearly the same extent as specific KD of either SSRP1 or SPT16.

Figure 4. Stability of SPT16 and SSRP1 proteins is reduced under conditions of mRNA depletion.(A)Lysates were prepared from HeLa cells treated with 100 ng/ml actinomycin D (ActD) or 50 μg/ml cyclohexamide (CHX) for the amount of time indicated above each lane and analyzed by western blotting with antibodies against SPT16, SSRP1, or β-actin.(B)Quantitation of the intensities of the bands in (A) corresponding to SPT16 and SSRP1 proteins normalized using the intensity of β-actin bands. The normalized intensity at ‘time 0’ was set at 1.0.
The FACT complex contains SSRP1 and SPT16 mRNAs
Based on the data described above, we proposed that the SSRP1 and/or SPT16 mRNA might be a component of the FACT complex, and either facilitate interaction of the protein subunits or maintain complex stability. To determine whether SSRP1 and/or SPT16 mRNAs are present in the FACT complex in vivo, we performed an RNA immunoprecipitation (RIP) experiment in HeLa cells. FACT complexes were immunoprecipitated from HeLa cell lysates using anti-SSRP1 antibody, and RNA that coprecipitated with the complexes was isolated and used as the template for RT-PCR with primers specific for SSRP1, SPT16, or control mRNAs. Since FACT is known to be involved in transcription and might, therefore, bind non-specifically to newly synthesized RNAs, we did not include a crosslinking step before immunoprecipitation (normally used to stabilize nucleic acid-protein binding). This experiment revealed that both SSRP1 and SPT16 mRNAs were present in FACT complexes immunoprecipitated with anti-SSRP1, while 18S rRNA and IL-8 mRNA (used as controls) were not (Fig. 5A). Presence of the SSRP1 and SPT16 mRNAs in the immunoprecipitates was not due to isolation of polypeptide chains bound to ribosome-RNA complexes in the process of translation, since SSRP1 and SPT16 mRNAs also co-precipitated with FACT when nuclear extracts depleted of ribosome-RNA complexes were used instead of total cell lysates (Fig. 5B).
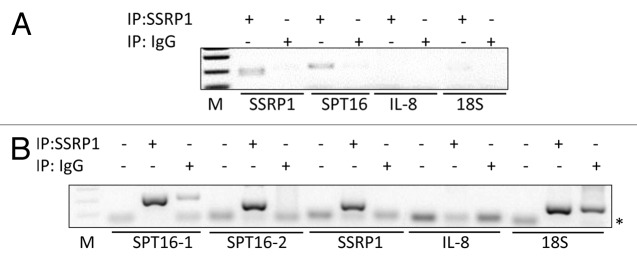
Figure 5. The FACT complex contains SSRP1 and SPT16 mRNAs. HeLa cell lysates were used for immunoprecipitation (IP) with anti-SSRP1 or IgG (non-specific isotype-matched control) antibodies. RNA isolated from the immunoprecipitated complexes was used as the template for RT-PCR with primer sets specific for SSRP1, SPT16, or IL-8 mRNAs or 18s rRNA.(A)Total cell extracts were used for immunoprecipitation.(B)Nuclear cell extracts were used for immunoprecipitation. In (B), ‘no-template’ controls (first lane in each set of three) and two different sets of SPT16 primers (SPT16-1and SPT16-2) were used. *Indicates the position of the primers on the gel. Note that an18S product was detected with both anti-SSRP1- and IgG-immunoprecipitated complexes, suggesting non-specific binding.
SSRP1 mRNA present in the FACT complex is needed to maintain stability of the SSRP1 and SPT16 protein subunits in the intracellular environment
Presence of the SSRP1 and SPT16 mRNAs in FACT complexes suggested that they might play a functional role in promoting interaction of the SSRP1 and SPT16 protein subunits and/or in preserving stability of the complex. To test these possibilities, we first used RNAi to deplete SSRP1 or SPT16 from cells and examined interaction of SSRP1 and SPT16 proteins by immunoprecipitation. If RNA is needed for the binding of two subunits, then we may observe presence of unbound subunits shortly after RNA depletion, while if RNA helps stabilization of the complex we will observe parallel reduction of both subunits in the complex. We observe parallel reduction of both subunits protein levels and no any unbound subunit up to 48 h after siRNA transfection, suggesting that complex formation between SSRP1 and SPT16 proteins was not affected by RNAi-mediated depletion of either SSRP1 or SPT16 mRNA (Fig. 6A and B). These results indicate that neither FACT subunit mRNA is required for formation of a complex between the SSRP1 and SPT16 proteins, which is consistent with demonstration of SSRP1-SPT16 protein-protein binding in the absence of mRNAs in previous in vitro FACT complex reconstitution experiments.3,20,21 Thus, we propose that SSRP1 mRNA is needed to maintain stability of the protein subunits of the complex in the cellular environment.
