

Abstract
Loss of neurotrophic support in the striatum caused by reduced brain-derived neurotrophic factor (BDNF) levels plays a critical role in Huntington's disease (HD) pathogenesis. BDNF acts via TrkB and p75 neurotrophin receptors (NTR), and restoring its signaling is a prime target for HD therapeutics. Here we sought to determine whether a small molecule ligand, LM22A-4, specific for TrkB and without effects on p75NTR, could alleviate HD-related pathology in R6/2 and BACHD mouse models of HD. LM22A-4 was administered to R6/2 mice once daily (5–6 d/week) from 4 to 11 weeks of age via intraperitoneal and intranasal routes simultaneously to maximize brain levels. The ligand reached levels in the R6/2 forebrain greater than the maximal neuroprotective dose in vitro and corrected deficits in activation of striatal TrkB and its key signaling intermediates AKT, PLCγ, and CREB. Ligand-induced TrkB activation was associated with a reduction in HD pathologies in the striatum including decreased DARPP-32 levels, neurite degeneration of parvalbumin-containing interneurons, inflammation, and intranuclear huntingtin aggregates. Aggregates were also reduced in the cortex. Notably, LM22A-4 prevented deficits in dendritic spine density of medium spiny neurons. Moreover, R6/2 mice given LM22A-4 demonstrated improved downward climbing and grip strength compared with those given vehicle, though these groups had comparable rotarod performances and survival times. In BACHD mice, long-term LM22A-4 treatment (6 months) produced similar ameliorative effects. These results support the hypothesis that targeted activation of TrkB inhibits HD-related degenerative mechanisms, including spine loss, and may provide a disease mechanism-directed therapy for HD and other neurodegenerative conditions.
Introduction
Huntington's disease (HD) is a neurodegenerative disorder characterized by striatal and cortical degeneration leading to progressive motor, cognitive, and psychiatric disturbances (Vonsattel and DiFiglia, 1998). It is caused by a mutation in the HTT gene causing expression of the huntingtin protein with an expanded polyglutamine stretch (Huntington's Disease Collaborative Research Group, 1993). Neuropathological hallmarks include intranuclear aggregates of mutant huntingtin and selective degeneration of striatal medium spiny neurons (MSNs; Vonsattel et al., 1985; DiFiglia et al., 1997).
Numerous mechanisms have been linked to HD neurodegeneration but their pathogenic contributions are unclear (Zuccato et al., 2010). However, multiple lines of evidence attribute a fundamental role to loss of neurotrophic support, primarily deficits in brain-derived neurotrophic factor (BDNF), which binds to the TrkB and p75 neurotrophin receptors (NTR; p75NTR). Mutant huntingtin has deleterious effects on BDNF gene expression causing decreased protein levels principally in striatum and cortex of HD patients and mouse models (Zuccato et al., 2010). Depleting endogenous BDNF produces an HD phenotype (Baquet et al., 2004; Canals et al., 2004; Strand et al., 2007), while overexpressing BDNF in HD mouse models has an ameliorative effect (Gharami et al., 2008; Xie et al., 2010). Consequently, developing BDNF-based strategies for HD therapeutics has been prioritized. Along these lines, preclinical testing has focused on identifying compounds that upregulate endogenous BDNF levels (Duan et al., 2004; Borrell-Pagès et al., 2006; Rigamonti et al., 2007; Apostol et al., 2008; DeMarch et al., 2008; Duan et al., 2008; Peng et al., 2008; Pineda et al., 2009; Simmons et al., 2009; Giampà et al., 2010; Zuccato et al., 2010; Mielcarek et al., 2011; Simmons et al., 2011). These studies reinforce the importance of BDNF as a therapeutic target; however, they used compounds that were not specific for BDNF or TrkB, which could lead to unwanted off-target effects.
An alternative approach to BDNF-based treatments is to develop small molecules that specifically activate TrkB. The rationale for targeting TrkB in HD is based not only on BDNF deficiencies but also on multiple linkages between the receptor's signaling and underlying HD degenerative mechanisms. Our laboratories identified small molecule TrkB ligands with structures similar to loop II of BDNF (Massa et al., 2010), a region that confers TrkB activation and specificity (Longo and Massa, 2013). One such ligand, [N,N′,N″-tris(2-hydroxyethyl)-1,3,5-benzenetricarboxamide)] (LM22A-4), binds to TrkB but not other Trks or p75NTR, and had a negative receptor binding screen (Cerep). The absence of p75NTR binding makes LM22A-4 distinct from a “BDNF mimetic” that would evoke a full range of BDNF-related activities. LM22A-4 prevents quinolinic acid-induced striatal neuron death, crosses the blood–brain barrier to activate TrkB and its associated signaling, including AKT and ERK (Massa et al., 2010), and ameliorates motor deficits and pathology in rodent models of traumatic brain injury, stroke, and Rett syndrome (Massa et al., 2010; Han et al., 2012; Schmid et al., 2012). This study was undertaken to determine whether specifically activating TrkB with LM22A-4 can alleviate HD-related striatal pathology, including huntingtin aggregates and dendritic spine loss, and motor disturbances, using R6/2 and BACHD HD mouse models.
Materials and Methods
The procedures used for this study were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and with protocols approved by the Institutional Animal Care and Use Committee of Stanford University. These protocols include efforts to minimize animal suffering and number of mice used.
All mice were group housed (3–5 mice/cage) and received cotton nestlets and rodent chow ad libitum. In rare instances, when fighting occurred among cagemates, the aggressor was removed and housed singly. Tail DNA was used for genotyping via real-time (RT) PCR and CAG repeat numbers were measured with ABI GeneMapper 4.0 by Laragen.
R6/2 mice.
Male R6/2 mice and their wild-type (WT) littermates were purchased from The Jackson Laboratory [B6CBA-TgN (HD exon1)62; JAX stock number: 006494]. These mice are transgenic for the 5′ end of the human HD gene carrying 100–150 glutamine (CAG) repeats. R6/2 mice used in this study had an average of 131 ± 2.3 (mean ± SD) CAG repeats; LM22A-4 and vehicle-treated R6/2 mice did not differ in CAG repeat length. Mice were maintained with a 12 h light/dark cycle (on 6:00 A.M., off 6:00 P.M.). R6/2 mice and their WT littermates received wet mush food and HydroGel (ClearH2O) daily starting at the time of overt motor symptoms (∼8 weeks old).
BACHD mice.
Male BACHD mice and their WT littermates were also purchased from The Jackson Laboratory (stock number: 008197). These mice are a bacterial artificial chromosome-mediated transgenic model that expresses full-length human mutant huntingtin with 97–98 CAG repeats (Gray et al., 2008). BACHD mice used in this study all had 98 CAG repeats. Mice were maintained on a reverse 12 h light/dark cycle (on 6:00 P.M., off 6:00 A.M.).
LM22A-4.
LM22A-4 was manufactured by and purchased from Ricerca Biosciences. Each preparation was characterized by HPLC, nuclear magnetic resonance spectrometry and liquid chromatography/mass spectrometry (LC-MS) and had a purity of >97%. It has a molecular weight of 339.14 and does not contain peptide bonds. In assays of neurotrophic activity, LM22A-4 had maximum levels of activity ∼80% of that of BDNF and an EC50 value <500 pm, a profile consistent with a potent partial agonist (Massa et al., 2010). An in-depth characterization of the ligand's pharmacokinetics and pharmacodynamics as well as its chemical structure is available in a published report from our laboratories (Massa et al., 2010). Preparations of LM22A-4 were characterized for bioactivity using hippocampal neuron survival assays, as previously described (Massa et al., 2010).
Analysis of LM22A-4 brain and plasma levels in R6/2 mice.
The CNS bioavailability of LM22A-4, at the doses and routes of administration to be used in this study, was assessed in 10-week-old R6/2 mice. LM22A-4 in saline solution (0.9% sodium chloride in distilled water; Sigma) was administered once via intranasal (5 mg/kg, 0.5 ml/kg) and intraperitoneal (50 mg/kg, 10 ml/kg) routes simultaneously to maximize brain levels. To control for blood contamination of brain tissue, atenolol, a compound with minimal blood–brain barrier penetration, was delivered at 10 mg/kg in saline solution via oral gavage at the time of LM22A-4 administration. Mice were killed at 1 h or 3 h post dose (n = 4 mice/time point), and forebrain and plasma samples were collected. Brain and plasma concentrations of LM22A-4 and atenolol were determined by reverse phase LC with triple-quadrupole tandem mass spectroscopic detection (LC-MS/MS) by Absorption Systems. Test accuracy was verified by generating a standard curve using known amounts of LM22A-4 added to blank brain extract.
Study design and drug treatments.
We tested the effects of LM22A-4 in both R6/2 and BACHD mice. R6/2 mice are useful for drug testing because they develop symptoms rapidly and reliably, and BACHD mice better represent the genetic component of HD with a slower disease progression making them better suited for targeting early degenerative mechanisms. Both of these models have been recommended for preclinical therapeutic testing based on systematic behavioral analyses comparing numerous available HD murine models (Menalled et al., 2009).
The 2 × 2 study designs (transgenic/WT × LM22A-4/vehicle) were used using groups of male R6/2 or BACHD mice. Vehicle control groups received sterile saline solution and experimental groups received LM22A-4 in saline solution at 5 mg/kg (0.5 ml/kg) intranasally and 50 mg/kg (10 ml/kg) intraperitoneally, simultaneously. These LM22A-4 doses were chosen based on LC-MS/MS data on brain penetration and doses that produced biological effects in previous in vitro (Massa et al., 2010) and in vivo (Schmid et al., 2012) studies. All mice received one intranasal instillation and one intraperitoneal injection per day 5–6 d per week and were weighed weekly. On days motor behavior was assessed, dosing occurred after behavior tests were conducted. Treatment for R6/2 mice started at 4 weeks of age and continued for 7 weeks and, for BACHD mice, began at 2 months of age and continued for 6 months. Mice were treated in cohorts (n = 10/group) to render behavior testing more manageable.
One cohort of R6/2 mice was used to assess the effects of LM22A-4 on survival and was dosed throughout their life span. Mice were killed if they could not right themselves within 30 s of being placed on their sides. Spontaneous deaths that occurred overnight were recorded the next morning. Mice used to assess survival were not used for histology.
Behavior testing.
Behavior testing and dosing were performed by experimenters that were blind to the treatment and genotype conditions. Testing of R6/2 mice was performed during the light phase of the light/dark cycle while the BACHD mice were tested during the dark phase. Mice were transported from the housing room to the behavior procedure room ∼1 h before testing.
Rotarod and clasping performance.
R6/2 mice were tested on a rotarod (Med Associates) once a week at 8, 9, and 10 weeks old, while BACHD mice were tested at 2, 5, and 8 months. Test sessions consisted of two 300 s trials (intertrial interval (ITI) ∼15 min) with acceleration from 4 to 40 rpm. The latency to fall from the rotarod was recorded and the trials were averaged. For R6/2 mice, clasping behavior was tested once a week at 8, 9, and 10 weeks old: mice were suspended by the tail for 60 s and the latency for the hindlimbs or all four paws to clasp for at least 3 s was recorded.
Vertical pole descent test.
The vertical pole descent test was conducted on R6/2 mice at 9 weeks old and BACHD mice at 5 months old using methods similar to those described previously (Matsuura et al., 1997; Hickey et al., 2008; Simmons et al., 2011). Mice were placed at the top of a coarse, vertical wooden pole (diameter: 1 cm; height: 55 cm) in a cage with bedding and allowed to descend. They were first given a practice run and then four test trials (ITI ∼2 min); the time to reach the cage bottom was recorded. If the mouse did not descend or dropped or slipped down the pole without climbing, a default value of 60 s was recorded. The descent times of the four tests were averaged.
Wire hang.
R6/2 mice were subjected to the wire hang test once at 10 weeks old. They were allowed to grasp, using all limbs, a threaded metal rod (diameter 0.4 cm) that was positioned horizontally 6 inches above their home cage. Mice received three trials (ITI ∼5 min) during which they were allowed to hang until their grip failed or 60 s had elapsed. Latency to fall was recorded and the trial scores were averaged.
Activity chamber.
Gross locomotor activity was assessed in R6/2 mice at 6 weeks old and BACHD mice at 8 months old in an activity chamber (43.2 × 43.2 cm; Med Associates) inside a sound-attenuating arena under red light. Mice were placed in the center of the chamber and allowed to explore for 30 min while being tracked by an automated beam system (Activity Monitor software, Med Associates). Distance moved and velocity was recorded.
