

Abstract
Aims
Endothelial cells (ECs) control vascular permeability by forming a monolayer that is sealed by extracellular junctions. Various mediators modulate the endothelial barrier by acting on junctional protein complexes and the therewith connected F-actin cytoskeleton. Different Rho GTPases participate in this modulation, but their mechanisms are still partly resolved. Here, we aimed to elucidate whether the opening and closure of the endothelial barrier are associated with distinct localized RhoA activities at the subcellular level.
Methods and results
Live fluorescence resonance energy transfer (FRET) microscopy revealed spatially distinct RhoA activities associated with different aspects of the regulation of endothelial monolayer integrity. Unstimulated ECs were characterized by hotspots of RhoA activity at their periphery. Thrombin receptor activation in the femoral vein of male wistar rats and in cultured ECs enhanced RhoA activity at membrane protrusions, followed by a more sustained RhoA activity associated with cytoplasmic F-actin filaments, where prolonged RhoA activity coincided with cellular contractility. Unexpectedly, thrombin-induced peripheral RhoA hotspots were not spatially correlated to the formation of large inter-endothelial gaps. Rather, spontaneous RhoA activity at membrane protrusions coincided with the closure of inter-endothelial gaps. Electrical impedance measurements showed that RhoA signalling is essential for this protrusive activity and maintenance of barrier restoration.
Conclusion
Spontaneous RhoA activity at membrane protrusions is spatially associated with closure, but not formation of inter-endothelial gaps, whereas RhoA activity at distant contractile filaments contributes to thrombin-induced disruption of junctional integrity. Thus, these data indicate that distinct RhoA activities are associated with disruption and re-annealing of endothelial junctions.
Keywords: Rho GTPase, FRET microscopy, Cytoskeleton, Endothelial function, Vasoactive agents
1. Introduction
Endothelial cells (ECs) form a barrier between blood and surrounding tissue and actively regulate the exchange of nutrients and proteins between these compartments.1,2 During inflammation, exposure to ischaemia or angiogenic stimuli, endothelial cell–cell interactions become loosened resulting in the generation of intercellular gaps that allow uncontrolled paracellular passage of fluids, solutes, and proteins.1 Although vascular leakage has been associated with many diseases,3 no curative treatments are available.
The endothelial barrier function is regulated at the level of junctional proteins, which are connected to the F-actin cytoskeleton via protein complexes.1,2 In addition, in vivo the glycocalyx forms part of the endothelial barrier to macromolecules.4 Among several regulators, the Rho GTPase family members Cdc42, Rac, and RhoA play a critical role in regulating permeability of the endothelial barrier under basal conditions and in response to external stimuli through the regulation of adhesive and contractile forces.2,5–8 These Rho GTPases modulate the organization and dynamics of the actin cytoskeleton by activating downstream signalling that induces the protruding activity of lamella during initiation and expansion of cell–cell contacts or alternatively that mediate cell contraction in response to endothelial permeability-increasing mediators.2 Rac1 and Cdc42 are generally thought to orchestrate the spatial control of filopodia and lamellipodia contributing to barrier improvement, whereas RhoA facilitates the organization of F-actin fibres and causes barrier disruption.2 Recent studies, however, suggest a more complex involvement of Rho GTPases in positive and negative barrier regulation.9
Over the past decade, the small GTPase RhoA has been implicated as a potent regulator of endothelial barrier function. Activation of RhoA by vasoactive substances such as endotoxin, VEGF, and thrombin results in a loss of endothelial barrier integrity both in vitro10 and in vivo.5,11,12 This RhoA activation might be preceded by a decrease in Rac1 activity.8 Furthermore, as a key modulator of the F-actin cytoskeleton RhoA plays a major role in fundamental processes such as cell motility, cell shape, membrane trafficking, ruffling, and cell contraction.13 Organization of the actin cytoskeleton is also an important determinant of junction stability. RhoA through its targets including Rho kinase induces the phosphorylation of myosin light chain at the F-actin filaments resulting in enhanced actomyosin interaction and stress fibre (SF) formation.10 The resulting increase in junctional tension promotes junctional disassembly and loss of endothelial barrier integrity. In apparent contrast to its classical barrier-disruptive effects, RhoA-mediated signalling also has barrier-protective effects, as was evidenced by macromolecule permeability assays.7 Such opposing activities provide a potential obstacle for future vascular leakage therapy aimed at inhibiting RhoA signalling to improve conditions of increased permeability.
In ECs, activation of RhoA by thrombin or VEGF is accompanied by an increase in membrane-associated RhoA and a decrease in cytosolic RhoA as evidenced by cell-fractionation assays.14,15 Furthermore, upon stimulation with thrombin, a shift in localization of the phosphorylation of MYPT1, a surrogate marker for RhoA/Rho kinase activity from cell–cell junctional regions in non-stimulated conditions towards F-actin SFs was observed.7 These data suggested that activation of RhoA can occur at multiple intracellular locations and that the intracellular localization of active RhoA determines its biological effect. Indeed, evidence that Rho GTPase activation is highly spatially and temporally regulated16–19 has been provided by studies using fluorescent biosensors in migrating cells. For example, specific interference with localized activation of RhoA at the leading edge by knockdown of RhoGEF-H1 expression in HeLa cells prevented directional migration.17 At the rear of a migrating cell, RhoA mediates the contractile response, that is, necessary for tail retraction, whereas at the front of a migrating cell, RhoA is associated with protrusive activity.18,19 However, such spatio-temporal information is not available under confluent conditions, where ECs are non-polarized in the x–y plane, and most of what we know about Rho GTPase signalling is based on ‘classic’ Rho GTPase tools that do not faithfully reflect this level of regulation.20 Therefore, a key question that still remains to be investigated is, whether a spatial association exists between local RhoA activation and AJ disruption followed by inter-endothelial gap formation or whether RhoA acts more indirectly at sites remote from newly formed gaps.
The aim of the present study was to elucidate how the different subcellular localizations of RhoA activation are associated with the regulation of endothelial monolayer integrity. We hypothesized that basal RhoA activity would be suppressed in mature endothelial junctions favouring barrier stabilization, but that upon stimulation with thrombin, RhoA activity at the cell periphery would result in a localized contractile process resulting in disruption of endothelial junctions and the formation of inter-endothelial gaps. To this end, we studied the spatio-temporal dynamics of RhoA activation in confluent EC monolayers utilizing fluorescence resonance energy transfer (FRET) RhoA biosensors.21
2. Methods
2.1. General
Sources of reagents, use of rat femoral veins and human umbilical vein endothelial cells (HUVECs), and detailed description of the methods can be found in the Supplementary material online, Materials and Methods. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996), with the principles outlined in the Declaration of Helsinki, and was undertaken with approval of the local ethics committee (DEC/dierexperimentencommissie) regarding animal and human research. Male Wistar rats (n = 6, 300–360 g; Harlan, Horst, The Netherlands) were anaesthetized with pentobarbital (ip 60 mg/kg) and ketamine HCl (im 70 mg/kg) with a pentobarbital maintenance dose (ip 30 min/15 mg/kg). Adequacy of anaesthesia was monitored by continuous measurement of mean arterial pressures (100–120 mmHg), and heart rates (340–380 bpm) via a catheter in the carotid artery. At the end of the experiment, 4% formaldehyde (Sigma) in saline was infused during 10 min for fixation of the veins.