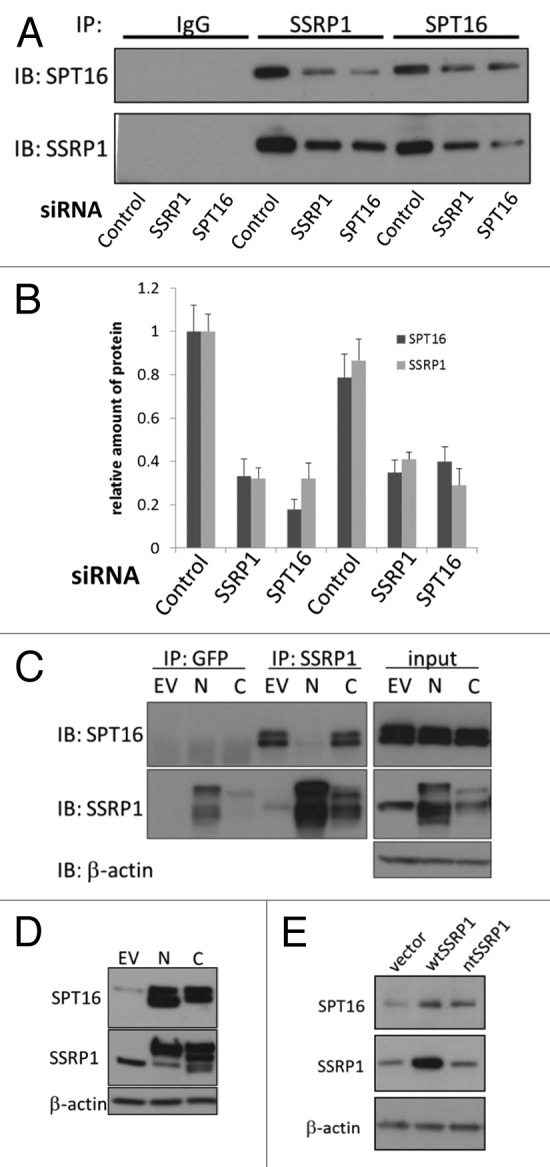
Figure 6. The stabilizing effect of SSRP1 mRNA on SPT16 protein levels does not require direct interaction of the two proteins. (A and B) Reduction of mRNAs of SSRP1 and SPT16 leads to stoichiometric reduction of both proteins in the complex.(A)Western blotting of immunoprecipitates of SSRP1 and SPT16 or control IgG in HeLa cells transfected with the indicated siRNAs.(B)Quantitation of data shown in (A). Band intensities of SSRP1 and SPT16 in control siRNA-transfected cells immunoprecipitated with anti-SSRP1 antibodies were set as 1.0.(C)HeLa cells were transfected with the empty EGFP fusion vector (no protein fused to EGFP, EV), or with constructs producing SSRP1 with EGFP fused to either its N or C terminus (N and C, respectively). At 48 h after transfection, extracts of the cells were used for immunoprecipitation with either GFP or SSRP1 antibodies. Immunoprecipitates were immunoblotted with antibodies, shown on the right. GFP-C-term-SSRP1 was not immunoprecipitated with anti-GFP antibodies (in multiple experiments), although it was precipitated with antiSSRP1. GFP is also visible in these cells.(D)Populations of HeLa cells transduced with the constructs indicated at the top of the figure were expanded and sorted for GFP expression. After establishing and expanding ~100% GFP-positive cell populations (~2 wk), cell extracts were analyzed by western blotting to assess expression of exogenous SSRP1 variants and associated changes in SPT16 protein levels. Note that it was difficult to obtain a stable cell population overexpressing SSRP1 constructs, including wild type SSRP1 (see “Discussion”). However, in all cases in which SSRP1 protein expression was increased after cell expansion, SPT16 protein levels were found to be increased as well. (E) Overexpression of a mutant SSRP1 mRNA that cannot be translated leads to increased SPT16 protein levels. WI38 cells were transduced with either an empty expression vector (vector), or vectors directing expression of the wild type SSRP1 mRNA (wt) or a mutant SSRP1 mRNA with a stop codon inserted after the first methionine (nt). After puromycin selection and expansion of transduced cells, total cell extracts were analyzed by western blotting with the indicated antibodies.
To test this possibility, we evaluated the relative importance of SPT16 protein binding to SSRP1 mRNA vs. SSRP1 protein for stability of the SPT16 protein. This was done by creating a set of GFP-tagged SSRP1 constructs differing in the ability to bind to SPT16 protein due to the presence of GFP on either C or N-termini. HeLa cells were transduced with these different constructs, and 48 h later, used in immunoprecipitation assays to assess SSRP1-SPT16 protein-protein binding (Fig. 6C). In addition, GFP-positive transduced cells were sorted and expanded to allow evaluation of the effect of the overexpressed SSRP1 constructs on endogenous SPT16 protein levels (Fig. 6D). Like wild type SSRP1, recombinant GFP-tagged at the C-terminus (‘C-GFP-SSRP1’) full-length SSRP1 can bind to SPT16 protein because the N-terminal SPT16-binding domain of SSRP1 is preserved (refs.22 and 23 and Fig. 6C). In contrast, the SPT16-binding activity of this domain is eliminated in recombinant full-length SSRP tagged with GFP at the N-terminus (‘N-GFP-SSRP1’) (Fig. 6C). Despite this difference in SPT16 binding, transduction of either tagged construct into HeLa cells resulted in a similar increase in SPT16 protein levels (Fig. 6D). This result indicates that the positive effect of SSRP1 expression on protein levels of SPT16 does not involve binding between the SSRP1 and SPT16 proteins.
To directly test the hypothesis that SSRP1 mRNA, not SSRP1 protein, stabilizes SPT16 protein, we created a construct directing expression of a mutant SSRP1 mRNA that cannot be translated. To achieve this, we modified the full-length SSRP1 mRNA to include a stop codon immediately after the codon for the first methionine of the SSRP1 protein. The mRNA for this mutant is identical to the wild type SSRP1 mRNA except for the presence of the introduced stop codon, but does not produce any protein. When we expressed this SSRP1 mutant into cells, we observed an increase in endogenous SPT16 protein levels similar to that seen in cells transduced with a wild-type SSRP1 expression vector (Fig. 6E). This result unequivocally demonstrates that presence of SSRP1 mRNA is, in itself (without SSRP1 protein), sufficient to stabilize the SPT16 protein.