Quantitative RT-PCR for huntingtin RNA.
A separate cohort of WT and R6/2 mice were dosed with vehicle or LM22A-4, as described above (5 mg/kg, i.n. and 50 mg/kg, i.p.), for 2 weeks starting at 6 weeks of age (n = 2 WT mice and 3–4 R6/2 mice/group). One hour after their last injection, mice were deeply anesthetized with avertin (2,2,2-tribromoethanol), brains were removed, and the striatum was dissected and flash frozen at −80°C until use. Total RNA was isolated from striatum using TRIzol reagent (Invitrogen), according to the manufacturer's instructions, and reverse-transcribed (2 μg) into single-stranded cDNA using a High Capacity RNA-to-cDNA kit (Invitrogen; PN4387406). cDNA products were used as templates for PCR amplification using the following primers that recognize both mouse and human huntingtin (Wang et al., 2008): HD1S (forward) 5′-ATGGCGvACCCTGGAAAAGCT-3′ and HD40A (reverse) 5′-TGCTGCTGGAAGGACTTGAG-3′ [Integrated DNA Technologies (IDT)]. The internal control was GAPDH, which was detected with the following primers (IDT): 33935 (forward) 5′-TGCACCACCACCTGCTTAG-3′ and 35093 (reverse) 5′-GATGCAGGGATGATGTTC-3′. cDNA (5 μl for a final concentration of 10 ng) was added to Power SybrGreen mix (Invitrogen; PN4367659) containing 200 nm of each forward and reverse primer. Water was added to bring total volume to 20 μl. All samples were run in triplicate. Quantitative RT-PCR (qRT-PCR) was conducted using an Applied Biosystems StepOne RT-PCR system. Huntingtin RNA levels were calculated and normalized to the internal control (GAPDH) and then expressed relative to the WT vehicle group.
Western blot analyses.
Two hours after their last injection with LM22A-4 or vehicle, one cohort of R6/2 and WT mice (n = 8–10 mice/group) was deeply anesthetized with avertin and their brains rapidly removed. One hemisphere was used for Western blotting, the other for Golgi staining. The striatum was dissected from the former hemisphere and flash frozen at −80°C until use. Striatal tissue was homogenized and sonicated in radioimmunoprecipitation assay lysis buffer containing protease and phosphatase inhibitors [50 mm Tris, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% NP-40, 10% glycerol, 1 mm phenylmethanesulfonylfluoride, 10 mm sodium fluoride, 1 mm sodium orthovanadate, 10 mm sodium pyrophosphate, 50 mm β-glycerophosphate, and a protease inhibitor cocktail (Roche)]. Lysates were centrifuged at 14,000 ×g for 10 min, supernatant was collected, and protein concentrations were determined using Cytoskeleton Red Assay reagents (Cytoskeleton). Protein samples (25 μg/lane) were electrophoresed through a NuPAGE 4–12% Bis-Tris Gel with MOPS or MES SDS running buffer (Invitrogen) and transferred to polyvinylidene difluoride membranes.
To assess TrkB, AKT, ERK, PLCγ, and CREB phosphorylation, membranes were probed for a phospho-protein and then stripped and reprobed for the corresponding total protein. Antibodies used were as follows: rabbit monoclonal phospho-TrkBY817 (Epitomics), rabbit polyclonal phospho-TrkBY515 (Novus Biologicals), rabbit polyclonal phospho-TrkBY705 (Signalway Antibody), rabbit polyclonal TrkB (Millipore), mouse monoclonal phospho-ERK (Thr202/Tyr204) and phospho-AKT (Ser473) and rabbit polyclonal ERK and AKT (Cell Signaling Technology), rabbit polyclonal phospho-phospholipase C-γ (PLCγ)Y783 (Epitomics) and mouse monoclonal PLCγ (BD PharMingen), rabbit monoclonal phospho-cAMP response element (CRE)-binding protein (CREB; Cell Signaling Technology), and mouse monoclonal CREB (Cell Signaling Technology).
These striatal lysates were also used to assess levels of mature BDNF (mBDNF; ∼14–15 kDa; Lee et al., 2001; Matsumoto et al., 2008) and dopamine- and cAMP-regulated phosphoprotein with a molecular weight of 32 kDa (DARPP-32) using rabbit anti-BDNF (Santa Cruz Biotechnology) and rabbit anti-DARPP-32 (Epitomics), respectively. For BDNF blots, recombinant human BDNF (PreproTech) was loaded on the same gels as the samples to verify mBDNF bands.
To control for variations in protein loading, all blots were also stripped and reprobed with α-tubulin monoclonal antibody (Sigma). Western blot signals were detected with an enhanced chemiluminescence detection system (GE Healthcare). For quantification, immunoreactive bands were manually outlined, and densities were measured using Un-Scan-It gel software (v6.1; Silk Scientific). For each blot, the densities of phospho-protein-immunoreactive bands were expressed as a fraction of the band for the total protein in the same lane and TrkB, mBDNF, and DARPP-32 were expressed as a fraction of tubulin in the same lane. Samples were run 2–6 separate times, data were normalized to the WT vehicle group of that gel, and these results were averaged.
Modified Golgi staining.
Mice were deeply anesthetized with avertin and their brains were rapidly removed and immersed in modified Golgi-Cox staining solution (developed and provided by Deqiang Jing and Francis Lee at Cornell University) for 8 d at room temperature in the dark. They were then transferred to 30% sucrose in dH2O at 4°C for 72 h; this solution was changed after the first 12 h. Brains were then cut, using a vibratome, into 150 μm sections immersed in 30% sucrose at room temperature. Sections were mounted onto slides coated with 0.3% gelatin. After drying briefly, slides were dipped in 40% sucrose three times and allowed to air dry for 72 h in the dark. After dH2O washes, sections were stained with developing solution, washed again, dehydrated through graded ethanols, immersed in HistoClear, and then coverslipped using DPX mounting medium.
Immunocytochemistry.
Mice were deeply anesthetized with avertin and transcardially perfused with 4% paraformaldehyde in 0.1 m PB, pH 7.4. Brains were postfixed (1–2 h), cryoprotected in 20% sucrose/PB, and sectioned (40 μm, coronal) using a freezing microtome. Free-floating sections were processed for immunohistochemical localization of DARPP-32 (1:1000; Millipore AB1656), parvalbumin (PV; 1:750; Abcam), huntingtin protein (1:200; clone EM48, Millipore), or ubiquitin (1:500; Dako) using the Vectastain Elite ABC kit (Vector Laboratories) and visualized with a diaminobenzidine (DAB) substrate kit (Vector Laboratories) or 0.05% DAB (Sigma) in Tris-buffered saline, pH 7.5, with 0.03% H2O2. Immunofluorescent staining was used to visualize IBA-1 (1:1000; Wako).
Microscopy and quantitative analyses of striatal histology.
All analyses were performed blind to the genotype and treatment of the mice. For all histological analyses, the dorsolateral striatum was examined at rostral to mid-caudal levels (+1.18 to +0.02 mm relative to bregma; Paxinos and Franklin, 2008), as HD pathology progresses from dorsal to ventral and anterior to posterior in this region (Vonsattel et al., 1985). Sampling fields were placed ventral to and abutting the corpus callosum. For the analysis of nuclear huntingtin staining, the primary motor cortex was also examined (+0.98 to +0.86 mm relative to bregma; Paxinos and Franklin, 2008) with the sample field (see below) in cortical layers 4/5. Images were acquired with a Zeiss Axio Imager M2 microscope, AxioCam HRc camera, and Axiovision or Microlucida (MBF Bioscience) software. In some instances, immunostaining was done in multiple sets and quantifications were normalized to the WT vehicle group of that staining set.
To assess nuclear huntingtin accumulation, all immunoreactive nuclei and intranuclear aggregates within a sample field of 250 × 250 μm (0.063 mm2) were manually traced in the striatum and cortex in one section per mouse (n = 7–9/group) while viewed with a 100× oil objective using Neurolucida v.10 image analysis software (MBF Bioscience). Numbers of aggregates and their areas were measured. The intensity of nuclear huntingtin immunostaining was measured in two sample fields (90 × 70 μm) per hemisphere of two sections per mouse (8 fields/mouse total). Within a sample field, a contour was drawn around each nucleus (containing both diffuse and aggregate staining) for density profiling and the background within the sample field was subtracted using the four corners subtraction interpolation function of the UN-Scan IT v.6.1 densitometry software (Silk Scientific).
DARPP-32, IBA-1, and PV immunostaining was analyzed in 4–6 striatal sections per mouse (n = 9–13/group). For area of DARPP-32 and IBA-1 staining, one sample field was imaged, at 10× and 20×, respectively, per section in each hemisphere (total of 8–12 fields/mouse). Since PV-containing cells are relatively sparse in the striatum, four nonoverlapping sample fields in each hemisphere per section were imaged at 10× to cover most of the striatum (∼32 fields total/mouse). Immunostaining was quantified using Image Pro Plus v6.3 (Media Cybernetics). Thresholds were set manually to identify densely immunolabeled cells and their processes that were distinct from labeling in the neuropil. The same image exposure times and threshold settings were used for sections from each treatment group. The area occupied by the immunostained cells and processes was expressed as a percentage of the total area analyzed and normalized by the WT vehicle group. The number of PV-stained somas was also counted in the striatal fields described above using the AutoNeuron function of Neurolucida software, which reconstructs neurons using a tracing algorithm to differentiate somas from processes based on the image background and size constraints. This unbiased approach was used and then checked for accuracy by an investigator blind to experimental groups.
In addition, a more extensive analysis of PV- and DARPP-32-immunostained neurites was conducted in R6/2 and BACHD mice, respectively, by manually tracing neurons in a sample field delineated by nine squares (250 × 250 μm each) in the dorsolateral region of one striatal section (approximately bregma 0.26–0.14 mm). Immunoreactive neurites were analyzed with Neurolucida using an automated scanning procedure (meander scan) affording unbiased nonoverlapping field selection. Neurons, and neurites originating from them, were manually traced under a 40× objective and focusing through the Z-plane. For DARPP-32, all neurons in every third meander scan field were traced (569 ± 11 MSNs per BACHD mouse; mean ± SEM). For PV, all neurons in all fields were traced (45 ± 2 neurons per R6/2 mouse). Neurolucida Branched Structure Analysis was used to compute the volume and length of the neurites.
Dendritic spine density was obtained by manually tracing Golgi-stained MSNs in the dorsolateral striatum while viewing and adjusting focus at 100× using Neurolucida. Neurons that minimally overlapped adjacent neurons were selected. All branches from one dendritic tree of each neuron were traced and 2–3 neurons were analyzed per mouse (n = 8/group).
The volume of the striatum was estimated in every eighth coronal 40 μm section between 1.34 and −0.22 mm relative to bregma in mice of each treatment group (n = 9 mice/group) using the Cavalieri method within StereoInvestigator (MBF Bioscience).
Statistical analyses.
GraphPad Prism v.5 was used to determine significance using a one-way ANOVA with a Newman–Keuls post hoc test and/or a Student's t test for paired comparisons. A repeated-measures (RM) ANOVA was used for body weight and rotarod analyses. If the variance significantly differed, as assessed with the Bartlett's test, a t test with Welch's correction was used to assess statistical significance. Statistical outliers were removed if values were 2 SDs from the mean (criteria determined a priori). Results are expressed as group mean ± SEM and statistical significance was set at p ≤ 0.05.
Results
LM22A-4 brain levels in R6/2 mice
Previous pharmacokinetic studies indicated that, 1 h after LM22A-4 was administered at 10 or 50 mg/kg intraperitoneally, brain concentrations were <100 nm (Massa et al., 2010; Schmid et al., 2012), which is the concentration that produced maximal cell survival effects in vitro (Massa et al., 2010). Similarly, when LM22A-4 was administered at 5 mg/kg intranasally, low brain concentrations were obtained (18 ± 5 and 79 ± 69 nm at 0.5 and 1 h, respectively; below detectable limit at 3 h; mean ± SEM). Thus, we investigated whether delivering LM22A-4 via intraperitoneal and intranasal routes simultaneously would produce forebrain levels of the compound >100 nm in R6/2 mice. To this end, a single dose of 50 mg/kg LM22A-4 intraperitoneally coadministered with 5 mg/kg intranasally was delivered to R6/2 mice, and 1 or 3 h post dosing (n = 4/time point), forebrain and plasma samples were collected for LC-MS/MS. The mean concentration of LM22A-4 in forebrain, after correction for blood contamination, was 367 ± 77 nm at 1 h after dosing and 17 ± 2 nm by 3 h. Although brain-to-plasma ratios were low (0.3 ± 0.2 and 0.1 ± 0.1 after 1 and 3 h, respectively; mean ± SEM), the brain concentrations obtained were similar to or exceeded those at which LM22A-4 is biologically active and maximally neuroprotective in in vitro and in vivo assays of TrkB function (Massa et al., 2010).