Umbilical cords were obtained from The Department of Obstetrics of the Amstelland Hospital in Amstelveen, The Netherlands. ECs were cultured on gelatin-coated dishes and 1 h before the experiments started, growth factors, and serum were withdrawn from the cultures and replaced by 1% human serum albumin.11
2.2. FRET imaging
To measure RhoA activity, FRET imaging was performed as previously described21 with some minor modifications described below. Using Amaxa Nucleofector technology, HUVECs were transfected with the probes Raichu-RhoA, Raichu-rho-binding domain (RBD)-X, or Raichu-RhoA/KRas-CT, whereas Raichu is short for ‘Ras and interacting protein chimeric unit’. These probes were a kind gift of Prof M. Matsuda, Osaka University, Japan.
Raichu-RhoA contains truncated RhoA as well as a RBD located between the fluorogenic groups YFP and CFP. In addition, an authentic RhoA CAAX box is fused to CFP. Activation of RhoA results in a FRET signal, enabling the monitoring of the balance between rhoGEF and rhoGAP activities at the subcellular level and thus serving as a surrogate marker for RhoA activation. Alternatively, Raichu-RhoA/KRas-CT was used. This is essentially the same construct as Raichu-RhoA, but this probe is located preferentially at the plasma membrane due to the carboxyl terminal region of K-Ras4B instead of the RhoA tail. This selective location improves the signal-to-noise ratio at the plasma membrane. For imaging of endogenous RhoA, Raichu-RBD-X was used, which lacks the RhoA-fragment between the two fluorophores, and, therefore, is useful to visualize the activation of endogenous RhoA. However, Raichu-RBD-X is an ‘inverse’ FRET probe. It presents with increased FRET intensity when endogenous RhoA is inactive, meaning that RhoA activation is seen as a decrease in FRET intensity. Raichu-RBD-X thus gives a less favourable signal-to-noise ratio when compared with Raichu-RhoA, but in contrast to Raichu-RhoA, also reflects the contribution of rhoGDI in the regulation of RhoA activity.21
During three-channel FRET analysis, RhoA activity was determined as the calculated FRET intensity corrected for the amount of probe present (FRETc/A). Three-channel FRET microscopy was performed with a Zeiss Axiovert 200 Marianas™ inverted microscope under the control of the Slidebook software. Briefly, RhoA activity was followed for 15 min with interval times of 30 s. The time point of thrombin stimulation (1 U/mL) is indicated per experiment.
Alternatively, FRET was analysed by photobleaching of the acceptor molecule in fixed cells as was described in the Supplementary material online, Materials and Methods section. This type of FRET is less prone to artefacts caused by bleed-through of light from one channel to another.
2.3. ECIS measurements
Electrical impedance measurements were performed as described by Opp et al.22 using the electrical cell-substrate impedance sensing system (ECIS, Applied BioPhysics, Troy, NY, USA). ECs were seeded on gelatin-coated 8W1E gold-electrodes and impedance of the confluent cell layer was measured in the presence or absence of the indicated inhibitors. Different properties of the impedance signal were used to characterize barrier integrity23 and micromotion of the ECs.
2.4. In vivo imaging of RhoA activity
Thrombin receptor activating peptide (TRAP) was infused according to Chintala et al.24 in a cannulated femoral vein. Veins were perfusion-fixed, permeabilized, dissected, and stained for F-actin and active RhoA using GST-tagged Rho-GTP-binding domains (RBDs).25
2.5. Statistical analysis
All data presented are representative for three to five independent experiments, unless otherwise indicated. For basic statistical analysis, the SPSS software was used. Data were compared by Student's t-test. P-values of <0.05 were considered to be significant.
3. Results
3.1. Spontaneous and thrombin-induced formation and closure of inter-endothelial gaps
To study the dynamics of the regulation of endothelial monolayer integrity, confluent ECs were imaged by differential interference contrast (DIC) microscopy. DIC time-lapse imaging revealed a continuous remodelling of cell–cell interactions under basal conditions. Remarkably, many tiny inter-endothelial gaps appeared and disappeared spontaneously (Figure 1A), with an average opening time of <3 min (see also Table 2). After thrombin (1 U/mL) stimulation, much larger gaps formed in the monolayer, which often persisted for >1 h (Figure 1B). Similarly, these gaps closed through dynamic protrusive membrane activities.
Figure 1.

Spontaneous and thrombin-induced formation and closure of inter-endothelial gaps. Live cell imaging of endothelial monolayers was performed by DIC Microscopy. Images were taken every 10 s. HUVECs were cultured on gelatin-coated ibidi µ-slides and grown to confluence for 48 h. The smaller images present the enlargements of the white frames at different time points. For identification, outlines of individual ECs are shown on the right. Areas coloured in black present inter-endothelial gaps. (A) DIC time-lapse imaging shows the formation of tiny inter-endothelial gaps (arrow) in basal, post-confluent EC layers. These gaps form and close spontaneously, with an average opening time of 168 s. Black arrows point to distinct gaps. (B) The addition of thrombin (1 U/mL, at 00:01:40) to the confluent EC layer stimulates cell contraction, which finally results in the formation of large inter-endothelial gaps at multiple sides of the cell that have prolonged opening times of over 30 min. After stimulation, the cells actively counteract the barrier opening through dynamic protrusive membrane structures (such as lamellipodia, ruffles, waves, and protrusions) that finally fill the gap.
Table 2.
Long-time inhibition of Rho kinase leads to prolonged endothelial gap opening
| Gap opening timea | Basal | Y-27632b |
|---|---|---|
| 10–99 s | 39 | 8 |
| 100–999 s | 15 | 12 |
| >1000 s | 4 | 2 |
| n | 58 | 22 |
| Mean ± SDc | 168 ± 354 s | 349 ± 458 s |
aInter-endothelial gaps were categorized in three groups by their opening time.
bRho kinase was blocked for 24 h by incubation with Y-27632 (10 µM).
cMean and standard deviation of the exact opening times per condition.