The stability of SSRP1 and SPT16 mRNAs is enhanced in the presence of FACT complex proteins
Having demonstrated that SSRP1 and SPT16 mRNAs bind to the FACT complex and enhance stability of its protein components, we next investigated whether this association has any effect on the mRNAs themselves. The most likely mechanism by which a nuclear protein complex could affect an mRNA would be through protection of the mRNA from degradation in the nucleus. Although there was no reduction in SSRP1 or SPT16 mRNA levels at short time points (0–48 h) after transfection of siRNAs targeting the opposite subunit (Fig. 2D and E), we designed an experiment to test this at later time points. Cells were transduced with shRNAs targeting SSRP1, SPT16 or GFP (control) and selected in puromycin for 3 d. In these cells, in contrast to cells transiently transfected with siRNAs, we observed reduced levels of SPT16 mRNA following SSRP1 KD and reduced levels of SSRP1 mRNA following SPT16 KD by RT-PCR, quantitative RT-PCR (q-RT-PCR) and Northern hybridization (Fig. 7A–C). While statistically significant, these reductions in mRNA levels were more modest than changes observed in protein levels (SPT16 and SSRP1 mRNA levels were reduced ~60% and ~40%, respectively, in cells with KD of the opposite subunit (Fig. 7C). To determine whether the observed reductions in mRNA levels were due to reduced transcription or enhanced mRNA degradation, we treated shRNA-transduced/puromycin-selected cells with actinomycin D to block transcription and then measured SSRP1 and SPT16 mRNA levels at different time points by Northern hybridization. In control (shGFP-expressing) cells, the half-life of both SSRP1 and SPT16 mRNAs was between 4 and 8 h, but in shSSRP1-expressing cells, the half-life for SPT16 mRNA was reduced to between 2 and 4 h. The half-life of SSRP1 mRNA was not changed upon expression of shSPT16, which is consistent with the generally lesser impact of SPT16 on SSRP1 as compared with the effect of SSRP1 on SPT16 (Fig. 7D).

Figure 7. Stability and translation of SSRP1 and SPT16 mRNAs depends on the presence of the FACT complex. (A–C) Cells were transduced with lentiviral vectors directing expression of the indicated shRNAs and used for the experiments at the indicated times after a short selection with puromycin (3 d). (A)RT-PCR of total RNA from HT1080 cells.(B)Northern hybridization of total RNA from HeLa cells. (C) Quantitative RT-PCR of total RNA from HeLa cells.(D)Northern blot hybridization of total RNA obtained from HeLa cells transduced with the indicated shRNAs at different time points after the start of treatment with actinomycin D (100ng/ml). (E)HeLa cells were transfected with siRNAs targeting GFP, SSRP1, or SPT16 and, 48 h later, incubated for 2 h with 35S-labeled methionine. Cells were then lysed and used for immunoprecipitation (IP) with anti-SSRP1, anti-SPT16, or control (IgG) antibodies.
If the half-life of an mRNA is reduced we should also observe reduction in this mRNA translation. Translation of SPT16 and SSRP1 mRNAs was monitored using 35S metabolic labeling of cells followed by immunoprecipitation. This was compared in HeLa cells transfected with control siRNA (i.e., expressing normal levels of endogenous FACT) or with siRNAs targeting either SSRP1 or SPT16 to deplete the total amount of FACT complex present in the cells. We found that translation of SPT16 was suppressed if the total amount of FACT complex was reduced via siRNA-mediated KD (Fig. 7E). The effect of SPT16 siRNA on SSRP1 translation was not obvious (similar to control SSRP1 band of slightly lower molecular weight appeared in this case as seen in Fig. 7E, insets). These results indicate that SPT16 mRNA is more stable and more readily translated in the presence of FACT complex (i.e., under conditions of association of the SSRP1 and SPT16 mRNAs with the complex).
Discussion
The results of this study provide new information regarding the molecular mechanisms that regulate FACT levels in cells, which is the most basic level of control for FACT activity. The previously described tissue-, differentiation state-, and tumor-related differences in FACT subunit (SSRP1 and SPT16) levels13,14,16 suggest that tight regulation of the complex is likely important for development and health of multicellular organisms. Indeed, FACT has been identified as a prospective target for anticancer treatment13 and prognostic marker of aggressive cancers.14 An improved understanding of endogenous mechanisms controlling FACT levels may enable rational design of strategies for inhibition of FACT as a cancer therapy.
Most functional and biochemical studies of FACT suggest that its two subunits, SSRP1 and SPT16, act as a single chromatin remodeling complex to regulate various nuclear processes including transcription and replication.3,22,24,25 Our studies of SSRP1 and SPT16 mRNA and protein expression also support the notion that the two subunits act as a single unit—levels of the 2 subunits were found to be highly concordant on both the mRNA and protein levels in different cells, tissues and conditions.16 The work reported here shows that this concordance derives from complex reciprocal regulation between SSRP1 and SPT16 mRNAs and proteins.