Deficits in TrkB activation and signaling in R6/2 striatum are prevented by LM22A-4
Since levels of TrkB are decreased in the striatum of HD patients and multiple mouse models of the disease (Zuccato and Cattaneo, 2007), we determined the levels of full-length TrkB protein in the striatum of 11- to 12-week-old R6/2 and WT mice treated for 7 weeks with LM22A-4 or vehicle (n = 8–10 mice/group). Striatal TrkB levels were reduced by 28 ± 3% in vehicle-treated R6/2 mice compared with WTs, and treatment with LM22A-4 did not affect levels of the receptor (Fig. 1A). TrkB phosphorylation at Y817, the tyrosine residue associated with PLCγ docking (Reichardt, 2006), is reduced in the striatum of the R6/1 HD mouse model (Gharami et al., 2008) but is not altered in YAC128 mice (Gharami et al., 2008; Xie et al., 2010). To determine whether TrkB activation is reduced in R6/2 striatum, phosphorylation was assessed at three TrkB tyrosine residues (Y705, Y515, and Y817) chosen on the basis of availability of phospho-specific antibodies and on the signaling pathways associated with these sites. Relative to WTs, R6/2 mice exhibited a 47 ± 9% decrease in striatal TrkB phosphorylation at Y705 (Fig. 1B). This residue resides in the TrkB autoregulatory loop of the kinase domain and its phosphorylation can further activate the receptor (Huang and Reichardt, 2001). The other phospho-sites examined were unchanged (Fig. 1C,D). Next, we investigated whether LM22A-4 could engage its target and promote TrkB activation. Striatal TrkB phosphorylation was increased by the ligand at Y705 in R6/2 mice, but a change in phosphorylation at Y515 or Y817 was not detected at the 2 h post-LM22A-4 injection time point chosen for analysis. In WT mice, TrkB phosphorylation was increased at site Y515 with LM22A-4 administration, as shown previously (Massa et al., 2010), but not at Y705 or Y817 (Fig. 1B–D).
Figure 1.

LM22A-4 activates TrkB and its associated downstream signaling in striatum of R6/2 mice. Representative Western blots of striatal homogenates from 11- to 12-week-old WT or R6/2 mice treated with vehicle (Veh) or LM22A-4 (LM-4) for 7 weeks. For all quantitation, n = 8–10 mice/group were used and values were normalized to the WT Veh group run on the same gel. Immunobands from one mouse per group and corresponding densitometric group analyses are shown. A–D, Full-length TrkB levels (A) and its phosphorylation (p) at tyrosine residues Y705 (B), Y515 (C), and Y817 (D) are depicted. Samples from each mouse were run in three to four replicates. TrkB levels were lower in striatum of R6/2 mice relative to WTs (***p ≤ 0.001 vs WT Veh) regardless of LM-4 treatment. TrkB phosphorylation (pTrkB) at Y705 (**p ≤ 0.01 vs WT Veh), but not Y515 or Y817, was decreased in Veh-treated R6/2 mice compared with WTs. LM-4 inhibited the reduction in Y705 phosphorylation (+p ≤ 0.05 vs R6/2 Veh). LM-4 also increased phosphorylation at Y515 in WT mice (#p ≤ 0.05 vs WT Veh; two-tailed Student's t test). E–G, Western blots showing phosphorylation of AKT (E), ERK (F), and PLCγ (G), the three main downstream signaling pathways of TrkB. Samples from each mouse were run in four to six replicates. Phosphorylation of AKT, PLCγ, and ERK was decreased in Veh-treated R6/2 mice compared with WTs (**p ≤ 0.01 and ***p ≤ 0.001 vs WT Veh). LM-4 prevented the deficits in AKT and PLCγ (+p ≤ 0.05 and ++p ≤ 0.01 vs R6/2 Veh) but not ERK phosphorylation. H, Western blot showing phosphorylation of CREB at Ser133. Samples from each mouse were run in duplicate. pCREB was reduced by 36 ± 10% in R6/2 mice given Veh (*p < 0.05 vs WT Veh; two-tailed Student's t test) and LM-4 prevented this decrease (+p < 0.05 vs R6/2 Veh; two-tailed Student's t test); there was also an increase in pCREB in the WT LM-4 group that did not reach statistical significance (p = 0.07 vs WT Veh). I, Western blot showing mBDNF protein levels in striatum. Samples from each mouse were run in triplicate. mBDNF levels are reduced in striatum of R6/2 Veh mice (***p ≤ 0.001 vs WT Veh) and this difference was attenuated by LM-4 treatment (+p ≤ 0.05 vs R6/2 Veh; two-tailed Student's t test). For all analyses, results are expressed as the mean ± SEM and, for comparison purposes, some nonadjacent lanes of Western blots were moved together and separated by spaces. Statistical significance was determined by ANOVA with Newman–Keuls post hoc testing, unless otherwise noted.
We also investigated LM22A-4's effects on the three major downstream signaling pathways associated with TrkB activation: PI3K/AKT, PLCγ/PKC, and MAPK/ERK1/2, all of which have altered activity levels in HD patients and/or mouse models (Colin et al., 2005; Giralt et al., 2009; Bodai and Marsh, 2012). A marked reduction in AKT activation was evident in the striatum of R6/2 mice given vehicle compared with WTs (Fig. 1E) and LM22A-4 treatment ameliorated this decrease. Activation of ERK was also significantly decreased in the vehicle-treated R6/2 striatum compared with WTs, although the levels were highly variable (Fig. 1F; p = 0.001 Bartlett's test for equal variance). LM22A-4 did not significantly affect ERK activation. PLCγ activity was reduced in R6/2 mice and was increased with LM22A-4 administration (Fig. 1G). This increase occurred despite the lack of a detectable increase in phosphorylation at Y817 on TrkB at the 2 h time point examined.
Activating AKT, ERK, and PLCγ pathways via TrkB signaling has been shown to phosphorylate CREB, which promotes transcription by recruiting CREB-interacting proteins to the CREs on CREB target genes, such as BDNF (Finkbeiner et al., 1997; Du and Montminy, 1998; Bonni et al., 1999; Shaywitz and Greenberg, 1999). Mutant huntingtin impairs CREB signaling and CRE-mediated transcription, which has been linked to the BDNF deficits associated with HD (Steffan et al., 2000; Zuccato et al., 2001; Gines et al., 2003; DeMarch et al., 2008). Previous studies have shown that striatal levels of BDNF protein and/or mRNA are reduced in HD patients (Ferrer et al., 2000; Gauthier et al., 2004) and numerous HD mouse models (Zuccato et al., 2001; Luthi-Carter et al., 2002; Duan et al., 2003; Gines et al., 2003; Spires et al., 2004a; Lynch et al., 2007; Simmons et al., 2009), including R6/2 mice (Simmons et al., 2011). Although most BDNF within the striatum is cortically derived, low levels of BDNF transcripts are present, possibly via BDNF mRNA transport to dendrites, and translated locally (Altar et al., 1997; Luthi-Carter et al., 2002; An et al., 2008). Thus, whether increasing TrkB signaling with LM22A-4 could also alleviate deficits in CREB activation and BDNF levels in striatum was investigated. Phosphorylation (Ser 133) of CREB was reduced by 36 ± 10% in vehicle-treated R6/2 mice compared with the WT vehicle group and LM22A-4 prevented this decrease (Fig. 1H). pCREB was also elevated in WT mice given LM22A-4 though this increase did not reach statistical significance. Given that LM22A-4 prevented pCREB reduction, striatal BDNF levels were also assessed. The band corresponding to the mature form of the protein (mBDNF; 14–15 kDa; Matsumoto et al., 2008) was measured, as this form activates TrkB signaling (Huang and Reichardt, 2001). Striatal mBDNF was reduced by 37 ± 5% in R6/2 mice given vehicle compared with WTs (Fig. 1 I), as shown previously with Western immunoblotting in R6/2 mice and other HD mouse models (Lynch et al., 2007; Apostol et al., 2008; Gharami et al., 2008; Simmons et al., 2009, 2011). LM22A-4 treatment partially restored these levels in R6/2 mice but had no effect in WTs.
Diffuse and aggregated huntingtin in nuclei of striatal and cortical neurons is reduced by LM22A-4
Intranuclear aggregates of the mutant huntingtin protein are characteristic of HD pathology in humans (DiFiglia et al., 1997) and mouse models (Mangiarini et al., 1996; Davies et al., 1997; Menalled et al., 2003; Gray et al., 2008; Zuccato et al., 2010). In numerous brain areas, the mutant protein is aberrantly processed and translocated from the cytoplasm to the nucleus, where it is present diffusely and also in dense, compact aggregates. The size of these aggregates was reduced in the striatum of R6/1 mice with transgenic overexpression of BDNF (Gharami et al., 2008). We examined the effects of LM22A-4 on the area and intensity of nuclear huntingtin immunostaining in the R6/2 striatum and cortex. The number and size of the densely stained intranuclear aggregates appeared diminished in R6/2 mice given LM22A-4 compared with vehicle-treated controls, as did the intensity of the diffuse nuclear huntingtin staining (Fig. 2A). Quantitative analyses confirmed these observations (Fig. 2B,C). Compared with vehicle treatment, treating R6/2 mice with LM22A-4 significantly reduced the number of neuronal nuclei containing diffuse huntingtin staining in both the striatum and cortex, as well as the area occupied by stained nuclei and their staining intensity (Fig. 2B). In addition, the total area occupied by the intranuclear huntingtin aggregates was reduced by 57 ± 10% in striatum and 41 ± 9% in cortex due to decreases in the number of aggregates and the mean area per aggregate (Fig. 2C).
Figure 2.

LM22A-4 reduces diffuse and aggregated huntingtin in nuclei of striatal and cortical neurons of R6/2 mice. A, Representative photomicrographs showing nuclear huntingtin immunostaining in striatum of R6/2 mice treated with vehicle (Veh; top) or LM22A-4 (LM-4; bottom). Diffuse huntingtin immunostaining was lighter and surrounded the huntingtin aggregates (arrows), which appeared smaller and less numerous in R6/2 mice given LM-4 (n = 9 mice) compared with those given Veh (n = 7 mice). Scale bar, 5 μm. Quantitative analyses of a sample field (0.063 mm2) in striatum (Str) and cortex (Cx) of one section per mouse confirmed these observations. B, The total area, number, and intensity of nuclei containing diffuse huntingtin staining decreased with LM-4 treatment in both brain areas (+p ≤ 0.05, ++p ≤ 0.01, and +++p ≤ 0.001 vs R6/2 veh). C, The total area occupied by huntingtin aggregates was reduced in Str and Cx due to a decrease in aggregate number and size (+p ≤ 0.05, ++p ≤ 0.01, and +++p ≤ 0.001 vs R6/2 veh). Results are normalized to the R6/2 vehicle group and expressed as mean ± SEM. Statistical significance was determined with a two-tailed Student's t test.
Striatal transgene expression and atrophy are not affected by LM22A-4 in R6/2 mice
The reduction in the number of immunopositive nuclei and area occupied by intranuclear huntingtin staining seen with LM22A-4 treatment could occur if the ligand inhibits expression of the huntingtin transgene or increases the volume of the striatum. Thus, we examined whether LM22A-4 alters levels of huntingtin transcripts or striatal volume in R6/2 mice. The level of transgene expression in striatum of R6/2 mice, which express exon 1 of human huntingtin under the control of the human HD promoter (Mangiarini et al., 1996; Davies et al., 1997), was determined with qRT-PCR using primers specific for mouse and human huntingtin (Wang et al., 2008) relative to endogenous GAPDH. As expected, R6/2 mice given vehicle had huntingtin transcript levels that were 2.5 times higher than WTs (Fig. 3A). LM22A-4 did not affect huntingtin transcript levels in R6/2 mice.
Figure 3.