Thus, junctional remodelling in endothelial monolayers is a continuous and dynamic process. Inter-endothelial gaps form and close either spontaneously or upon stimulation with thrombin, but under the latter conditions, gaps are bigger and have prolonged opening times.
3.2. Subcellular distribution of RhoA activity in baseline and thrombin-stimulated ECs
To investigate the spatio-temporal relationship between RhoA activity and the dynamics of endothelial monolayer integrity, RhoA activity was visualized with a high temporal and spacial resolution through live cell FRET imaging using a Raichu-RhoA biosensor. This biosensor was used first to determine the subcellular distribution of RhoA activity under basal as well as under thrombin-stimulated conditions, to study RhoA activity associated with F-actin filaments, and finally to assess the role of RhoA activity in the dynamics of inter-endothelial gaps.
Post-confluent, growth factor-deprived ECs showed a low level of RhoA activity, which was homogenously distributed throughout the cell. Some accumulations of RhoA were found in peri-nuclear areas (Figure 2A) and in membrane protrusions (arrows), otherwise, RhoA activity was low at the cell–cell contact sites. Imaging of subconfluent ECs confirmed that RhoA activity was repressed where the adjacent cells had been connected through newly formed contacts (Supplementary material online, Movie S1).
Figure 2.
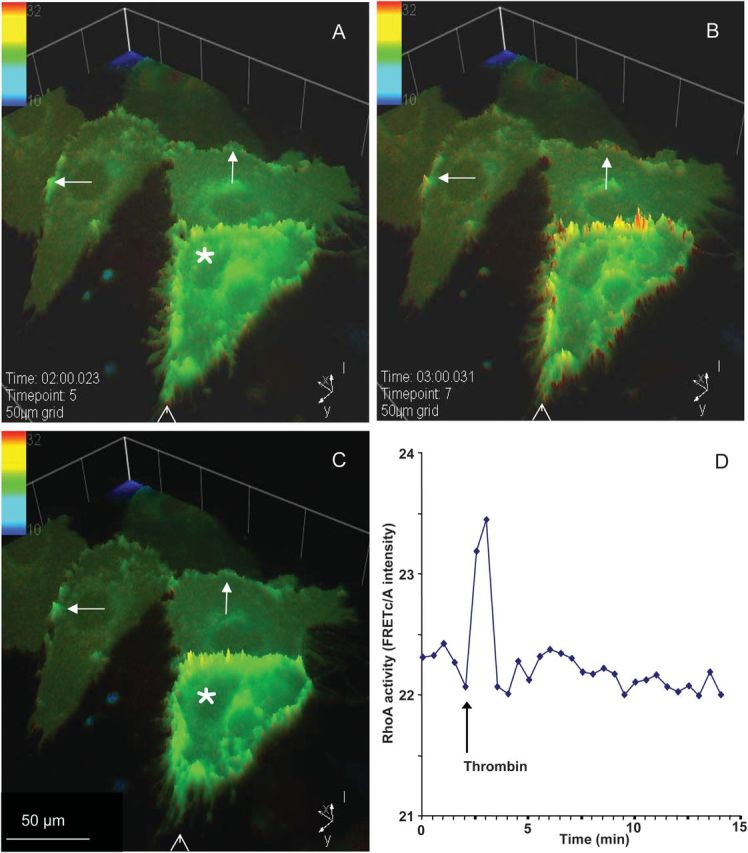
Thrombin stimulation leads to rapid, but short lasting enhancement of RhoA activity at cell margins. After 2 min of baseline recording, cells were stimulated with thrombin. (A–C) Intensity plot of RhoA activity before (A) and after thrombin stimulation (B and C), at indicated time points. The addition of thrombin caused varying effects. EC with a high RhoA activity started to contract and formed inter-endothelial gaps, whereas in cells with transient increased RhoA activity in membrane protrusions (arrows), no contractions or the loss of cell–cell contacts were observed. Contraction of the cell marked with an asterisk can be observed by comparing the position of this cell relative to the arrow head. (D) Quantitative analysis of thrombin-induced RhoA activity time course shows a transient increase in overall RhoA activity after stimulation with thrombin.
After thrombin challenge, RhoA activity at the cell margins displayed a considerable heterogeneity between individual cells. Thrombin caused an immediate increase in RhoA activity, either at the entire cell periphery, or, particularly, in protrusive structures (Figure 2B). ECs with high RhoA activity throughout the membrane lost cell–cell contacts and started contraction, as evidenced by the decrease in the cell area (Figure 2A and C, asterisk). In contrast, ECs showing occasional hotspots of RhoA activity in the protrusions kept their cell–cell interactions intact. Quantitative analysis showed a transient increase in overall RhoA activity for ∼1 min, immediately after thrombin stimulation (Figure 2D).
Many marginal areas that also displayed protrusions, exhibit short but clear increases in RhoA activity shortly after thrombin stimulation (Figure 3A). Pre-incubation with the RhoA inhibitor C3-transferase completely abrogated these FRETc/A intensity changes (Figure 3A). Separate quantitative analysis of the changes in RhoA activity indicated that RhoA activity at the cell margins was higher than in the cytosol, both under basal as well as under thrombin-stimulated conditions (Figure 3B). Verification of these data by FRET analysis based on a linear unmixing algorithm in fixed cells showed that thrombin induced a similar and significant transient increase in RhoA activity at the cell periphery but not in the cell body of rat lung microvascular ECs (Supplementary material online, Figure S2).
Figure 3.