FACT subunit co-regulation was first indicated by our finding that RNAi-mediated knock-down (KD) of SSRP1 led not only to the expected decreases in SSRP1 mRNA and protein, but also to a dramatic decrease in SPT16 protein levels. SPT16 KD had a similar, although consistently weaker and more gradual, effect on SSRP1 protein levels (Fig. 2). The most obvious explanation for these results was that the stability of each subunit protein depends on presence of the other and, therefore, reduced expression of one via RNAi leads to a decrease in the other. However, this hypothesis was in conflict with the observation that both SSRP1 and SPT16 proteins are extremely stable (Fig. 1F). To explain why an otherwise stable complex disappears rapidly upon reduction of mRNA levels of one of the subunits, we proposed that the mRNA itself plays a role in stabilization of the complex (i.e., the subunit proteins).
Involvement of an mRNA in stabilization of a protein or protein complex is not a widely observed mode of protein regulation; indeed, stabilization of mRNAs by proteins is a much more common phenomenon. Nevertheless, there are several published reports demonstrating that binding of an mRNA to a protein alters protein function or stability. For example, the noncoding 5S rRNA was shown to play a critical role in regulating Mdmx protein stability26 and, in another study, the p53 mRNA was shown to promote stability of the p53 protein through interaction with the p53 negative regulator, Mdm2 protein.27
In this study, we used a number of complementary approaches to test the somewhat unorthodox hypothesis that FACT subunit mRNAs play a role in stability of FACT subunit proteins, and all of the obtained results support the hypothesis. First, using Actinomycin D to block transcription, protein half-life analysis showed that the stability of SSRP1 and SPT16 proteins is dependent upon the presence of RNA (Fig. 4). Second, immunoprecipitation experiments demonstrated specific physical association of SSRP1 and SPT16 mRNAs (but not unrelated RNAs) with FACT complex subunits in living cells (Fig. 5). Third, comparison of the effects of SSRP1 variants either able or unable to bind SPT16 on SPT16 protein levels confirmed that interaction of SSRP1 and SPT16 proteins does not affect SPT16 protein stability (Fig. 6C and D). Finally, the most direct experiment showed that overexpression of an SSRP1 variant containing a stop codon after the first methionine (thus, producing SSRP1 mRNA but no protein) was just as effective as wild type SSRP1 in inducing increased SPT16 protein levels (Fig. 6E). Taken together, these results establish that the mRNAs of both FACT subunits are components of the FACT complex and that stability of FACT subunit proteins is dependent upon their interactions with the mRNAs, not the corresponding proteins.
In all of our experiments, the effect of SSRP1 presence on SPT16 protein levels was notably stronger than the effect of SPT16 on SSRP1 protein levels. In the model of SSRP1-SPT16 co-regulation built from our data (Fig. 8), this identifies SSRP1 mRNA expression as the key driver of the mechanism that controls levels of both FACT subunits. This role for SSRP1 mRNA is supported by findings from our previous analysis of FACT elevation in the process of in vitro transformation: while both SSRP1 and SPT16 were found to be elevated in transformed cells on protein levels, only the mRNA of SSRP1 was increased.14 In line with this is the much higher frequency of SSRP1 mRNA elevation in multiple human tumors as compared with SPT16 mRNA.14 Therefore, a critical next step in this area will be to characterize the promoter of SSRP1 and determine mechanisms controlling transcription of SSRP1 mRNA.

Figure 8. Proposed scheme of the regulation of FACT complex subunits in mammalian cells (see “Discussion” for details).
Through analysis of FACT expression in normal tissues, we established that FACT is expressed at very high levels during embryonic development and in undifferentiated progenitor and stem cells of adult organisms.16 We and others also observed that levels of FACT subunits (particularly SPT16) are quickly and effectively reduced upon induction of differentiation.15-19 In theory, rapid and complete elimination of the activity of a complex can occur if the complex subunits are unstable and regulated at the level of transcription: transcription termination will be quickly followed by degradation of the proteins. However, if such a hypothetical complex is in high demand during a particular stage of development, expression of high levels of unstable protein subunits would be extremely energy consuming. On the other hand, if a complex is very stable, its production might require less energy, but its activity could not be effectively turned off in a rapid manner simply by halting transcription. Our proposed model for regulation of the FACT complex solves the problem of how to minimize energy consumption required for protein production while ensuring that protein levels (and, therefore, activity) can be quickly and effectively reduced. The FACT complex is very stable, but only in the presence of FACT subunit mRNAs. When transcription of the mRNAs is reduced, the stability of the protein complex is significantly reduced and its levels drop rapidly (Fig. 8).
While presence of SSRP1 and SPT16 mRNAs in the FACT complex is beneficial for the stability of the proteins in the complex, the association also has a beneficial effect on the mRNAs themselves. Presence of FACT proteins enhances the stability and efficacy of translation of the mRNAs (Fig. 7). This suggests that when FACT protein levels are low, new FACT complex formation may be hindered due to rapid degradation of FACT mRNAs. This aspect of our model of FACT regulation is supported indirectly by the difficulties we experienced in trying to obtain stable ectopic overexpression of FACT subunits in cells: only a few of our attempts were successful. Limitation of FACT mRNA translation in the absence of FACT proteins may provide an extra ‘security measure’ to ensure that FACT activity remains off when it is not needed (i.e., in normal differentiated cells of adult tissues). The likely importance of this mode of regulation is underscored by the association of FACT re-expression with tumorigenesis.