Huntingtin transcript levels and striatal atrophy are not affected by LM22A-4 in R6/2 mice. A, Data from qRT-PCR using primers recognizing both human and mouse huntingtin (htt) are shown (n = 2 for WTs and 3–4 R6/2 mice/group). Htt RNA levels were calculated and normalized to the internal control (GAPDH) and then expressed relative to the WT vehicle (Veh) group. Relative levels of htt transcript are increased in R6/2 striatum (***p = 0.001 vs WTs) and are not altered by LM22A-4 (LM-4). B, The volume of the striatum was significantly decreased in the Veh-treated R6/2 mice compared with the WTs (*p ≤ 0.05). LM-4 treatment did not prevent striatal atrophy in R6/2 mice (n = 9 mice/group). Results are expressed as mean ± SEM. Statistical significance was determined by ANOVA with Newman–Keuls post hoc testing.
Mutant huntingtin expression causes substantial atrophy of the striatum in HD patients (Vonsattel et al., 1985) and mouse models (van Dellen et al., 2000a; Hockly et al., 2002; Canals et al., 2004; Chopra et al., 2007; Giampà et al., 2009), which may be reversed with BDNF overexpression (Gharami et al., 2008). We determined whether treatment with LM22A-4 could prevent this gross morphological change. The volume of the striatum decreased by 22 ± 5% in vehicle-treated R6/2 mice compared with WT vehicle group (Fig. 3B), which is consistent with previous reports (Hockly et al., 2002; Chopra et al., 2007; Giampà et al., 2009; Simmons et al., 2011). The striatal volume of R6/2 mice given LM22A-4 did not differ from those given vehicle.
LM22A-4 ameliorates reduced DARPP-32 and PV levels in striatal neurons
Within the striatum of HD patients, GABAergic MSNs are particularly susceptible to degeneration (Vonsattel et al., 1985; Vonsattel and DiFiglia, 1998). These cells are enriched with the dopamine signaling protein, DARPP-32, levels of which are decreased in the striatum of HD patients and mouse models and can be upregulated by BDNF (Mangiarini et al., 1996; Bibb et al., 2000; van Dellen et al., 2000b; Bogush et al., 2007; Gharami et al., 2008). Thus, we investigated the effects of LM22A-4 on striatal DARPP-32 in R6/2 mice. The area occupied by DARPP-32-immunostained cells in the striatum was significantly lower in vehicle-treated R6/2 mice compared with the WTs (Fig. 4A,B). Moreover, Western blot analyses of striatal extracts showed that DARPP-32 levels were decreased by 61 ± 5% in the R6/2-vehicle group compared with WTs (Fig. 4C). R6/2 mice treated with LM22A-4 showed small but significant increases in both the area occupied by DARPP-32 immunostaining and protein levels (Fig. 4).
Figure 4.

Decreases in striatal DARPP-32 levels are ameliorated by LM22A-4 in R6/2 mice. A, Representative DARPP-32 immunostaining in striatum of WT (left) and R6/2 mice (right) treated with vehicle (Veh; top) or LM22A-4 (LM-4; bottom) is shown. Scale bar, 80 μm. The immunostaining appeared lighter, especially in the neuropil, in R6/2 mice given Veh (n = 10) compared with the WT veh (n = 12), WT LM-4 (n = 10) and R6/2 LM-4 (n = 13) groups. B, Quantitative analyses showed that the decrease in area occupied by DARPP-32 immunostained cells and processes in Veh-treated R6/2 mice (*p ≤ 0.05 vs WT Veh) was prevented with LM-4 treatment (+p ≤ 0.05 vs R6/2 Veh). The area occupied by the immunostained cells and processes was expressed as a percentage of the total area analyzed and normalized by the WT Veh group. C, Representative Western blot of striatal homogenates from 11- to 12-week-old WT or R6/2 mice treated with Veh or LM-4 shows that DARPP-32 protein levels are decreased in the R6/2 Veh group compared with WTs (***p ≤ 0.001) and that LM-4 ameliorated this deficit (+p ≤ 0.05; two-tailed Student's t test). Immunobands from two mice per group are shown. For quantitation, samples from each mouse (n = 8–10 mice/group) were run in triplicate, values were normalized to the WT Veh group, and runs for each mouse were averaged. Results are expressed as mean ± SEM. Statistical significance was determined by ANOVA and Newman–Keuls post hoc test, unless otherwise noted.
Another set of striatal neurons that degenerates in HD is the PV-containing GABAergic interneurons (Harrington and Kowall, 1991). Of the striatal interneurons, those containing PV are the most vulnerable to HD degeneration (Harrington and Kowall, 1991) and correspondingly have the highest incidence of intranuclear huntingtin aggregates (Kosinski et al., 1999) and a deficiency in excitotoxicity-induced activated CREB, a protein that is neuroprotective in these cells (Giampà et al., 2006, 2009). Furthermore, PV-containing interneurons are regulated by BDNF (Cicchetti et al., 2000; Berghuis et al., 2004; Grosse et al., 2005; Eto et al., 2010) and although they constitute only 3–5% of striatal cells (Kawaguchi et al., 1995), they provide strong inhibitory control of neurons projecting to the cortex (Koós and Tepper, 1999), a connection that is disrupted in HD (Cepeda et al., 2013). Therefore the effects of LM22A-4 on this cell type were investigated. The area occupied by PV-immunostained cell bodies and processes was significantly decreased in R6/2 mice compared with WTs (Fig. 5A,B). However, the number and somal area of PV-containing cells was similar [number (mean ± SD): 57 ± 32 and 52 ± 9 cells/mm2; area: 69 ± 13 and 63 ± 11 μm2, for WT veh vs R6/2 veh, respectively]. Thus, the decrease in total area appeared to be predominately due to degeneration of parvalbuminergic neurites. Direct quantitative analyses of the neurites of PV-stained cells showed that the percentage of striatal PV cells lacking immunoreactive neurites was higher in vehicle-treated R6/2 mice than WTs (Fig. 5C). Of the neurite-containing PV cells, neurite number was unaltered (data not shown) but neurites on branch orders 1–4 had significantly decreased volumes in R6/2 mice given vehicle, and showed a decrease, although not statistically significant, in the length of third- and fourth-order branches compared to WTs (Fig. 5D,E). Treating R6/2 mice with LM22A-4 decreased the percentage of cells lacking PV-stained neurites and restored neurite volume and length to WT levels (Fig. 5C–E).
Figure 5.

Degeneration of neurites from parvalbuminergic interneurons in R6/2 striatum is prevented by LM22A-4. A, Reconstructed drawings from Neurolucida tracings of PV-immunostained interneurons in striatum. Scale bar, 10 μm. B, Quantitative analyses showed that the decrease in area occupied by PV-immunostained neurons and neurites seen in vehicle (Veh)-treated R6/2 mice (*p ≤ 0.05 vs WT Veh) was prevented with LM22A-4 (LM-4) treatment (+p ≤ 0.05 vs R6/2 Veh; n = 9/group). The area occupied by the immunostained cells and processes was expressed as a percentage of the total area analyzed and normalized by the WT Veh group. C, R6/2 mice given Veh had a higher percentage of soma that lacked PV-stained neurites than WT mice (*p ≤ 0.05) and LM-4 prevented this deficit (+p ≤ 0.05 vs R6/2 Veh). D, E, PV-stained neurites of R6/2 vehicle mice also had reduced volumes on branch orders 1–4 (*p ≤ 0.05 vs WT Veh) and were shorter on branch orders 3–4, although this difference did not reach statistical significance. LM-4 increased the volume and length of the PV-stained neurites (+p ≤ 0.05 vs R6/2 Veh). Results are expressed as mean ± SEM. Statistical significance was determined by ANOVA with Newman–Keuls post hoc testing.
LM22A-4 decreases inflammation in the striatum of R6/2 mice
Inflammatory reactions, specifically microglial activation and/or cytokine release, have been described in brains of HD patients (Sapp et al., 2001; Pavese et al., 2006; Simmons et al., 2007; Silvestroni et al., 2009) and mouse models (Bjorkqvist et al., 2008; Bouchard et al., 2012; Franciosi et al., 2012; Kwan et al., 2012), including R6/2 mice (Simmons et al., 2007; Wacker et al., 2009), and may contribute importantly to the progression of the disease (Möller, 2010). We evaluated the effect of LM22A-4 on inflammation, as assessed with immunostaining for IBA-1, a protein expressed by activated or proliferating microglia (Ridley et al., 1992; Imai et al., 1996; Ito et al., 1998), that is increased in R6/2 brains (Simmons et al., 2007; Wacker et al., 2009). The area occupied by IBA-1-stained microglia increased by 73 ± 13% in R6/2 mice given vehicle compared with the WT vehicle group (Fig. 6). R6/2 mice treated with LM22A-4 had significantly reduced IBA-1 staining area compared with those given vehicle.
Figure 6.

Inflammation is decreased by LM22A-4 in the R6/2 striatum. Immunostaining for IBA-1, a microglial marker, is shown in the striatum of a vehicle (Veh)-treated WT (top left) and R6/2 (top right) mouse, and an LM22A-4 (LM-4)-treated R6/2 mouse (bottom left). Scale bar, 80 μm. Quantitation showed that the area occupied by IBA-1 immunostaining increased significantly in Veh-treated R6/2 mice compared with WT mice (***p ≤ 0.001 vs WT Veh) and that LM-4 ameliorated this effect (+p ≤ 0.05; n = 9/group). The area occupied by the immunostained cells and processes was expressed as a percentage of the total area analyzed and normalized by the WT Veh group. Results are expressed as the mean ± SEM. Statistical significance was determined with an ANOVA and Newman–Keuls post hoc test.
Dendritic spine pathology of MSNs is prevented by LM22A-4
We also investigated the effect of LM22A-4 on dendritic spine density, as marked spine loss is evident on MSNs of HD patients (Graveland et al., 1985; Ferrante et al., 1991) and mouse models (Guidetti et al., 2001; Spires et al., 2004b; Xie et al., 2010; Lerner et al., 2012), including R6/2 mice (Klapstein et al., 2001; Heck et al., 2012), and BDNF deficiencies are associated with this deficit (Zuccato and Cattaneo, 2007; Xie et al., 2010). Dendritic branch orders 2 and higher were analyzed, as these are the most affected in HD mice (Klapstein et al., 2001; Lerner et al., 2012). Spine density was decreased by 25 ± 5% in vehicle-treated R6/2 mice relative to WTs. This reduction was similar to that described in the literature for R6/2 mice (Klapstein et al., 2001; Heck et al., 2012). Treatment with LM22A-4 prevented the decrease in spines at each branch order examined (Fig. 7A–C).
Figure 7.

LM22A-4 prevents dendritic spine loss on R6/2 striatal MSNs. A, Reconstructed drawings from Neurolucida tracings of one dendritic tree of Golgi-stained MSNs in striatum. Scale bar, 10 μm. B, Spines on the fourth dendritic branch of MSNs from each of the four experimental groups are shown. Scale bar, 2.5 μm. C, Spine density on dendrites of MSNs was reduced in vehicle (Veh)-treated R6/2 mice (n = 7–9 mice/group; **p ≤ 0.01 vs WT Veh). This decrease was prevented by LM22A-4 (LM-4) on each branch order examined (*p ≤ 0.05 vs WT Veh and +p ≤ 0.05 vs R6/2 Veh; two-tailed Student's t tests; left graph) and this protective effect was particularly evident when data from branch orders 2–5 were combined (+++p < 0.001 vs R6/2 Veh; right graph). Results are expressed as mean ± SEM. Statistical significance was determined by ANOVA with Newman–Keuls post hoc testing.
LM22A-4's effects on body weight, life span, and gross motor disturbances in R6/2 mice
Body weight was assessed in R6/2 and WT mice that received LM22A-4 or vehicle treatment for 7 weeks starting at 4 weeks of age (Fig. 8A). R6/2 mice given vehicle weighed significantly less than WTs by the fourth week of treatment and throughout the rest of the study. R6/2 mice treated with LM22A-4 also weighed less than WTs starting at the fourth treatment week; however, their body weights were slightly (9 ± 2%) but significantly greater than R6/2-vehicle mice by the last treatment weeks (Fig. 8A). LM22A-4 did not affect WT body weight compared with those given vehicle. Furthermore, LM22A-4 did not significantly affect the survival of R6/2 mice in Kaplan-Meier analysis (Fig. 8B). R6/2 mice given LM22A-4 lived an average of 105 ± 24 d (mean ± SD) compared with 101 ± 28 d for those that received vehicle. In addition, WT mice treated with LM22A-4 did not have shorter life spans compared with WT mice treated with vehicle, suggesting a lack of toxicity of the ligand.
Figure 8.