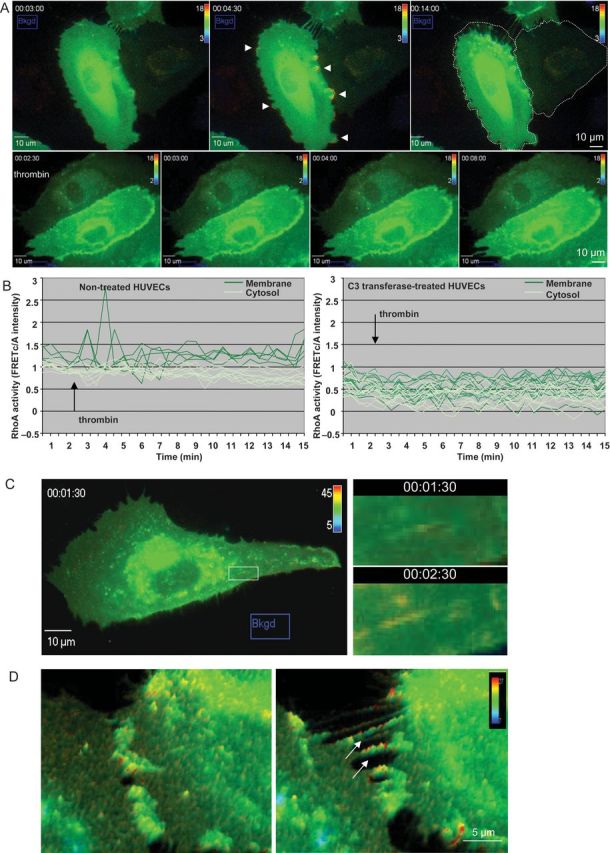
Thrombin-enhanced RhoA activity at the cell margins and at F-actin filaments. Raichu-RhoA transfected HUVECs were grown on Delta T dishes. During time-lapse imaging with an interval time of 30 s non-treated and C3-tranferase pre-treated cells were stimulated with thrombin. Three-channel corrected FRET analysis was normalized for the acceptor concentration. RhoA activity is displayed as a pseudo-colour thermal map corresponding to the scale shown in the right upper corner. Red corresponds to high RhoA activity and blue to low RhoA activity. (A) Upper panels: images of RhoA activity are shown just prior to thrombin stimulation (left panel), 1½ min after stimulation (middle panel) and 11 min after stimulation (right panel) of non-treated cells. The arrow heads indicate the increase in RhoA activity at the cell margins. The original outline of the cell in the right panel (dashed line) indicates the extent of the cell contraction. See also Supplementary material online, Movie S2. Control cells imaged in the same conditions do not contract and lack RhoA activity hotspots in the cell margins (Supplementary material online, Movie S3). Lower panels: thrombin stimulation of C3-transferase pre-treated cells occurred at t = 2½ min. Images of RhoA activity are shown (from left to right) just prior to thrombin stimulation, ½, 1½ and 5½ min after thrombin stimulation. (B) Quantity-time analysis of thrombin-induced RhoA activity. RhoA activity was quantified in individual regions at the cell margins (dark green lines) and in the cytosol (light green lines). Note that thrombin-mediated RhoA activity increased only in the regions near the cell margins of non-treated cells (left panel). (C) Thrombin induces RhoA activity at F-actin SFs. Raichu-RhoA transfected HUVECs were grown on Delta T dishes. The upper panel shows RhoA activity displayed as a pseudo-colour thermal map just before (left panel) and 2 min after (right panel) stimulation with thrombin. The bottom panels are an enlargement of the white boxes in the upper panels. (D) RhoA activity at finger-like bridges occurs prior to full separation of ECs. Raichu-RhoA transfected HUVECs were grown on Delta T dishes. During time-lapse imaging with a time interval of 30 s, the cells were stimulated with thrombin at t = 2½ min. Localization of RhoA activity before (left) and after (right) thrombin stimulation. The arrows indicate the finger-like bridges prior to separation of the cells.
To verify that thrombin also mediates endogenous RhoA activation at the cell margins, ECs were also transfected with an ‘inverse’ FRET probe showing increased FRET intensity when endogenous RhoA is inactive.21 Imaging showed a similar degree of RhoA activation upon thrombin stimulation, but with lower sensitivity (data not shown).
Thus, basal RhoA activity is found at sites of membrane protrusions in post-confluent endothelial layers. Thrombin evokes a fast and transient increase of RhoA activity. This RhoA activation is prominent at the cell margins, lasts for ∼1 min, but varies from cell to cell. Cells with a high level of RhoA activity show contraction and disruption of cell–cell contacts, whereas ECs with localized protrusive RhoA activity maintain junctional contacts.
3.3. Thrombin increases RhoA activity at F-actin filaments
Based on the previous reports which indicated the presence of RhoA/Rho kinase on contractile SFs, we sought out to determine whether RhoA activation was associated with F-actin filaments.7,13 Live cell imaging of these actin filaments was feasible in cells with a low RhoA biosensor expression (Figure 3C upper panels). The enlarged images show that increased RhoA activity was localized at cytosolic fibre-like structures in thrombin-stimulated cells.
To show that RhoA activity is needed for F-actin rearrangement, we compared the RhoA activity at the cell margin with little movement (Supplementary material online, Figure S3, region 1) to RhoA activity at the contracting site (arrow and region 2). Quantification shows that upon thrombin stimulation, RhoA activity increased at the membrane for ∼1 min in non-motile cells (graph, purple line). At the contracting filament, thrombin-mediated RhoA activity rapidly increased and remained high for >5 min (graph, orange line).
Interestingly, during the contraction of the cells, RhoA activity was observed at the remaining finger-like bridges next to the inter-endothelial gaps (Figure 3D arrows). After fixation and F-actin counterstaining of thrombin-stimulated ECs, co-localization of RhoA biosensor and actin was observed at these finger-like bridges between the ECs (Supplementary material online, Figure S3C).
These data indicate that continuous RhoA activity at the fibre-like structures is associated with rearranging F-actin filaments.
3.4. RhoA and inter-endothelial gap formation
An important question is whether RhoA activity is involved in the dynamics of inter-endothelial gaps, which was first addressed by investigating their spatio-temporal relationship, followed by a functional analysis of the role of RhoA signalling in gap dynamics.
In contrast to our initial expectations, RhoA activity was not increased at sites were gaps spontaneously formed. Surprisingly, a transient increase in RhoA activity was visible before and during the closure of these gaps at sites of protrusive activity filling the gaps (Figure 4A).
Figure 4.