Our data indicate that the two FACT subunits are expressed and maintained in cells only in concert and as a complex. However, separate functions for the two subunits are described in two publications. The first study analyzed global gene expression profiles in lung adenocarcinoma cells upon inhibition of either SSRP1 or SPT16 expression using shRNA, and found that there is a set of genes that are differentially dependent on either SSRP1 or SPT16 expression.12 The second paper demonstrated that SSRP1, but not SPT16, is involved in regulating spindle formation during mitosis.28 It should be noted, however, that neither of these studies unequivocally established that FACT subunits act independently. The results of the first study might be due to differences in the degree of downregulation of target genes or off-target effects of the shRNAs. The second study, although it did show that only SSRP1 function is needed for proper spindle formation, did not exclude the passive presence of the SPT16 subunit. Neither study evaluated the effect of KD of one FACT subunit on the protein level of the other subunit; however, our own testing of the cells used in these studies showed that they displayed co-regulation similar to all other cells we have tested (data not shown). Therefore, whether only one (as claimed by the reports) or both (as predicted from our results) FACT subunits were actually “targeted” in these studies is unclear.
In summary, we have identified a complex and unusual mechanism of regulation of cellular FACT complex levels that involves interactions of SSRP1 and SPT16 mRNAs and proteins. This mechanism of inter-regulation leads to rapid and effective downregulation of FACT protein levels when FACT mRNA levels decline and provides a “safety mechanism” that hinders production of new FACT subunit proteins when FACT complex levels are low. Our identification of these aspects of FACT regulation will guide future studies including those aimed at mapping interacting sites of the FACT mRNAs and proteins, and determining how expression of SPT16 mRNA and SSRP1 mRNA (the key controller) is regulated, how normal FACT regulatory mechanisms are disrupted in tumor cells, and how FACT can best be targeted for treatment of cancer.
Materials and Methods
Reagents
Etoposide, actinomycin D, cycloheximide, and tamoxifen were purchased from Sigma Aldrich Corp. Bortezomib was obtained from LC Laboratories.
Cell lines
HeLa, HT1080, Wi38, MCF7, MCF10A, and MDA-MB231 cell lines were purchased from ATCC and maintained according to instructions from ATCC. C2C12 mouse myoblasts were kindly provided by Dr Asoke Mal (Roswell Park Cancer Institute). C2C12 cells were maintained for 48 h before differentiation in DMEM supplemented with 20% FBS; differentiation was induced by changing the medium to DMEM containing 2% horse serum (Invitrogen, Life Science Technologies). Primary human BJ fibroblasts expressing 4-OH-tamoxifen (4-OHT)-inducible oncogenic H-RasV12 (BJ-RasV12ER cells) were obtained from Dr Reuven Agami (The Netherlands Cancer Institute).
Vectors and expression constructs
Mission shRNA lentiviral vectors were purchased from Sigma Aldrich Corp (http://www.sigmaaldrich.com/life-science/functional-genomics-and-rnai/shrna/shrna-search-and-order.html) and included: shControl-shRNA against GFP; shRNAs against SSRP1: shSSRP1–2 (#2 in Fig. 2A); SHGLYC-TRCN0000019270, shSSRP1–4 (#1 in Fig. 2A); SHGLYC-TRCN0000019272 and shSSRP1–5 (#3 in Fig. 2A); SHGLYC-TRCN0000019273; and shRNAs against SPT16: shSPT16–3 (#2 in Fig. 2A); SHGLYC-TRCN0000001259, and shSPT16–4 (#1 in Fig. 2A) SHGLYC-TRCN0000001260. Unless otherwise indicated, shSSRP1–2 and shSPT16-4 were used. N-GFP-SSRP1 construct was previously described.13 C-GFP-SSRP1 was generated through subcloning of full length human SSRP1 lacking stop codon in frame with EGFP into the same pLM-CMV lentiviral vector (kind gift of Peter Chumakov, Cleveland Clinic). Non-translatable SSRP1 was generated from the pReceiver-Lv105-SSRP1 construct (Genecopeia; http://www.genecopoeia.com/product/search/detail.php?prt=1&cid=&key=B0096) by introducing a mutation encoding a stop codon after the start codon of SSRP1 using Quick Change Lightning Site-Directed Mutagenesis kit (Agilent Technologies; http://www.genomics.agilent.com/en/Site-Directed-Mutagenesis/QuikChange-Lightning/?cid=AG-PT-175&tabId=AG-PR-1162). The primers used for mutagenesis were forward: 5′–AAA GCA GGC TCC ACC ATC TAG GAG ACA CTG GAG TTC-3′ and reverse: 5′–GAA CTC CAG TGT CTC CTA GAT GGT GGA GCC TGC TTT-3′).
siRNA transfection
Transfection of HT1080 cells with siRNAs was performed using Lipofectamine 2000 following the manufacturer’s protocol (Invitrogen Corp). siRNAs targeting SSRP1(On-Target plus SMART pool, cat# L-011783-00) and SPT16 (On-Target plus SMART pool, cat# L-009517-00) and siCONTROL non-targeting siRNA (cat# D-001210-01) were purchased from Thermo Scientific Dharmacon (http://www.thermoscientificbio.com/rnai-and-custom-rna-synthesis/sirna/on-targetplus-sirna/search-gene/). Six hours after transfection, cells were seeded into new plates for use in experiments.