Effects of LM22A-4 on body weight, life span, and motor performance in R6/2 mice. A, Body weights of vehicle (Veh)-treated R6/2 mice were significantly less than WT mice given Veh or LM22A-4 (LM-4) starting at 7 weeks of age (fourth treatment week) and progressively decreased with aging. This decline was delayed by LM-4 treatment from the fifth to seventh treatment weeks (n = 35–37 mice/group).***p ≤ 0.001 vs WT Veh and +p ≤ 0.05 vs R6/2 Veh, two-way RM-ANOVA, with Bonferroni post-testing. B, Kaplan–Meier analysis curve showing that LM-4 does not affect survival of R6/2 mice (n = 10/group). C, Time to descend a vertical pole was longer for Veh-treated R6/2 mice than WTs (***p ≤ 0.001 vs WT Veh); LM-4 decreased descent time in R6/2 mice (++p ≤ 0.01 vs R6/2 Veh; n = 18/group). D, Hanging duration was shorter for R6/2 mice given Veh than WTs (***p ≤ 0.001 vs WT Veh); LM-4-treated R6/2 mice remained hanging longer than R6/2 Veh mice (+++p ≤ 0.001; n = 9–10/group). E, Rotarod performance was impaired in R6/2 mice (*p ≤ 0.05 and ***p ≤ 0.001 vs WT Veh) and was not affected by LM-4 treatment (n = 19–20/group). Results are expressed as mean ± SEM. Statistical significance was determined with an ANOVA and Newman–Keuls post hoc test, unless otherwise noted.
Before testing for the effects of LM22A-4 on motor ability, we first investigated whether the compound affected overall activity levels. The activity of mice allowed to explore ad libitum an open chamber was measured after 2 weeks of dosing. During the first 10 min of exploration, all mice, regardless of genotype or treatment, traveled similar distances at a similar velocity (data not shown) indicating that LM22A-4 did not generally affect activity levels.
R6/2 mice have a distinct gross motor phenotype that reliably emerges at ∼6 weeks of age and includes tremor, hindlimb clasping, and handling/stress-induced seizures (Mangiarini et al., 1996; Hickey et al., 2005; Stack et al., 2005). We evaluated the effect of LM22A-4 on the latter two motor disturbances. Clasping was assessed with the tail suspension test once a week from ages 8 to 10 weeks old. Fewer R6/2 mice treated with LM22A-4 clasped (4 of 10 mice) than those given vehicle (8 of 9 mice) at 8 weeks of age (p = 0.03, 2 × 2 contingency table, Fisher's exact test). However, this effect was not seen at 9 or 10 weeks old and clasp latency was not affected by LM22A-4 treatment (mean of three tests, R6/2 veh: 25 ± 2 s; R6/2 LM22A-4: 28 ± 3 s). The number of seizures associated with handling at the time of dosing was also assessed throughout treatment. R6/2 mice given vehicle and those that received LM22A-4 had a similar mean number of seizures over treatment weeks 2–7 (4.2 ± 1.7 vs 3.6 ± 1.0, respectively, n = 10/group). However, LM22A-4 showed a tendency to delay the onset of seizures. During treatment weeks 2–4, 44% of the R6/2 mice given vehicle had seizures while only 17% of those treated with LM22A-4 seized (p = 0.06, Student's t test). This difference was eliminated by treatment weeks 5–7, with seizures observed in 52 ± 5% of R6/2 vehicle mice and 53 ± 8% of R6/2 LM22A-4 mice.
LM22A-4 improves motor ability in R6/2 mice
The therapeutic efficacy of LM22A-4 on motor performance was evaluated in R6/2 mice using tests that assessed particular aspects of movement. The vertical pole descent test is a sensorimotor assay of downward climbing that is particularly sensitive to striatal damage (Matsuura et al., 1997). Performance on this test significantly differed between the groups when tested at 9 weeks of age. R6/2 mice given vehicle took ∼2 times longer to climb down the pole compared with WT mice (Fig. 8C). Treatment of R6/2 mice with LM22A-4 significantly improved their descent time. LM22A-4 also improved the hanging endurance/grip strength of R6/2 mice, in the wire hang test. Almost all of the WT mice held onto the metal rod for the duration of the test, while vehicle-treated R6/2 mice fell at much earlier times (Fig. 8D). R6/2 mice treated with LM22A-4 remained hanging 2 ± 0.5 (mean ± SD) times longer than those given vehicle. LM22A-4 did not affect motor balance and coordination in rotarod testing (Fig. 8E).
Striatal neuropathology is ameliorated by LM22A-4 in BACHD mice
Given the positive results in R6/2 mice, we investigated whether LM22A-4 could produce similar effects in the BACHD mouse model of HD, which expresses full-length mutant huntingtin and is a better replicate of the genetic component of the disease (Menalled, 2005; Gray et al., 2008; Menalled et al., 2009). These mice develop a subtle HD phenotype at a slower rate than R6/2 mice, which could afford greater discrimination of TrkB ligand effects. Behavioral deficits emerge at ∼2 months of age, while cortical BDNF expression decreases at 6 months, and, at 12 months, huntingtin aggregates and reduced striatal volume appear (Gray et al., 2008; Menalled et al., 2009; Pouladi et al., 2012).
We treated BACHD mice with LM22A-4 for 6 months beginning at 2 months of age. As little is known about pathology in these mice at 8 months of age, we first investigated whether striatal BDNF and TrkB levels are altered at this age and then determined suitable endpoints for discerning drug effects. Western immunoblotting was used to assess changes in BDNF and TrkB levels in striatum of BACHD mice due to mutant huntingtin and/or LM22A-4 treatment. In vehicle-treated BACHD mice, protein levels of mBDNF were 16 ± 5% lower (Fig. 9A) than in WTs. As most striatal BDNF is cortically derived and this transport is disrupted in HD (Altar et al., 1997; Zuccato and Cattaneo, 2007), this result agrees with a previous study showing that BDNF mRNA levels are reduced in cortex of BACHD mice at 6 months of age (Gray et al., 2008). TrkB levels were similar in WT and BACHD mice (Fig. 9B). At the 2 h postinjection time point examined, LM22A-4 did not significantly alter striatal mBDNF or TrkB levels.
Figure 9.

Mature BDNF levels are decreased while TrkB levels are unaltered in striatum of 8-month-old BACHD mice. Representative Western blots from BACHD mice (treated for 6 months beginning at 2 months of age) showing striatal protein levels of (A) mBDNF (∼14–15 kDa) and (B) TrkB. Immunobands from one mouse per group and corresponding densitometric analyses are shown. Corresponding tubulin immunobands from the stripped and reprobed blots are shown at the bottom of each gel panel. For quantitation, samples from each mouse (n = 5–9 mice/group) were run in duplicate, values were normalized to the WT vehicle group, and runs for each mouse were averaged. mBDNF protein levels are lower in striatum of vehicle-treated BACHD mice compared with WTs (*p ≤ 0.05). Striatal levels of TrkB were unaltered in BACHD mice. LM22A-4 (LM-4) did not significantly affect mBDNF or TrkB levels. Results are expressed as mean ± SEM, and, for comparison purposes, some nonadjacent lanes of Western blots were moved together and separated by spaces. Statistical significance was determined with an ANOVA with a Newman–Keuls post hoc test.
Intranuclear huntingtin aggregates were not detectable with EM48 or ubiquitin immunostaining in BACHD mice at 8 months of age (data not shown). Striatal DARPP-32 protein levels appeared to decrease slightly, though not significantly, in BACHD mice at 10 months of age (Pouladi et al., 2012). Similarly, we found that the area and intensity of neuronal DARPP-32 immunostaining did not differ in BACHD and WT mice (data not shown). Since neurites are particularly susceptible to degeneration, we also determined whether DARPP-32-containing processes were affected by mutant huntingtin. DARPP-32-containing neurites exhibited decreased surface area and length in vehicle-treated BACHD mice compared with WTs, which did not reach statistical significance (Fig. 10A–C). However, LM22A-4 treatment did significantly increase both measures in BACHD mice compared with those given vehicle.
Figure 10.
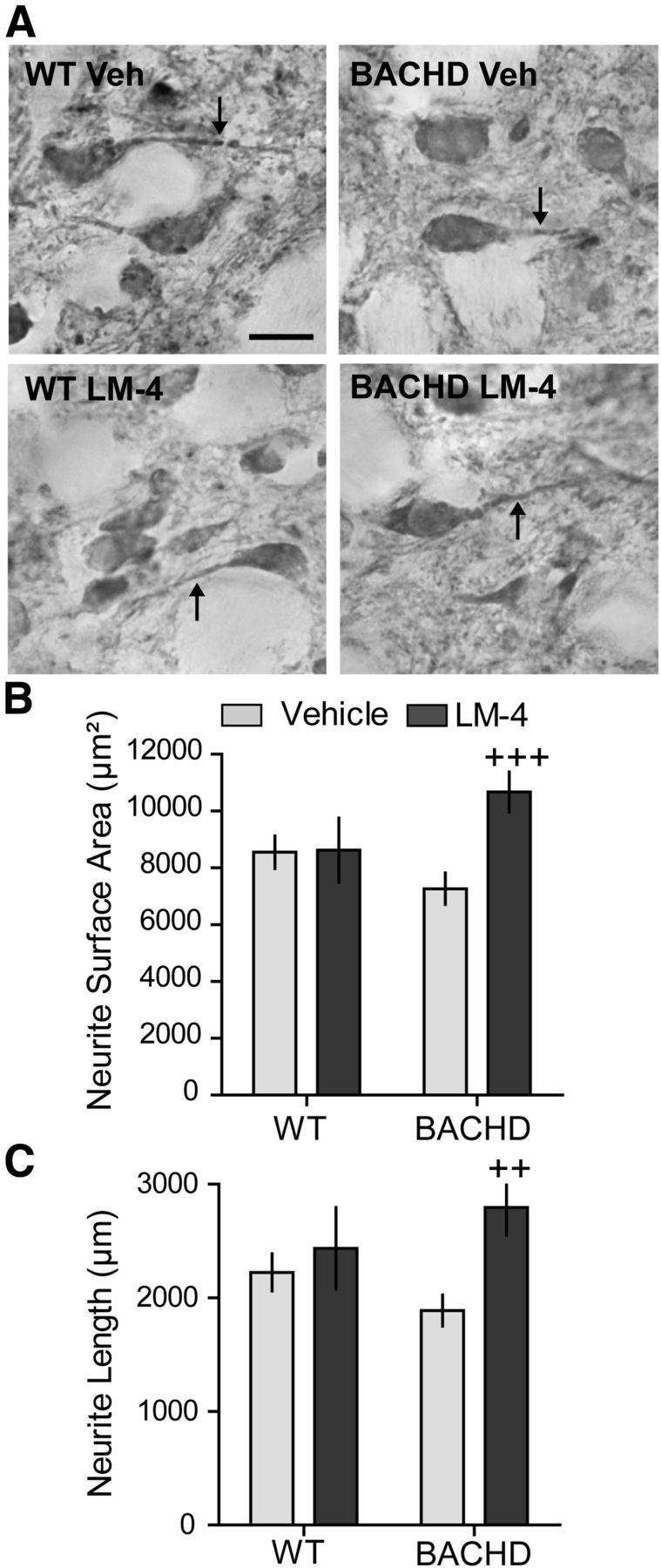
LM22A-4 ameliorates degeneration of striatal DARPP-32 neurites in BACHD mice. A, DARPP-32 immunostaining in striatum of WT (left) and BACHD mice (right) treated with vehicle (Veh; top) or LM22A-4 (LM-4; bottom) is shown. Scale bar, 10 μm. The neurites of DARPP-32-stained neurons (A, arrows) of BACHD mice tend to have a smaller surface area (B) and shorter length (C) than those of WT mice (n = 8–9 mice/group; ANOVA p = 0.04, F = 3.24; p = 0.07, Student's t test WT Veh vs BACHD Veh). Treating BACHD mice with LM-4 significantly increased the surface area and length of the neurites containing DARPP-32 (++p = 0.005 and +++p = 0.001 vs BACHD veh, two-tailed Student's t tests).
Changes in the electrophysiology of corticostriatal circuits arise at 2–6 months old in BACHD mice (Gray et al., 2008; Spampanato et al., 2008; André et al., 2011). Therefore, we investigated whether potentially related structural changes were evident by assessing dendritic spine density (Fig. 11A), which has not been investigated in these mice. The density of dendritic spines on MSNs differed significantly between groups (n = 9–10 mice/group) with a 30 ± 6% decrease in vehicle-treated BACHD mice on dendritic branches third-order and higher compared with WTs (Fig. 11B, right graph). For both third- and fourth-order branches, vehicle-treated BACHD mice had significantly fewer spines than WTs and this decrease was prevented by LM22A-4 treatment (Fig. 11B, left graph).