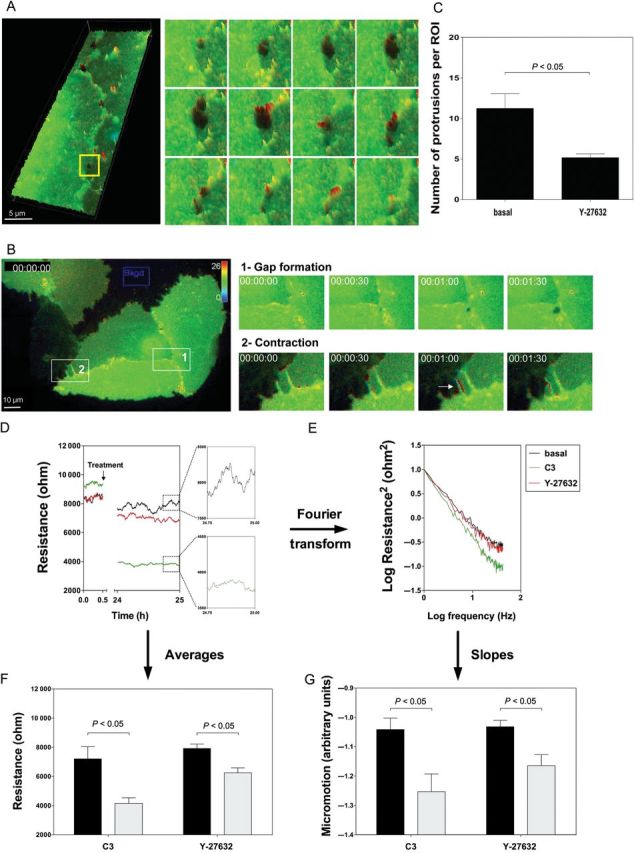
RhoA is involved in the closure, but not in the formation of small inter-endothelial gaps. (A) RhoA activity was measured in confluent HUVEC monolayers 48 h after transfection with Raichu-RhoA/kRas-CT via FRET microscopy (interval of 30 s). RhoA activity is displayed as a pseudo-colour thermal map. Red corresponds to high and blue to low RhoA activity. The enlargements of the yellow square on the right represent the 3D visualizations of RhoA activity during the spontaneous formation and the closure of a small gap in the basal EC layer. Three-dimensional images were rendered with the Slidebook software. Upper panels: RhoA activity during gap formation is at baseline value. Middle panels: increased RhoA activity at one side of the gap initiates gap closure. Lower panels: RhoA activity decreases after gap closure. Similar observations were made in five independent experiments. (B) Raichu-RhoA/kRas-CT transfected HUVECs confirm the lack of thrombin-mediated RhoA activity prior to inter-endothelial gap formation. Raichu-RhoA/kRas-CT transfected HUVECs were grown on Delta T dishes. During time-lapse imaging with an interval time of 30 s, thrombin was added at t = 0 min. RhoA activity is displayed as a pseudo-colour thermal map. The scale for the map is shown in the right upper corner. Red corresponds to high RhoA activity and blue to low RhoA activity. Owing to this construct, the signal-to-noise ratio at the membrane is improved. Note the lack of RhoA activity prior to the thrombin-mediated inter-endothelial gap formation (enlarged area 1), whereas at sites of contraction, RhoA activity is increased (enlarged area 2, arrow). (C) Inhibition of Rho kinase leads to decreased protrusive membrane activity. DIC image stacks were taken with a distance of 0.28 µm in the z-direction between every image and a 40-times magnification. Typical examples of such images are shown in Supplementary material online, Figure S4. The quantification of the whole region of interest (ROI, 100 × 100 µm) per stack series shows a significant decrease in protrusion activity after Rho kinase inhibition. Data are presented as mean ± SEM. (D–G) Barrier integrity and motility within the confluent EC layer significantly decreases after Rho A/Rho kinase inhibition. ECs were inoculated in electrode-containing ECIS wells and grown to confluence (∼72 h). Electrical resistance was measured continuously (dt = 1 s), before and 24 h after addition of the RhoA inhibitor C3-transferase (1 µg/mL) or the Rho kinase inhibitor Y-27632 (10 µM). Resistance of the endothelial barrier was measured as a parameter describing barrier integrity. With Fast Fourier Transformation, the biological noise caused by cellular movement on the measurement electrodes, was analysed as a measure of micromotion. (D) The time series of the monolayer shows a decrease in absolute resistance after incubation with the inhibitors indicating a loss in endothelial barrier integrity, whereas the untreated control remained stable at ∼8000 ohm. The data are representative for four experiments. (E) Log–log power spectrum of the Fourier transformed resistance values. The slopes of the curves are used to characterize the biological noise of the signals and thereby quantify the cellular micromotion (for details, see Supplementary material online, Methods). Slopes are calculated by a linear curve fit. (F) Since the resistance values remain stable during the measured periods, averages were calculated to perform statistics. The blockage of RhoA/Rho kinase causes a significant drop in resistance. Data are presented as mean ± SEM. (G) The inhibition of RhoA and Rho kinase shows significant motion-lowering effects on the endothelium. Data are presented as mean ± SEM.
To test whether thrombin-mediated RhoA activation near the cell margins is involved in the formation of these large gaps, FRETc/A intensity at places where gaps appeared after thrombin stimulation were quantified. In line with the basal conditions, no increased RhoA activity was found prior to gap formations (Table 1). These data were verified using Raichu-RhoA/KRas-CT, a RhoA biosensor preferentially localizing at the plasma membrane.21 Careful examination of thrombin-activated ECs transfected with this probe confirmed that RhoA activity was not elevated prior to the formation of inter-endothelial gaps (Figure 4B), whereas at sites of contraction, RhoA activity increased. Similarly, no RhoA activation was observed prior to the formation of VEGF-induced inter-endothelial gaps (Supplementary material online, Figure S5).
Table 1.
Coincidence of RhoA activity with inter-endothelial gap closure but not gap formation
| Control | Thrombin | |
|---|---|---|
| Lack of RhoA activity coinciding with gap formation | 6 (4) | 9 (5) |
| RhoA activity coinciding with gap closure | 17 (3) | 11 (4) |
The table shows the number of gaps that opened without prior RhoA activity as well as the number of gaps where RhoA activity was observed prior to closure of the gap, both for spontaneously formed gaps as well as for thrombin-induced gaps. Gap number (number of experiments).
The presence of RhoA activity at membrane protrusions where gaps closed suggested a role for RhoA-dependent membrane protrusions in the process of gap closure. Inhibition of Rho kinase attenuated the number of membrane protrusions (Figure 4C and Supplementary material online, Figure S4). As inhibition of RhoA/Rho kinase also largely prevented the formation of thrombin-induced gaps, thrombin-induced gaps were inaccessible for investigation of the role of RhoA in their closure. Spontaneous gaps still formed under those conditions. Their opening times doubled when Rho kinase was inhibited, indicating the involvement of Rho kinase in the closure of inter-endothelial gaps (Table 2). To test whether the observed RhoA activity at membrane protrusions contributes to the rapid motility of the cell periphery known as micromotion22 and re-annealing of cell–cell interactions, the cell impedance was monitored. Resistance, which is part of the impedance, served as a measure for EC barrier function, whereas micromotion was reflected by fluctuations in the electrical signal. Pharmacological inhibition of either RhoA or its downstream target Rho kinase hampered the cellular micromotion (Figure 4E and G). Under those conditions, the endothelium was no longer able to maintain barrier integrity as evidenced by a net decrease in electrical resistance (Figure 4D and F).
These data show a novel role of active RhoA in the closure of inter-endothelial gaps by mediating initial protrusive membrane activity that results in filling of the gaps.