Western blot
Protein extracts were prepared by lysing cells in Cell Culture Lysis Reagent (Promega). Protein concentrations were determined with the Bio-Rad Dc protein assay kit. Equal protein amounts were run on 4–20% gradient precast gels (Invitrogen Corp) and transferred to Immobilon-P membranes (Millipore Corp). The membranes were blocked in 5% nonfat milk-TBS-T buffer and incubated with primary antibodies for 1.5 h at room temperature. The following antibodies were used: anti-p53 (monoclonal mouse DO1, cat# sc-126, Santa Cruz Biotechnologies; http://www.scbt.com/datasheet-126-p53-do-1-antibody.html), anti-SSRP1 (mouse monoclonal 10D1, cat# 609702, BioLegend, Inc; http://www.biolegend.com/purified-anti-ssrp1-antibody-1978.html), and anti-Spt16 (monoclonal mouse 8D2, cat# 607002, BioLegend, Inc; http://www.biolegend.com/purified-anti-spt16-fact140-complex-antibody-2003.html). To verify equal protein loading and transfer, anti-β-actin (Sigma) antibodies were used. Anti-MHC (myosin heavy chain) antibody was kindly provided by Asoke Mal (Roswell Park Cancer Institute) and was used to confirm myotube differentiation of C2C12 myoblasts. Horseradish peroxidase-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology. Quantitation of the data was performed using QuantityOne® software from Bio-Rad Laboratories, Inc or Image J software from NCI.
Northern blot
Total RNA was extracted from cells using TRIzol (Invitrogen). Five μg of RNA was separated by electrophoresis on a 1% agarose-formaldehyde gel and blotted onto a Hybond N+ membrane (Amersham Biosciences). The membrane was dried, and RNA was UV-crosslinked. Membranes were hybridized for 16 h with specific [32P]-labeled probes: 600 bp oligonucletide probes for SSRP1, SPT16 and tubulin were generated using PCR with specific primers. The DNA oligonucleotides were labeled using a Random primed DNA labeling kit (Roshe) in the presence of [α-32P]dCTP. After hybridization, the filters were washed with 3 changes (5 min each) of 2 × SSC, 0.1% SDS at 55 °C, and 3 changes (5 min each) of 1 × SSC, 0.1% SDS at 55 °C. Membranes were exposed to X-ray film for 16 h at −80 °C
RT-PCR
Total RNA was isolated by TRIzol reagent according to the manufacturer’s protocol. First-strand cDNA was synthesized from 2 µg total RNA using the iScript cDNA Synthesis kit (Bio-Rad; http://www.bio-rad.com/prd/en/CA/LSR/PDP/M87EWZESH/iScript-cDNA-Synthesis-Kit) in a 40-µl reaction according to the manufacturer’s protocol. PCR was performed using 2 µl of first-strand cDNA and the AccuPrime Pfx DNA Polymerase kit (Invitrogen Corp; http://www.invitrogen.com/site/us/en/home/References/protocols/nucleic-acid-amplification-and-expression-profiling/pcr-protocol/accuprime-pfx-dna-polymerase.html) with conditions of 95 °C for 2 min followed by 21 cycles of 95 °C for 30 sec, 50 °C for 30 sec and 72 °C for 30 sec. Primer pairs included: SPT16 (forward: 5′-GTGGAAAAGGCCATTGAAGA; reverse: 5′-GTGATAGCCCCAAAGTGCAT), SSRP1 (forward: 5′-AGGCAAGAATGAGGTGACA; reverse: 5′-TACATCCGCCTTTGACAACA), and GAPDH (forward: 5′-GCCTTCCGTGTCCCCACTGC; reverse: 5′-GGCTGGTGGTCCAGGGGTCT).
Quantitative RT-PCR
Total RNA and first-strand cDNA was synthesized as described above for standard RT-PCR. Quantitative real-time RT-PCR (qRT-PCR) was performed using 1 µl of first-strand cDNA with primers purchased from Applied Biosystems: SSRP1-Hs00172629_m1, SUPT16H (SPT16)-Hs00200446_m1, and 18S-Hs99999901_s1. qRT-PCR was performed using TaqMan gene Expression Master Mix (Applied Biosystems; http://www.invitrogen.com/site/us/en/home/brands/Applied-Biosystems.html) and the default parameters of the 7900HT sequence detection system (ABI PRISM; Applied Biosystems). To compare gene expression between samples, the threshold cycle (CT) value was normalized using the mean CT for the reference gene, 18S rRNA (rRNA). The normalized mRNA level was defined as ΔCT = CT (mean for test gene) − CT (mean for the reference gene). All reactions were performed in triplicate, and the experiments were repeated at least twice. The results are presented as the mean of at least 2 experiments.
RNA immunoprecipitation
HeLa cells were lysed in cold NP40 lysis buffer containing protease inhibitors for 5 min on ice (producing ‘total cell extract’) and then centrifuged at 1000 rpm for 10 min. The nuclear pellet was lysed in RIP buffer (20 mM Hepes, pH 7.5, 150 mM NaCl, 2.5 mM MgCl2, 250 mM sucrose, 0.05% NP40, 0.5% Triton X-100) containing RNASIN (Promega), 1 mM DTT and protease inhibitors, on ice for 15 min (producing ‘nuclear extract’). Total cell extracts or nuclear extracts were pre-cleared by rotation with protein A/G agarose (equilibrated in RIP buffer) in the presence of yeast tRNA (30 μg per 1 ml) at 4 °C for 1 h. Pre-cleared lysates were incubated with anti-SSRP1 or mouse IgGb2 (non-specific isotype-matched control) antibodies overnight at 4 °C, followed by 2 h incubation with protein A/G agarose. The immunoprecipitates were washed with RIP buffer 5 times and RNA was isolated by phenol/chloroform/isoamyl alcohol extraction and ethanol precipitation. The isolated RNA was DNase-treated and reverse-transcribed using an iScript cDNA Synthesis kit (Bio-Rad). PCR was performed using the AccuPrime Pfx DNA Polymerase kit (Invitrogen) and conditions of 95 °C for 2 min, 30 cycles of 95 °C for 30 sec, 55 °C for 30 sec and 72 o C for 30 sec. Primer pairs were as follows: SPT16 (SPT16-1 forward:5′-AGAAGCAAAGAGGCGATTGA; reverse: 5′-GATTGTGGCAATGTGAAACG), SPT16-2 (forward: 5′-GTGGAAAAGGCCATTGAAGA; reverse: 5′-GTGATAGCCCCAAAGTGCAT), SSRP1 (forward: 5′-CAGGCAAGAATGAGGTGACA; reverse: 5′-TACATCCGCCTTTGACAACA), 18S (forward: 5′-AAACGGCTACCACATCCAAG; reverse: 5′-CCTCCAATGGATCCTCGTTA), and IL8 (forward: 5′-GCAAGGTAACTCAGACAATTCCA; reverse: 5′-GCAAGCTAAGACTCTCCAGCA).