Figure 11.

LM22A-4 prevents deficits in dendritic spines of BACHD striatal MSNs. A, Reconstructed drawings from Neurolucida tracings of one dendritic tree of Golgi-stained MSNs in striatum of WT (left) and BACHD mice (right) treated with vehicle (Veh; top) or LM22A-4 (LM-4; bottom). Scale bar, 10 μm. B, Spine density of MSNs was reduced in Veh-treated BACHD mice on dendritic branch orders 3–5 (*p ≤ 0.05 vs WT Veh, ANOVA and Newman–Keuls post hoc test, right graph) particularly on third and fourth orders (*p ≤ 0.05 vs WT Veh, Student's t test, left graph); this reduction was prevented by LM-4 (+p ≤ 0.04 vs BACHD Veh, Student's t test; n = 9–10/group). Results are expressed as mean ± SEM.
Effects of LM22A-4 on body weight and motor performance in BACHD mice
BACHD mice given vehicle traveled less distance in an activity chamber than WT mice at 8 months of age (Fig. 12A), but their velocity was unchanged (data not shown). LM22A-4 prevented the hypoactivity of BACHD mice. Downward climbing ability of vehicle-treated BACHD mice was also impaired, as they took significantly longer to descend a pole than their WT littermates (Fig. 12B). LM22A-4 treatment reduced the climbing time of BACHD mice on the vertical pole test. As seen with the R6/2 mice, LM22A-4 did not affect deficits in rotarod performance (Fig. 12C). Wire hang was not performed on BACHD mice as a previous report found that their grip strength is unaltered (Menalled et al., 2009).
Figure 12.

Effects of LM22A-4 on motor performance and body weight in BACHD mice. A, Distance traveled during the first 10 min in an activity chamber was decreased in vehicle (Veh)-treated BACHD mice compared with WT mice (*p ≤ 0.05). This hypoactivity was prevented by LM22A-4 (LM-4) treatment (+p ≤ 0.05 vs BACHD Veh; n = 12–15/group). B, Downward climbing was also impaired in BACHD Veh mice (***p ≤ 0.001 vs WT Veh); LM-4 improved descent time of BACHD mice on the vertical pole test (+p ≤ 0.05 vs BACHD Veh; n = 15–22/group). C, Deficits in rotarod performance were seen at 5 (Test 2) and 8 (Test 3) months of age (both **p ≤ 0.01 vs WT Veh), but not at 2 months (Test 1); LM-4 had no effect on performance on this test (n = 15–22/group). D, Body weight was not affected by LM-4 treatment in WT or BACHD mice (n = 15–22/group). Results are expressed as mean ± SEM. Statistical significance was determined by ANOVA with Newman–Keuls post hoc testing.
From the first week of treatment at 2 months of age, vehicle-treated BACHD mice weighed significantly more than WT mice (Fig. 12D), as reported previously (Gray et al., 2008; Menalled et al., 2009; Pouladi et al., 2012). BACHD mice given LM22A-4 also weighed more than WTs beginning at the third treatment week until the end of the experiment, and their weights did not differ from vehicle-treated BACHD mice.
Discussion
Current HD therapeutics are directed at diminishing disease symptoms rather than factors contributing to its progression. A key contributor to HD pathogenesis is the loss of BDNF and its signaling via TrkB (Zuccato and Cattaneo, 2007). TrkB signaling intermediates largely overlap those responsible for neuronal survival and synapse integrity. Thus, targeting TrkB receptors with small molecule ligands could offer mechanism-based therapeutics that will not only offset effects of BDNF deficiencies, but also combat underlying HD degenerative mechanisms. We tested this hypothesis by chronically administering the small molecule TrkB ligand, LM22A-4, to two HD mouse models. LM22A-4 penetrated the CNS in sufficient amounts to activate TrkB, improve motor ability, and prevent neuropathology, including huntingtin aggregates in striatum and cortex, as well as striatal inflammation, dendritic spine deficits, decreased DARPP-32 levels, and degeneration of neurites on PV-containing cells. These results strongly support the idea that small molecule ligands specific for TrkB could be effective HD therapeutics.
A concern with using TrkB ligands for HD therapy is adequate expression of the target, as striatal TrkB levels are reduced in HD patients and mouse models, and may be present in insufficient amounts to activate downstream signaling (Zuccato and Cattaneo, 2007). Although striatal TrkB levels were reduced by ∼30% in R6/2 mice and a concomitant decrease in activation was observed, LM22A-4 restored TrkB phosphorylation and activated its major effectors: AKT, PLCγ, and CREB. Thus, TrkB receptors retained their capability to trigger downstream signaling and supported striatal function. Overall, these findings demonstrate LM22A-4 target engagement accompanied by an ameliorated HD phenotype.
The restorative effect of LM22A-4 on AKT, PLCγ, and CREB signaling could counteract many HD-related degenerative mechanisms. AKT is a prosurvival kinase that phosphorylates, thereby inhibiting, substrates involved in cell death processes (Datta et al., 1999), and is decreased in the HD striatum (Humbert et al., 2002; Colin et al., 2005). When activated, AKT decreases mutant huntingtin aggregation and toxicity directly by phosphorylating huntingtin or indirectly by increasing arfaptin2's promotion of huntingtin degradation via the proteasome (Humbert et al., 2002; Colin et al., 2005; Rangone et al., 2005). BDNF-induced activation of AKT also protects striatal neurons from excitotoxic degeneration (Brito et al., 2013). Furthermore, disrupted BDNF-TrkB-PLCγ signaling and decreased transcription of CREB-dependent genes, including BDNF, is associated with learning deficits in HD mice (Giralt et al., 2009, 2012). Thus, AKT, PLCγ, and CREB activation by LM22A-4 could potentially prevent diverse and fundamental HD pathologies.
The large decreases in diffuse nuclear huntingtin and aggregates seen with LM22A-4 treatment indicate that the ligand mitigates huntingtin oligomer production and/or accumulation. Numerous processes contribute to aggregate formation including aberrant cleavage of mutant huntingtin, protein misfolding, and reduced protein degradation due to disruptions in autophagy and/or ubiquitin-proteasome system (Zuccato et al., 2010). LM22A-4 may mediate its effects on huntingtin oligomers by increasing their clearance, as BDNF/TrkB signaling can facilitate ubiquitin-proteasome processing (Jia et al., 2008; Cheng et al., 2011) or by activating AKT, as discussed above. Although the debate concerning whether insoluble aggregates are toxic or neuroprotective is ongoing (Kuemmerle et al., 1999; Zuccato et al., 2010), reduced aggregate load due to silencing mutant huntingtin expression (Yamamoto et al., 2000), overexpressing the BDNF transgene (Gharami et al., 2008), or various therapeutic approaches (Dedeoglu et al., 2002; Ferrante et al., 2002; Duan et al., 2003; Nguyen et al., 2005; Chopra et al., 2007; DeMarch et al., 2008; Simmons et al., 2011) is associated with improved cognition and/or motor performance in HD mice. Thus, LM22A-4's effects on mutant huntingtin pathology may contribute to its restorative effects on motor ability.
LM22A-4 ameliorated deficits in two cell populations severely affected in the HD striatum: DARPP-32-expressing MSNs and PV-containing interneurons. DARPP-32 is important for dopamine function, and reduced BDNF expression in HD mice exacerbates dopamine deficits (Bibb et al., 2000; Pineda et al., 2005). Thus, upregulating DARPP-32 with LM22A-4 may alleviate disturbances in dopamine transmission that contribute to motor deficits in HD mice. BDNF upregulates DARPP-32 via AKT signaling (Stroppolo et al., 2001; Bogush et al., 2007) and, since LM22A-4 increases AKT activation, it may elevate DARPP-32 levels via this mechanism. BDNF also affects the electrophysiological function and survival of striatal PV-containing interneurons (Cicchetti et al., 2000; Berghuis et al., 2004; Grosse et al., 2005; Eto et al., 2010). These interneurons are sparse but integral to striatal function as they mediate “feedforward” inhibition from cortex to MSNs (Bennett and Bolam, 1994; Kawaguchi et al., 1995). Therefore, preserving PV-containing interneurons with LM22A-4 may be another way that the ligand positively affects motor performance.
Notably, LM22A-4 largely reduced dendritic spine pathology of MSNs. Spine density was decreased on striatal MSNs by 25–30% in both R6/2 and BACHD mice and restoring TrkB signaling with LM22A-4 almost completely prevented these deficits. This effect may be direct, since TrkB is located on the plasma membrane of striatal spines (Wu et al., 1996; Drake et al., 1999; Ma et al., 2012) and is thus situated to initiate cytoskeletal changes, such as activity-driven actin polymerization needed to form and/or stabilize spines (Rex et al., 2007; Lynch et al., 2008). This process is disrupted in hippocampus of HD knock-in mice, as is long-term potentiation (LTP), and is restored with BDNF or by upregulating the neurotrophin with ampakines (Lynch et al., 2007; Simmons et al., 2009). In striatum, BDNF and TrkB signaling promote LTP (Jia et al., 2010) and may increase spine density of MSNs in HD mice (Baquet et al., 2004; Spires et al., 2004b; Xie et al., 2010). Furthermore, AKT, PLCγ, and CREB signaling, which are increased by LM22A-4, have been linked to regulating spine formation, morphology, and/or function (Zhou et al., 2007; Cuesto et al., 2011; Majumdar et al., 2011). Together, these results suggest that LM22A-4's positive effect on MSN spine density contributes substantially to the preserved striatal function in HD mice. This finding is the first demonstration that a small molecule ligand increases spine density in the HD striatum. It is of particular significance since spinopathy is considered an underlying mechanism in numerous neurodegenerative conditions and spines have become prime therapeutic targets (Blanpied and Ehlers, 2004; Lynch et al., 2008; Ehrnhoefer et al., 2011).
R6/2 and BACHD mice treated with LM22A-4 showed improved performance on the pole test but not the rotarod. Pole test performance is susceptible to reduced striatal dopamine (Matsuura et al., 1997), while the rotarod involves motor coordination and cardiopulmonary endurance and may require dopamine more for task learning (Chagniel et al., 2012). BDNF modulates motor system plasticity and function (Kleim et al., 2006; Fritsch et al., 2010; McHughen et al., 2010) so it is also of interest that rotarod performance was not affected when cortical and cortically derived striatal BDNF was selectively eliminated (Baquet et al., 2004). Moreover, upregulating BDNF using amapkines improved pole test performance of R6/2 mice to a greater extent than the rotarod (Simmons et al., 2011). Thus, the differential dependence on dopamine and/or BDNF may explain why LM22A-4 selectively improved performance on these tasks.
Despite the ameliorative effects of LM22A-4 on striatal neuropathology and motor dysfunction, the compound did not extend the R6/2 life span. Neuronal death does not cause R6/2 lethality as neuron loss is nominal before their death (Turmaine et al., 2000); however, many other factors may shorten their life span including body weight loss, seizures, muscle atrophy, cardiac dysfunction, and diabetes (Hunt and Morton, 2005). The former two factors have been negatively associated with elevated BDNF (Scharfman, 2005; Chaldakov et al., 2009); however, they were slightly improved with LM22A-4 treatment. LM22A-4's lack of effect on life span is unlikely to involve peripheral toxicity as TrkB receptors show little or no expression in the adult periphery including neural and non-neural tissues (Klein et al., 1990; Yamamoto et al., 1996). Given its many positive effects, LM22A-4's inability to extend R6/2 life span may not be pertinent in considering TrkB ligands as HD therapeutics.
Further support for the hypothesis that small molecule targeting of TrkB can ameliorate HD pathologies is provided by a recent study demonstrating that 7,8-dihydroxyflavone (7,8,-DHF) and its synthetic derivative (4′-dimethylamino-7,8-DHF) increased TrkB (Y706) and MAPK phosphorylation, improved coordination on one motor test, decreased brain atrophy, maintained DARPP-32 levels, alleviated impaired neurogenesis, and extended survival in the N171–82Q HD mouse model (Jiang et al., 2013). Whether these compounds rescued AKT, PLCγ, or CREB signaling deficiencies or reduced HD cellular pathology, including huntingtin aggregates and spine loss, was not determined. Since flavones have antioxidant effects and can protect cells in the absence of TrkB (Chen et al., 2011), the specificity of the actions of these compounds to TrkB needs to be assessed.