3.5. In vivo stimulation of the endothelial thrombin receptor enhances RhoA activity at sides of cytosolic F-actin
To investigate the spatio-temporal changes in RhoA activity upon activation of the thrombin receptor PAR1 in vivo, rats were intravenously administered a TRAP. It is known that this PAR1-peptide recapitulates the PAR1-dependent pro-inflammatory effects of thrombin in ECs.26 Infusion of TRAP in the femoral vein evoked a marked swelling of the hindlimb (Figure 5A). As FRET technology in vivo27 does not provide spatial resolution at the subcellular level, we adapted an alternative approach that allows simultaneous visualization of F-actin and active RhoA in fixed tissue.25
Figure 5.
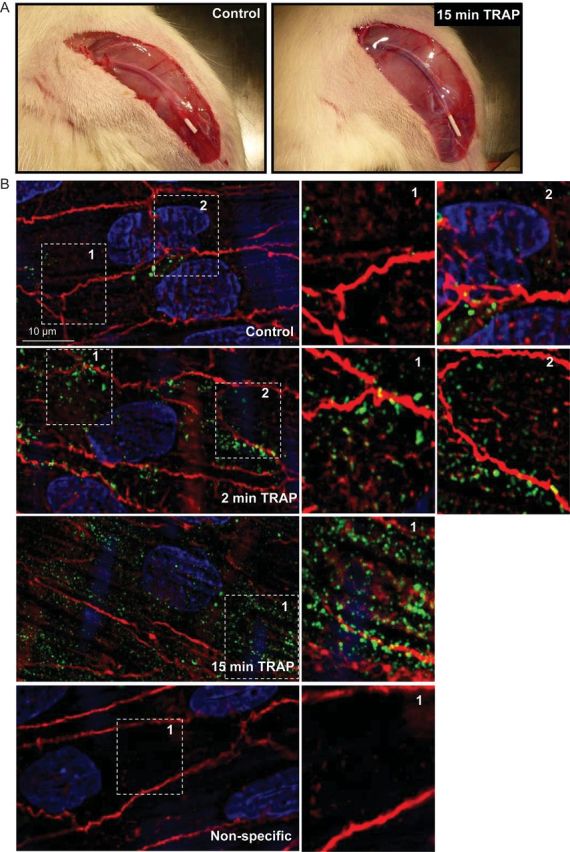
In vivo application of PAR1-activating peptide increases RhoA activity at F-actin cytoskeleton. (A) Infusion of TRAP (1 mg/kg) in the femoral vein evoked a marked swelling of the hindlimb. (B) Rat ECs of the femoral vein after in vivo administration of saline (top and bottom panel) or TRAP for two (second panel) or 15 min (third panel). The veins were fixed and permeabilized in vivo, then dissected, longitudinally cut opened and stained for active RhoA (green) using GST-tagged RBDs. Cells were counterstained for F-actin (red) and nuclei (blue) with rhodamine-phalloidin or DAPI. Enlargements of the white boxes are shown on the right.
Basal RhoA activity in situ showed a non-homogenous distribution, with some hotspots at the cell boundary (Figure 5B), as observed in cultured ECs at baseline. Two minutes after TRAP administration, active RhoA appeared in a spot-like patterns, enriched at the EC periphery. Fifteen minutes after TRAP infusion, an increase in active RhoA was present in the cell body, colocalizing with cytosolic F-actin filaments.
These data verify the time-dependent change in active RhoA localization within ECs upon thrombin stimulation, which were also observed in vitro. The short-term effect of PAR1 activation reveals that there may be an increase in RhoA activity at the cell periphery, which is followed by a shift of activity towards the cytosolic F-actin filaments 15 min after stimulation.
4. Discussion
The main finding of the present study is that RhoA activity was not spatially correlated to the formation of spontaneous or agonist-induced inter-endothelial gaps, but rather coincided with gap closure and contractile cytoskeletal elements. Specifically, four distinct RhoA activities were identified and associated with different aspects of regulation of endothelial monolayer integrity: (i) in non-stimulated endothelial monolayers, increased RhoA activity at the cell margins precedes the closure of transient, spontaneously formed gaps between ECs; (ii) thrombin stimulation rapidly increased RhoA activity at the cell margins in a non-homogenous fashion with hotspots in small finger-like membrane protrusions; (iii) thrombin increased RhoA activity along SFs in vitro and in vivo. Here, prolonged RhoA activity coincided with the rearrangement of the F-actin cytoskeleton; (iv) after thrombin stimulation, enhanced RhoA activity was visible at the remaining finger-like bridges around the inter-endothelial gap sites of contracting cells, prior to their separation.
We report here that in non-stimulated EC monolayers, RhoA activation occurs prior to gap closure, but does not precede the spontaneous formation of minute gaps between ECs. These data suggest an entirely novel role for RhoA in the maintenance of endothelial integrity and extend our previous observations that Rho kinase activity is needed to maintain endothelial barrier integrity.7 This barrier-promoting function of RhoA was evidenced by a decline in electrical impedance upon prolonged inhibition of either RhoA or Rho kinase, accompanied by a decrease in the subtle motility of the cell margins, known as micromotion. It remains to be investigated how RhoA returns to its inactive state upon gap closure. A likely scenario is that binding of p120ctn to the newly formed AJs mediates p190rhoGAP activation, which shuts down RhoA activity.28 Moreover, these minute changes in localized RhoA activity are reminiscent of the increased RhoA activity and actomyosin contractility at the contact edges that are required to drive expansion and completion of cell–cell adhesion in epithelial cells,29,30 where zones of RhoA activity are restricted to the periphery of contacting membranes driving the initiation, expansion, and completion of cell–cell adhesion.30 Interestingly, a specific exchange factor p114RhoGEF has been shown to be a component of a junction-associated Rho signalling module that drives spatially restricted activation of RhoA to regulate tight junction formation and epithelial morphogenesis, whereas the RhoGEF Ect2 promotes RhoA activity in the E-cadherin-based junctions.31 In addition, even an apicobasal Rac activity gradient has been proposed for epithelial junctions, which would be required for optimal establishment of tight junctions and apicobasal polarity.29,32 Similar mechanisms that occur in epithelial cells might also act in brain endothelial monolayer integrity, where tight junctions have a prominent contribution to barrier regulation. Most other endothelia have poorly developed tight junctions, but it is tempting to speculate that also in those endothelia one or more specific RhoGEFs exist regulating the closure of inter-endothelial gaps. Recent data, showing that the barrier stabilizer angiopoietin-1 recruits the RhoGEF Syx to endothelial junctions strongly point into this direction.33
Surprisingly, RhoA activity was not detected at sites where larger gaps between ECs formed upon stimulation with either thrombin or VEGF, although previous pharmacological inhibitor studies have clearly shown that the formation of gaps is dependent on RhoA activation.2 These data indicate that gap formation is a phenomenon that occurs secondary to RhoA activation at distant cellular regions. In line with these observations, Ngok et al.33 have shown that VEGF even stimulates the translocation of Syx from the junctions, although they did not formally prove lowered junctional RhoA activity. In ECs stimulated with thrombin, we found enhanced RhoA activity at the cell margins both in vitro as well as in vivo, particularly, at membrane protrusions. Similar activation patterns of RhoA were observed in endothelial cultures obtained from different vascular beds and different species, suggesting a universal role for localized RhoA activation. The protrusive activity disappeared several minutes after thrombin stimulation. Interestingly, activation of the RhoGEF-H1 has recently been shown to specifically contribute to the formation of comparable protrusive structures in migrating HeLa cells.17 The spatio-temporal relationship between RhoA and other small GTPases such as Rac and Cdc42 that also play a role in the formation of these ruffle-like structures remains to be investigated. Anon et al.34 showed that in epithelial monolayers closure of gaps >20 μm was Rac1 dependent, whereas the closure of smaller gaps was Rac independent. For those smaller gaps, a passive physical mechanism was proposed.