Immunoprecipitation
Cells were lysed in NP40 buffer, rotating at 4 °C for 15 min. Lysates were then centifuged at 14 000 × g for 15 min and supernatants were pre-cleared by rotating with protein A/G agarose beads at 4°C for 1 h. Cell lysates at 1 mg/ml were rotated overnight at 4 °C with anti- SSRP1 (cat# 609702, BioLegend, Inc, 5 μg antibody per 500 μl lysate). To capture immunocomplexes, lysates were rotated with protein A/G agarose beads for 1 h at 4 °C. Agarose beads were collected by centrifugation, washed 3 times with cold PBS and boiled in 60 μl of 2× loading buffer for 5 min. Supernatants were used for SDS-PAGE followed by autoradiography.
Metabolic labeling and immunoprecipitation
For metabolic labeling, cells were washed with methionine-free DMEM supplemented with 10% dialyzed FCS and 2mM glutamine and kept in this medium for 1 h before addition of 35[S]-methionine (50 μCi/ml, Perkin Elmer, cat# NEG772002MC). After 2 h of incubation, the 35[S]-methionine-containing medium was removed and cells were lysed in NP40 buffer with protease inhibitors. After rotation for 20 min at 4 °C, the extracts were centrifuged for 5 min at 13 000 rpm. Supernatants were used for immunoprecipitation. The extracts were incubated with SSRP1, SPT16, or IgGb2 antibodies from Biolegend (5 μg antibody per 500 μl of lysate at protein concentration of 1 mg/ml) overnight at 4 °C. Immunocomplexes were collected and analyzed as described above for “Immunoprecipitation”.
Acknowledgments
We would like to thank Bruce Specht for his constant enormous support of our research work; Dr Patricia Stanhope Baker for critical reading and editing of the manuscript; Dr Andrei Gudkov for critical discussion of the study. We also thank Dr Asoke Mal (Roswell Park Cancer Institute), and Dr Reuven Agami (The Netherlands Cancer Institute) for providing cells used in this work. This study was partially funded by a grant to KVG from Incuron, Inc.
Glossary
Abbreviations:
- FACT
facilitates chromatin transcription
- SSRP1
structure specific recognition protein 1
- SPT16
suppressor of Ty 16
- RNAi
RNA interference
- KD
knock down
- shRNA
small hairpin RNA
- siRNA
small interfering RNA
- CHX
cycloheximide
- ActD
actinomycin D
- BZM
bortezomib
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grant from INCURON, Inc to KVG.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25452
REFERENCES
- 1.Saunders A, Werner J, Andrulis ED, Nakayama T, Hirose S, Reinberg D, et al. Tracking FACT and the RNA polymerase II elongation complex through chromatin in vivo. Science. 2003;301:1094–6. doi: 10.1126/science.1085712. [DOI] [PubMed] [Google Scholar]
- 2.Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003;301:1090–3. doi: 10.1126/science.1085703. [DOI] [PubMed] [Google Scholar]
- 3.Birch JL, Tan BC, Panov KI, Panova TB, Andersen JS, Owen-Hughes TA, et al. FACT facilitates chromatin transcription by RNA polymerases I and III. EMBO J. 2009;28:854–65. doi: 10.1038/emboj.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan BC, Chien CT, Hirose S, Lee SC. Functional cooperation between FACT and MCM helicase facilitates initiation of chromatin DNA replication. EMBO J. 2006;25:3975–85. doi: 10.1038/sj.emboj.7601271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tan BC, Liu H, Lin CL, Lee SC. Functional cooperation between FACT and MCM is coordinated with cell cycle and differential complex formation. J Biomed Sci. 2010;17:11. doi: 10.1186/1423-0127-17-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Y, Wang TS. A coordinated temporal interplay of nucleosome reorganization factor, sister chromatin cohesion factor, and DNA polymerase alpha facilitates DNA replication. Mol Cell Biol. 2004;24:9568–79. doi: 10.1128/MCB.24.21.9568-9579.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumari A, Mazina OM, Shinde U, Mazin AV, Lu H. A role for SSRP1 in recombination-mediated DNA damage response. J Cell Biochem. 2009;108:508–18. doi: 10.1002/jcb.22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heo K, Kim H, Choi SH, Choi J, Kim K, Gu J, et al. FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol Cell. 2008;30:86–97. doi: 10.1016/j.molcel.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 9.Keller DM, Zeng X, Wang Y, Zhang QH, Kapoor M, Shu H, et al. A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Mol Cell. 2001;7:283–92. doi: 10.1016/S1097-2765(01)00176-9. [DOI] [PubMed] [Google Scholar]