In summary, these results validate the hypothesis that isolated targeting of TrkB, as distinct from the effects of BDNF via TrkB and p75NTR binding, can prevent HD-related degenerative mechanisms and motor function decline in multiple HD mouse models. They also encourage a small molecule strategy for treating HD and other neurodegenerative conditions. Subsequent studies will test if novel derivatives of the TrkB ligand with optimized bioavailability can improve cognitive and psychiatric disturbances and further ameliorate motor impediments and neuropathology.
Footnotes
This work was supported by the National Institutes of Health Grants R21 NS071024-01 and P30 NS069375-01A1, Taube Philanthropies, Koret Foundation, Jean Perkins Foundation, and the Veterans Administration.
F.M.L. is a founder of Pharmatrophix, a company focused on the development of neurotrophin receptor ligands.
References
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- An JJ, Gharami K, Liao GY, Woo NH, Lau AG, Vanevski F, Torre ER, Jones KR, Feng Y, Lu B, Xu B. Distinct role of long 3′ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell. 2008;134:175–187. doi: 10.1016/j.cell.2008.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- André VM, Cepeda C, Fisher YE, Huynh M, Bardakjian N, Singh S, Yang XW, Levine MS. Differential electrophysiological changes in striatal output neurons in Huntington's disease. J Neurosci. 2011;31:1170–1182. doi: 10.1523/JNEUROSCI.3539-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostol BL, Simmons DA, Zuccato C, Illes K, Pallos J, Casale M, Conforti P, Ramos C, Roarke M, Kathuria S, Cattaneo E, Marsh JL, Thompson LM. CEP-1347 reduces mutant huntingtin-associated neurotoxicity and restores BDNF levels in R6/2 mice. Mol Cell Neurosci. 2008;39:8–20. doi: 10.1016/j.mcn.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BD, Bolam JP. Synaptic input and output of parvalbumin-immunoreactive neurons in the neostriatum of the rat. Neuroscience. 1994;62:707–719. doi: 10.1016/0306-4522(94)90471-5. [DOI] [PubMed] [Google Scholar]
- Berghuis P, Dobszay MB, Sousa KM, Schulte G, Mager PP, Härtig W, Görcs TJ, Zilberter Y, Ernfors P, Harkany T. Brain-derived neurotrophic factor controls functional differentiation and microcircuit formation of selectively isolated fast-spiking GABAergic interneurons. Eur J Neurosci. 2004;20:1290–1306. doi: 10.1111/j.1460-9568.2004.03561.x. [DOI] [PubMed] [Google Scholar]
- Bibb JA, Yan Z, Svenningsson P, Snyder GL, Pieribone VA, Horiuchi A, Nairn AC, Messer A, Greengard P. Severe deficiencies in dopamine signaling in presymptomatic Huntington's disease mice. Proc Natl Acad Sci U S A. 2000;97:6809–6814. doi: 10.1073/pnas.120166397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, Raibon E, Lee RV, Benn CL, Soulet D, Magnusson A, Woodman B, Landles C, Pouladi MA, Hayden MR, Khalili-Shirazi A, Lowdell MW, Brundin P, Bates GP, Leavitt BR, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J Exp Med. 2008;205:1869–1877. doi: 10.1084/jem.20080178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpied TA, Ehlers MD. Microanatomy of dendritic spines: emerging principles of synaptic pathology in psychiatric and neurological disease. Biol Psychiatry. 2004;55:1121–1127. doi: 10.1016/j.biopsych.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Bodai L, Marsh JL. A novel target for Huntington's disease: ERK at the crossroads of signaling. The ERK signaling pathway is implicated in Huntington's disease and its upregulation ameliorates pathology. Bioessays. 2012;34:142–148. doi: 10.1002/bies.201100116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogush A, Pedrini S, Pelta-Heller J, Chan T, Yang Q, Mao Z, Sluzas E, Gieringer T, Ehrlich ME. AKT and CDK5/p35 mediate brain-derived neurotrophic factor induction of DARPP-32 in medium size spiny neurons in vitro. J Biol Chem. 2007;282:7352–7359. doi: 10.1074/jbc.M606508200. [DOI] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Borrell-Pagès M, Canals JM, Cordelières FP, Parker JA, Pineda JR, Grange G, Bryson EA, Guillermier M, Hirsch E, Hantraye P, Cheetham ME, Néri C, Alberch J, Brouillet E, Saudou F, Humbert S. Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase. J Clin Invest. 2006;116:1410–1424. doi: 10.1172/JCI27607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard J, Truong J, Bouchard K, Dunkelberger D, Desrayaud S, Moussaoui S, Tabrizi SJ, Stella N, Muchowski PJ. Cannabinoid receptor 2 signaling in peripheral immune cells modulates disease onset and severity in mouse models of Huntington's disease. J Neurosci. 2012;32:18259–18268. doi: 10.1523/JNEUROSCI.4008-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito V, Puigdellívol M, Giralt A, del Toro D, Alberch J, Ginés S. Imbalance of p75(NTR)/TrkB protein expression in Huntington's disease: implication for neuroprotective therapies. Cell Death Dis. 2013;4:e595. doi: 10.1038/cddis.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals JM, Pineda JR, Torres-Peraza JF, Bosch M, Martín-Ibañez R, Muñoz MT, Mengod G, Ernfors P, Alberch J. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington's disease. J Neurosci. 2004;24:7727–7739. doi: 10.1523/JNEUROSCI.1197-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Galvan L, Holley SM, Rao SP, André VM, Botelho EP, Chen JY, Watson JB, Deisseroth K, Levine MS. Multiple sources of striatal inhibition are differentially affected in Huntington's disease mouse models. J Neurosci. 2013;33:7393–7406. doi: 10.1523/JNEUROSCI.2137-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagniel L, Robitaille C, Lacharité-Mueller C, Bureau G, Cyr M. Partial dopamine depletion in MPTP-treated mice differentially altered motor skill learning and action control. Behav Brain Res. 2012;228:9–15. doi: 10.1016/j.bbr.2011.11.019. [DOI] [PubMed] [Google Scholar]
- Chaldakov GN, Tonchev AB, Aloe L. NGF and BDNF: from nerves to adipose tissue, from neurokines to metabokines. Riv Psichiatr. 2009;44:79–87. [PubMed] [Google Scholar]
- Chen J, Chua KW, Chua CC, Yu H, Pei A, Chua BH, Hamdy RC, Xu X, Liu CF. Antioxidant activity of 7,8-dihydroxyflavone provides neuroprotection against glutamate-induced toxicity. Neurosci Lett. 2011;499:181–185. doi: 10.1016/j.neulet.2011.05.054. [DOI] [PubMed] [Google Scholar]
- Cheng PL, Lu H, Shelly M, Gao H, Poo MM. Phosphorylation of E3 ligase Smurf1 switches its substrate preference in support of axon development. Neuron. 2011;69:231–243. doi: 10.1016/j.neuron.2010.12.021. [DOI] [PubMed] [Google Scholar]
- Chopra V, Fox JH, Lieberman G, Dorsey K, Matson W, Waldmeier P, Housman DE, Kazantsev A, Young AB, Hersch S. A small-molecule therapeutic lead for Huntington's disease: preclinical pharmacology and efficacy of C2–8 in the R6/2 transgenic mouse. Proc Natl Acad Sci U S A. 2007;104:16685–16689. doi: 10.1073/pnas.0707842104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchetti F, Prensa L, Wu Y, Parent A. Chemical anatomy of striatal interneurons in normal individuals and in patients with Huntington's disease. Brain Res Brain Res Rev. 2000;34:80–101. doi: 10.1016/S0165-0173(00)00039-4. [DOI] [PubMed] [Google Scholar]
- Colin E, Régulier E, Perrin V, Dürr A, Brice A, Aebischer P, Déglon N, Humbert S, Saudou F. Akt is altered in an animal model of Huntington's disease and in patients. Eur J Neurosci. 2005;21:1478–1488. doi: 10.1111/j.1460-9568.2005.03985.x. [DOI] [PubMed] [Google Scholar]
- Cuesto G, Enriquez-Barreto L, Caramés C, Cantarero M, Gasull X, Sandi C, Ferrús A, Acebes Á, Morales M. Phosphoinositide-3-kinase activation controls synaptogenesis and spinogenesis in hippocampal neurons. J Neurosci. 2011;31:2721–2733. doi: 10.1523/JNEUROSCI.4477-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/S0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- Dedeoglu A, Kubilus JK, Jeitner TM, Matson SA, Bogdanov M, Kowall NW, Matson WR, Cooper AJ, Ratan RR, Beal MF, Hersch SM, Ferrante RJ. Therapeutic effects of cystamine in a murine model of Huntington's disease. J Neurosci. 2002;22:8942–8950. doi: 10.1523/JNEUROSCI.22-20-08942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMarch Z, Giampà C, Patassini S, Bernardi G, Fusco FR. Beneficial effects of rolipram in the R6/2 mouse model of Huntington's disease. Neurobiol Dis. 2008;30:375–387. doi: 10.1016/j.nbd.2008.02.010. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Drake CT, Milner TA, Patterson SL. Ultrastructural localization of full-length trkB immunoreactivity in rat hippocampus suggests multiple roles in modulating activity-dependent synaptic plasticity. J Neurosci. 1999;19:8009–8026. doi: 10.1523/JNEUROSCI.19-18-08009.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998;273:32377–32379. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- Duan W, Guo Z, Jiang H, Ware M, Li XJ, Mattson MP. Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc Natl Acad Sci U S A. 2003;100:2911–2916. doi: 10.1073/pnas.0536856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Guo Z, Jiang H, Ladenheim B, Xu X, Cadet JL, Mattson MP. Paroxetine retards disease onset and progression in Huntingtin mutant mice. Ann Neurol. 2004;55:590–594. doi: 10.1002/ana.20075. [DOI] [PubMed] [Google Scholar]
- Duan W, Peng Q, Masuda N, Ford E, Tryggestad E, Ladenheim B, Zhao M, Cadet JL, Wong J, Ross CA. Sertraline slows disease progression and increases neurogenesis in N171–82Q mouse model of Huntington's disease. Neurobiol Dis. 2008;30:312–322. doi: 10.1016/j.nbd.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrnhoefer DE, Wong BK, Hayden MR. Convergent pathogenic pathways in Alzheimer's and Huntington's diseases: shared targets for drug development. Nat Rev Drug Discov. 2011;10:853–867. doi: 10.1038/nrd3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto R, Abe M, Kimoto H, Imaoka E, Kato H, Kasahara J, Araki T. Alterations of interneurons in the striatum and frontal cortex of mice during postnatal development. Int J Dev Neurosci. 2010;28:359–370. doi: 10.1016/j.ijdevneu.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Richardson EP., Jr Proliferative and degenerative changes in striatal spiny neurons in Huntington's disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci. 1991;11:3877–3887. doi: 10.1523/JNEUROSCI.11-12-03877.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington's disease. J Neurosci. 2002;22:1592–1599. doi: 10.1523/JNEUROSCI.22-05-01592.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Goutan E, Marín C, Rey MJ, Ribalta T. Brain-derived neurotrophic factor in Huntington disease. Brain Res. 2000;866:257–261. doi: 10.1016/S0006-8993(00)02237-X. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1047. doi: 10.1016/S0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- Franciosi S, Ryu JK, Shim Y, Hill A, Connolly C, Hayden MR, McLarnon JG, Leavitt BR. Age-dependent neurovascular abnormalities and altered microglial morphology in the YAC128 mouse model of Huntington disease. Neurobiol Dis. 2012;45:438–449. doi: 10.1016/j.nbd.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Fritsch B, Reis J, Martinowich K, Schambra HM, Ji Y, Cohen LG, Lu B. Direct current stimulation promotes BDNF-dependent synaptic plasticity: potential implications for motor learning. Neuron. 2010;66:198–204. doi: 10.1016/j.neuron.2010.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Gharami K, Xie Y, An JJ, Tonegawa S, Xu B. Brain-derived neurotrophic factor overexpression in the forebrain ameliorates Huntington's disease phenotypes in mice. J Neurochem. 2008;105:369–379. doi: 10.1111/j.1471-4159.2007.05137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampà C, DeMarch Z, D'Angelo V, Morello M, Martorana A, Sancesario G, Bernardi G, Fusco FR. Striatal modulation of cAMP-response-element-binding protein (CREB) after excitotoxic lesions: implications with neuronal vulnerability in Huntington's disease. Eur J Neurosci. 2006;23:11–20. doi: 10.1111/j.1460-9568.2005.04545.x. [DOI] [PubMed] [Google Scholar]