Thrombin-mediated RhoA activation was also localized at F-actin filaments. Previous observations using surrogate markers and inhibitors of Rho kinase activity in isolated SFs as well as in fixed cultured ECs and intact vessels established that Rho kinase is located along F-actin filaments and regulates contraction.7,12,13 However, we demonstrate for the first time in living cells that active RhoA is located along SFs during rearrangement (contraction) of the fibres. Thrombin-mediated cell contraction induced inter-endothelial gap formation after which RhoA activity increased at the remaining cell stretches prior to separation of the ECs. This RhoA-mediated cell separation resembles the detachment trailing portions of migrating cells for which it has been shown that RhoA and Rho kinase activity play an essential role.35
Systemic administration of TRAP induced a robust activation of RhoA in the murine endothelium in vivo. TRAP can induce massive leakage in vivo.36 Our own earlier studies demonstrated that thrombin in intact microvessels disrupted endothelial integrity in a RhoA/Rho kinase-dependent manner.5,12 Thus, the tight control of RhoA activity in the vasculature is likely to be essential for the proper regulation of the integrity of the endothelial barrier in vivo as well.
In conclusion, we have shown different subcellular locations of RhoA activity related to modulation of endothelial monolayer integrity. In non-stimulated ECs, RhoA activity was associated with improvement of endothelial barrier integrity. Thrombin-mediated RhoA activation at the ruffles may also temporarily enhance the barrier function. At other locations distal to the cell periphery, RhoA activation was shown to be involved in thrombin-mediated disruption of monolayer integrity. Here, thrombin-induced RhoA activation co-localization along the contracting F-actin filaments and at the final cell stretches of contracting cells, just prior to separation of the cell. Surprisingly, spatially elevated RhoA activity could not be associated with formation of gaps between ECs.
We propose that these subcellular localizations of RhoA activation are linked to processes regulating endothelial monolayer integrity and barrier function. Furthermore, the presented observations suggest that the general use of RhoA/Rho kinase blockers can have a positive effect in an acute pro-inflammatory setting like sepsis, characterized by increased permeability, but might be inadequate as a preventive or long-term therapy. It remains a future challenge to identify the regulating factors that drive the distinct RhoA activities, which would facilitate the development of novel therapies directed towards selective inhibition of the barrier-disruptive activities of RhoA.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by The Netherlands Heart Foundation (2003T032 and 2011T072 to G.P.v.N.A.); the European Union (EVGN contract LSHM-2003-503254); and the National Institutes of Health (NIH P01 HL60678 to R.D.M.).
Supplementary Material
Acknowledgements
We thank Jan van Bezu and Marten Engels for their excellent technical support.
Conflict of interest: none declared.
References
- 1.Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5:261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- 2.Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol. 2010;72:463–493. doi: 10.1146/annurev-physiol-021909-135833. [DOI] [PubMed] [Google Scholar]
- 3.Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature. 2005;437:497–504. doi: 10.1038/nature03987. [DOI] [PubMed] [Google Scholar]
- 4.Curry FE, Adamson RH. Vascular permeability modulation at the cell, microvessel, or whole organ level: towards closing gaps in our knowledge. Cardiovasc Res. 2010;87:218–229. doi: 10.1093/cvr/cvq115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gorovoy M, Neamu R, Niu J, Vogel S, Predescu D, Miyoshi J, et al. RhoGDI-1 modulation of the activity of monomeric RhoGTPase RhoA regulates endothelial barrier function in mouse lungs. Circ Res. 2007;101:50–58. doi: 10.1161/CIRCRESAHA.106.145847. [DOI] [PubMed] [Google Scholar]
- 6.Wójciak-Stothard B, Potempa S, Eichholtz T, Ridley AJ. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J Cell Sci. 2001;114:1343–1355. doi: 10.1242/jcs.114.7.1343. [DOI] [PubMed] [Google Scholar]
- 7.van Nieuw Amerongen GP, Beckers CML, Achekar ID, Zeeman S, Musters RJP, van Hinsbergh VWM. Involvement of Rho kinase in endothelial barrier maintenance. Arterioscler Thromb Vasc Biol. 2007;27:2332–2339. doi: 10.1161/ATVBAHA.107.152322. [DOI] [PubMed] [Google Scholar]
- 8.Spindler V, Schlegel N, Waschke J. Role of GTPases in control of microvascular permeability. Cardiovasc Res. 2010;87:243–253. doi: 10.1093/cvr/cvq086. [DOI] [PubMed] [Google Scholar]
- 9.Beckers CML, van Hinsbergh VWM, van Nieuw Amerongen GP. Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb Haemost. 2010;103:40–55. doi: 10.1160/TH09-06-0403. [DOI] [PubMed] [Google Scholar]
- 10.Essler M, Amano M, Kruse HJ, Kaibuchi K, Weber PC, Aepfelbacher M. Thrombin inactivates myosin light chain phosphatase via Rho and its target Rho kinase in human endothelial cells. J Biol Chem. 1998;273:21867–21874. doi: 10.1074/jbc.273.34.21867. [DOI] [PubMed] [Google Scholar]
- 11.Satchi-Fainaro R, Mamluk R, Wang L, Short SM, Nagy JA, Feng D, et al. Inhibition of vessel permeability by TNP-470 and its polymer conjugate, caplostatin. Cancer Cell. 2005;7:251–261. doi: 10.1016/j.ccr.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 12.van Nieuw Amerongen GP, Musters RJP, Eringa EC, Sipkema P, van Hinsbergh VWM. Thrombin-induced endothelial barrier disruption in intact microvessels: role of RhoA/Rho kinase-myosin phosphatase axis. Am J Physiol Cell Physiol. 2008;294:C1234–C1241. doi: 10.1152/ajpcell.00551.2007. [DOI] [PubMed] [Google Scholar]