- 10.Ikeda Y, Kinoshita Y, Susaki D, Ikeda Y, Iwano M, Takayama S, et al. HMG domain containing SSRP1 is required for DNA demethylation and genomic imprinting in Arabidopsis. Dev Cell. 2011;21:589–96. doi: 10.1016/j.devcel.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 11.Jimeno-González S, Gómez-Herreros F, Alepuz PM, Chávez S. A gene-specific requirement for FACT during transcription is related to the chromatin organization of the transcribed region. Mol Cell Biol. 2006;26:8710–21. doi: 10.1128/MCB.01129-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Zeng SX, Landais I, Lu H. Human SSRP1 has Spt16-dependent and -independent roles in gene transcription. J Biol Chem. 2007;282:6936–45. doi: 10.1074/jbc.M603822200. [DOI] [PubMed] [Google Scholar]
- 13.Gasparian AV, Burkhart CA, Purmal AA, Brodsky L, Pal M, Saranadasa M, et al. Curaxins: anticancer compounds that simultaneously suppress NF-κB and activate p53 by targeting FACT. Sci Transl Med. 2011;3:95ra74. doi: 10.1126/scitranslmed.3002530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia H, Miecznikowski JC, Safina A, Commane M, Ruusulehto A, Kilpinen S, et al. FACT is an “accelerator” of tumor transformation and potential marker and target of aggressive cancers. Cell Reports. 2013 doi: 10.1016/j.celrep.2013.06.013. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duroux M, Houben A, Růzicka K, Friml J, Grasser KD. The chromatin remodelling complex FACT associates with actively transcribed regions of the Arabidopsis genome. Plant J. 2004;40:660–71. doi: 10.1111/j.1365-313X.2004.02242.x. [DOI] [PubMed] [Google Scholar]
- 16.Garcia H, Fleyshman D, Kolesnikova K, Safina A, Commane M, Paszkiewicz G, et al. Expression of FACT in mammalian tissues suggests its role in maintaining of undifferentiated state of cells. Oncotarget. 2011;2:783–96. doi: 10.18632/oncotarget.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lolas IB, Himanen K, Grønlund JT, Lynggaard C, Houben A, Melzer M, et al. The transcript elongation factor FACT affects Arabidopsis vegetative and reproductive development and genetically interacts with HUB1/2. Plant J. 2010;61:686–97. doi: 10.1111/j.1365-313X.2009.04096.x. [DOI] [PubMed] [Google Scholar]
- 18.Hertel L, De Andrea M, Bellomo G, Santoro P, Landolfo S, Gariglio M. The HMG protein T160 colocalizes with DNA replication foci and is down-regulated during cell differentiation. Exp Cell Res. 1999;250:313–28. doi: 10.1006/excr.1999.4495. [DOI] [PubMed] [Google Scholar]
- 19.Hertel L, Foresta P, Barbiero G, Ying GG, Bonelli G, Baccino FM, et al. Decreased expression of the high-mobility group protein T160 by antisense RNA impairs the growth of mouse fibroblasts. Biochimie. 1997;79:717–23. doi: 10.1016/S0300-9084(97)86929-5. [DOI] [PubMed] [Google Scholar]
- 20.Tsunaka Y, Toga J, Yamaguchi H, Tate S, Hirose S, Morikawa K. Phosphorylated intrinsically disordered region of FACT masks its nucleosomal DNA binding elements. J Biol Chem. 2009;284:24610–21. doi: 10.1074/jbc.M109.001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winkler DD, Muthurajan UM, Hieb AR, Luger K. Histone chaperone FACT coordinates nucleosome interaction through multiple synergistic binding events. J Biol Chem. 2011;286:41883–92. doi: 10.1074/jbc.M111.301465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singer RA, Johnston GC. The FACT chromatin modulator: genetic and structure/function relationships. Biochem Cell Biol. 2004;82:419–27. doi: 10.1139/o04-050. [DOI] [PubMed] [Google Scholar]
- 23.Winkler DD, Luger K. The histone chaperone FACT: structural insights and mechanisms for nucleosome reorganization. J Biol Chem. 2011;286:18369–74. doi: 10.1074/jbc.R110.180778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orphanides G, Wu WH, Lane WS, Hampsey M, Reinberg D. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature. 1999;400:284–8. doi: 10.1038/22350. [DOI] [PubMed] [Google Scholar]
- 25.Reinberg D, Sims RJ., 3rd de FACTo nucleosome dynamics. J Biol Chem. 2006;281:23297–301. doi: 10.1074/jbc.R600007200. [DOI] [PubMed] [Google Scholar]
- 26.Li M, Gu W. A critical role for noncoding 5S rRNA in regulating Mdmx stability. Mol Cell. 2011;43:1023–32. doi: 10.1016/j.molcel.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Candeias MM, Malbert-Colas L, Powell DJ, Daskalogianni C, Maslon MM, Naski N, et al. P53 mRNA controls p53 activity by managing Mdm2 functions. Nat Cell Biol. 2008;10:1098–105. doi: 10.1038/ncb1770. [DOI] [PubMed] [Google Scholar]
- 28.Zeng SX, Li Y, Jin Y, Zhang Q, Keller DM, McQuaw CM, et al. Structure-specific recognition protein 1 facilitates microtubule growth and bundling required for mitosis. Mol Cell Biol. 2010;30:935–47. doi: 10.1128/MCB.01379-09. [DOI] [PMC free article] [PubMed] [Google Scholar]