- 13.Katoh K, Kano Y, Amano M, Onishi H, Kaibuchi K, Fujiwara K. Rho-kinase-mediated contraction of isolated stress fibers. J Cell Biol. 2001;153:569–584. doi: 10.1083/jcb.153.3.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birukova AA, Smurova K, Birukov KG, Kaibuchi K, Garcia JGN, Verin AD. Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc Res. 2004;67:64–77. doi: 10.1016/j.mvr.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 15.van Nieuw Amerongen GP, Koolwijk P, Versteilen A, van Hinsbergh VWM. Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arterioscler Thromb Vasc Biol. 2003;23:211–217. doi: 10.1161/01.atv.0000054198.68894.88. [DOI] [PubMed] [Google Scholar]
- 16.Ernkvist M, Luna Persson N, Audebert S, Lecine P, Sinha I, Liu M, et al. The Amot/Patj/Syx signaling complex spatially controls RhoA GTPase activity in migrating endothelial cells. Blood. 2009;113:244–253. doi: 10.1182/blood-2008-04-153874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nalbant P, Chang Y, Birkenfeld J, Chang Z, Bokoch GM. Guanine nucleotide exchange factor-H1 regulates cell migration via localized activation of RhoA at the leading edge. Mol Biol Cell. 2009;20:4070–4082. doi: 10.1091/mbc.E09-01-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pertz O, Hodgson L, Klemke RL, Hahn KM. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 2006;440:1069–1072. doi: 10.1038/nature04665. [DOI] [PubMed] [Google Scholar]
- 19.Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, et al. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461:99–103. doi: 10.1038/nature08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pertz O. Spatio-temporal Rho GTPase signaling – where are we now? J Cell Sci. 2010;123:1841–1850. doi: 10.1242/jcs.064345. [DOI] [PubMed] [Google Scholar]
- 21.Yoshizaki H, Ohba Y, Kurokawa K, Itoh RE, Nakamura T, Mochizuki N, et al. Activity of Rho-family GTPases during cell division as visualized with FRET-based probes. J Cell Biol. 2003;162:223–232. doi: 10.1083/jcb.200212049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Opp D, Wafula B, Lim J, Huang E, Lo J, Lo C. Use of electric cell-substrate impedance sensing to assess in vitro cytotoxicity. Biosens Bioelectron. 2009;24:2625–2629. doi: 10.1016/j.bios.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aman J, van Bezu J, Damanafshan A, Huveneers S, Eringa EC, Vogel SM, et al. Effective treatment of edema and endothelial barrier dysfunction with imatinib. Circulation. 2012;126:2728–2738. doi: 10.1161/CIRCULATIONAHA.112.134304. [DOI] [PubMed] [Google Scholar]
- 24.Chintala MS, Chiu PJ, Bernadino V, Tetzloff GG, Tedesco R, Sabin C, et al. Disparate effects of thrombin receptor activating peptide on platelets and peripheral vasculature in rats. Eur J Pharmacol. 1998;349:237–243. doi: 10.1016/s0014-2999(98)00200-3. [DOI] [PubMed] [Google Scholar]
- 25.Kiian I, Tkachuk N, Haller H, Dumler I. Urokinase-induced migration of human vascular smooth muscle cells requires coupling of the small GTPases RhoA and Rac1 to the Tyk2/PI3-K signalling pathway. Thromb Haemost. 2003;89:904–914. [PubMed] [Google Scholar]
- 26.McLaughlin JN, Shen L, Holinstat M, Brooks JD, Dibenedetto E, Hamm HE. Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J Biol Chem. 2005;280:25048–25059. doi: 10.1074/jbc.M414090200. [DOI] [PubMed] [Google Scholar]
- 27.Raina H, Zacharia J, Li M, Wier WG. Activation by Ca2+/calmodulin of an exogenous myosin light chain kinase in mouse arteries. J Physiol. 2009;587:2599–2612. doi: 10.1113/jphysiol.2008.165258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wildenberg GA, Dohn MR, Carnahan RH, Davis MA, Lobdell NA, Settleman J, et al. p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell. 2006;127:1027–1039. doi: 10.1016/j.cell.2006.09.046. [DOI] [PubMed] [Google Scholar]
- 29.Terry SJ, Zihni C, Elbediwy A, Vitiello E, Leefa Chong San IV, Balda MS, et al. Spatially restricted activation of RhoA signalling at epithelial junctions by p114RhoGEF drives junction formation and morphogenesis. Nat Cell Biol. 2011;13:159–166. doi: 10.1038/ncb2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamada S, Nelson WJ. Localized zones of Rho and Rac activities drive initiation and expansion of epithelial cell-cell adhesion. J Cell Biol. 2007;178:517–527. doi: 10.1083/jcb.200701058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ratheesh A, Gomez GA, Priya R, Verma S, Kovacs EM, Jiang K, et al. Centralspindlin and α-catenin regulate Rho signalling at the epithelial zonula adherens. Nat Cell Biol. 2012;14:818–828. doi: 10.1038/ncb2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mack NA, Porter AP, Whalley HJ, Schwarz JP, Jones RC, Khaja ASS, et al. β2-syntrophin and Par-3 promote an apicobasal Rac activity gradient at cell-cell junctions by differentially regulating Tiam1 activity. Nat Cell Biol. 2012;14:1169–1180. doi: 10.1038/ncb2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ngok SP, Geyer R, Liu M, Kourtidis A, Agrawal S, Wu C, et al. VEGF and Angiopoietin-1 exert opposing effects on cell junctions by regulating the Rho GEF Syx. J Cell Biol. 2012;199:1103–1115. doi: 10.1083/jcb.201207009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anon E, Serra-Picamal X, Hersen P, Gauthier NC, Sheetz MP, Trepat X, et al. Cell crawling mediates collective cell migration to close undamaged epithelial gaps. Proc Natl Acad Sci USA. 2012;109:10891–10896. doi: 10.1073/pnas.1117814109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alblas J, Ulfman L, Hordijk P, Koenderman L. Activation of Rhoa and ROCK are essential for detachment of migrating leukocytes. Mol Biol Cell. 2001;12:2137–2145. doi: 10.1091/mbc.12.7.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Korhonen H, Fisslthaler B, Moers A, Wirth A, Habermehl D, Wieland T, et al. Anaphylactic shock depends on endothelial Gq/G11. J Exp Med. 2009;206:411–420. doi: 10.1084/jem.20082150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.