- Giampà C, Middei S, Patassini S, Borreca A, Marullo F, Laurenti D, Bernardi G, Ammassari-Teule M, Fusco FR. Phosphodiesterase type IV inhibition prevents sequestration of CREB binding protein, protects striatal parvalbumin interneurons and rescues motor deficits in the R6/2 mouse model of Huntington's disease. Eur J Neurosci. 2009;29:902–910. doi: 10.1111/j.1460-9568.2009.06649.x. [DOI] [PubMed] [Google Scholar]
- Giampà C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington's disease. PLoS One. 2010;5:e13417. doi: 10.1371/journal.pone.0013417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gines S, Seong IS, Fossale E, Ivanova E, Trettel F, Gusella JF, Wheeler VC, Persichetti F, MacDonald ME. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington's disease knock-in mice. Hum Mol Genet. 2003;12:497–508. doi: 10.1093/hmg/ddg046. [DOI] [PubMed] [Google Scholar]
- Giralt A, Rodrigo T, Martín ED, Gonzalez JR, Milà M, Ceña V, Dierssen M, Canals JM, Alberch J. Brain-derived neurotrophic factor modulates the severity of cognitive alterations induced by mutant huntingtin: involvement of phospholipaseCgamma activity and glutamate receptor expression. Neuroscience. 2009;158:1234–1250. doi: 10.1016/j.neuroscience.2008.11.024. [DOI] [PubMed] [Google Scholar]
- Giralt A, Puigdellívol M, Carretón O, Paoletti P, Valero J, Parra-Damas A, Saura CA, Alberch J, Ginés S. Long-term memory deficits in Huntington's disease are associated with reduced CBP histone acetylase activity. Hum Mol Genet. 2012;21:1203–1216. doi: 10.1093/hmg/ddr552. [DOI] [PubMed] [Google Scholar]
- Graveland GA, Williams RS, DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington's disease. Science. 1985;227:770–773. doi: 10.1126/science.3155875. [DOI] [PubMed] [Google Scholar]
- Gray M, Shirasaki DI, Cepeda C, André VM, Wilburn B, Lu XH, Tao J, Yamazaki I, Li SH, Sun YE, Li XJ, Levine MS, Yang XW. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008;28:6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse G, Djalali S, Deng DR, Höltje M, Hinz B, Schwartzkopff K, Cygon M, Rothe T, Stroh T, Hellweg R, Ahnert-Hilger G, Hörtnag H. Area-specific effects of brain-derived neurotrophic factor (BDNF) genetic ablation on various neuronal subtypes of the mouse brain. Brain Res Dev Brain Res. 2005;156:111–126. doi: 10.1016/j.devbrainres.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Charles V, Chen EY, Reddy PH, Kordower JH, Whetsell WO, Jr, Schwarcz R, Tagle DA. Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp Neurol. 2001;169:340–350. doi: 10.1006/exnr.2000.7626. [DOI] [PubMed] [Google Scholar]
- Han J, Pollak J, Yang T, Siddiqui MR, Doyle KP, Taravosh-Lahn K, Cekanaviciute E, Han A, Goodman JZ, Jones B, Jing D, Massa SM, Longo FM, Buckwalter MS. Delayed administration of a small molecule tropomyosin-related kinase B ligand promotes recovery after hypoxic-ischemic stroke. Stroke. 2012;43:1918–1924. doi: 10.1161/STROKEAHA.111.641878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington KM, Kowall NW. Parvalbumin immunoreactive neurons resist degeneration in Huntington's disease striatum. J Neuropathol Exp Neurol. 1991;50:309. doi: 10.1097/00005072-199107000-00006. [DOI] [PubMed] [Google Scholar]
- Heck N, Betuing S, Vanhoutte P, Caboche J. A deconvolution method to improve automated 3D-analysis of dendritic spines: application to a mouse model of Huntington's disease. Brain Struct Funct. 2012;217:421–434. doi: 10.1007/s00429-011-0340-y. [DOI] [PubMed] [Google Scholar]
- Hickey MA, Gallant K, Gross GG, Levine MS, Chesselet MF. Early behavioral deficits in R6/2 mice suitable for use in preclinical drug testing. Neurobiol Dis. 2005;20:1–11. doi: 10.1016/j.nbd.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Hickey MA, Kosmalska A, Enayati J, Cohen R, Zeitlin S, Levine MS, Chesselet MF. Extensive early motor and nonmotor behavioral deficits are followed by striatal neuronal loss in knock-in Huntington's disease mice. Neuroscience. 2008;157:280–295. doi: 10.1016/j.neuroscience.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockly E, Cordery PM, Woodman B, Mahal A, van Dellen A, Blakemore C, Lewis CM, Hannan AJ, Bates GP. Environmental enrichment slows disease progression in R6/2 Huntington's disease mice. Ann Neurol. 2002;51:235–242. doi: 10.1002/ana.10094. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert S, Bryson EA, Cordelières FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F. The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev Cell. 2002;2:831–837. doi: 10.1016/S1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- Hunt MJ, Morton AJ. Atypical diabetes associated with inclusion formation in the R6/2 mouse model of Huntington's disease is not improved by treatment with hypoglycaemic agents. Exp Brain Res. 2005;166:220–229. doi: 10.1007/s00221-005-2357-z. [DOI] [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun. 1996;224:855–862. doi: 10.1006/bbrc.1996.1112. [DOI] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res. 1998;57:1–9. doi: 10.1016/S0169-328X(98)00040-0. [DOI] [PubMed] [Google Scholar]
- Jia JM, Chen Q, Zhou Y, Miao S, Zheng J, Zhang C, Xiong ZQ. Brain-derived neurotrophic factor-tropomyosin-related kinase B signaling contributes to activity-dependent changes in synaptic proteins. J Biol Chem. 2008;283:21242–21250. doi: 10.1074/jbc.M800282200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Gall CM, Lynch G. Presynaptic BDNF promotes postsynaptic long-term potentiation in the dorsal striatum. J Neurosci. 2010;30:14440–14445. doi: 10.1523/JNEUROSCI.3310-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Peng Q, Liu X, Jin J, Hou Z, Zhang J, Mori S, Ross CA, Ye K, Duan W. Small molecule TrkB receptor agonists improve motor function and extend survival in a mouse model of Huntington's disease. Hum Mol Genet. 2013;22:2462–2470. doi: 10.1093/hmg/ddt098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, Chesselet MF, Levine MS. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington's disease transgenic mice. J Neurophysiol. 2001;86:2667–2677. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- Kleim JA, Chan S, Pringle E, Schallert K, Procaccio V, Jimenez R, Cramer SC. BDNF val66met polymorphism is associated with modified experience-dependent plasticity in human motor cortex. Nat Neurosci. 2006;9:735–737. doi: 10.1038/nn1699. [DOI] [PubMed] [Google Scholar]
- Klein R, Martin-Zanca D, Barbacid M, Parada LF. Expression of the tyrosine kinase receptor gene trkB is confined to the murine embryonic and adult nervous system. Development. 1990;109:845–850. doi: 10.1242/dev.109.4.845. [DOI] [PubMed] [Google Scholar]
- Koós T, Tepper JM. Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat Neurosci. 1999;2:467–472. doi: 10.1038/8138. [DOI] [PubMed] [Google Scholar]
- Kosinski CM, Cha JH, Young AB, Mangiarini L, Bates G, Schiefer J, Schwarz M. Intranuclear inclusions in subtypes of striatal neurons in Huntington's disease transgenic mice. Neuroreport. 1999;10:3891–3896. doi: 10.1097/00001756-199912160-00031. [DOI] [PubMed] [Google Scholar]
- Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntington aggregates may not predict neuronal death in Huntington's disease. Ann Neurol. 1999;46:842–849. doi: 10.1002/1531-8249(199912)46:6<842::AID-ANA6>3.0.CO%3B2-O. [DOI] [PubMed] [Google Scholar]
- Kwan W, Magnusson A, Chou A, Adame A, Carson MJ, Kohsaka S, Masliah E, Möller T, Ransohoff R, Tabrizi SJ, Björkqvist M, Muchowski PJ. Bone marrow transplantation confers modest benefits in mouse models of Huntington's disease. J Neurosci. 2012;32:133–142. doi: 10.1523/JNEUROSCI.4846-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948. doi: 10.1126/science.1065057. [DOI] [PubMed] [Google Scholar]
- Lerner RP, Trejo Martinez Ldel C, Zhu C, Chesselet MF, Hickey MA. Striatal atrophy and dendritic alterations in a knock-in mouse model of Huntington's disease. Brain Res Bull. 2012;87:571–578. doi: 10.1016/j.brainresbull.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo FM, Massa SM. Small-molecule modulation of neurotrophin receptors: a strategy for the treatment of neurological disease. Nat Rev Drug Discovery. 2013;12:507–525. doi: 10.1038/nrd4024. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, Woods AM, Chan EY, Kooperberg C, Krainc D, Young AB, Tapscott SJ, Olson JM. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet. 2002;11:1911–1926. doi: 10.1093/hmg/11.17.1911. [DOI] [PubMed] [Google Scholar]
- Lynch G, Kramar EA, Rex CS, Jia Y, Chappas D, Gall CM, Simmons DA. Brain-Derived Neurotrophic Factor Restores Synaptic Plasticity in a Knock-In Mouse Model of Huntington's Disease. J Neurosci. 2007;27:4424–4434. doi: 10.1523/JNEUROSCI.5113-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Chen LY, Gall CM. The substrates of memory: defects, treatments, and enhancement. Eur J Pharmacol. 2008;585:2–13. doi: 10.1016/j.ejphar.2007.11.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Savas JN, Chao MV, Tanese N. Quantitative analysis of BDNF/TrkB protein and mRNA in cortical and striatal neurons using alpha-tubulin as a normalization factor. Cytometry A. 2012;81:704–717. doi: 10.1002/cyto.a.22073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar D, Nebhan CA, Hu L, Anderson B, Webb DJ. An APPL1/Akt signaling complex regulates dendritic spine and synapse formation in hippocampal neurons. Mol Cell Neurosci. 2011;46:633–644. doi: 10.1016/j.mcn.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/S0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN, Nehama D, Rajadas J, Longo FM. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest. 2010;120:1774–1785. doi: 10.1172/JCI41356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, Rauskolb S, Polack M, Klose J, Kolbeck R, Korte M, Barde YA. Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat Neurosci. 2008;11:131–133. doi: 10.1038/nn2038. [DOI] [PubMed] [Google Scholar]
- Matsuura K, Kabuto H, Makino H, Ogawa N. Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J Neurosci Methods. 1997;73:45–48. doi: 10.1016/S0165-0270(96)02211-X. [DOI] [PubMed] [Google Scholar]
- McHughen SA, Rodriguez PF, Kleim JA, Kleim ED, Marchal Crespo L, Procaccio V, Cramer SC. BDNF val66met polymorphism influences motor system function in the human brain. Cereb Cortex. 2010;20:1254–1262. doi: 10.1093/cercor/bhp189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB. Knock-in mouse models of Huntington's disease. NeuroRx. 2005;2:465–470. doi: 10.1602/neurorx.2.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington's disease with 140 CAG repeats. J Comp Neurol. 2003;465:11–26. doi: 10.1002/cne.10776. [DOI] [PubMed] [Google Scholar]
- Menalled L, El-Khodor BF, Patry M, Suárez-Fariñas M, Orenstein SJ, Zahasky B, Leahy C, Wheeler V, Yang XW, MacDonald M, Morton AJ, Bates G, Leeds J, Park L, Howland D, Signer E, Tobin A, Brunner D. Systematic behavioral evaluation of Huntington's disease transgenic and knock-in mouse models. Neurobiol Dis. 2009;35:319–336. doi: 10.1016/j.nbd.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielcarek M, Benn CL, Franklin SA, Smith DL, Woodman B, Marks PA, Bates GP. SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington's disease. PLoS One. 2011;6:e27746. doi: 10.1371/journal.pone.0027746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller T. Neuroinflammation in Huntington's disease. J Neural Transm. 2010;117:1001–1008. doi: 10.1007/s00702-010-0430-7. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Hamby A, Massa SM. Clioquinol down-regulates mutant huntingtin expression in vitro and mitigates pathology in a Huntington's disease mouse model. Proc Natl Acad Sci U S A. 2005;102:11840–11845. doi: 10.1073/pnas.0502177102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavese N, Gerhard A, Tai YF, Ho AK, Turkheimer F, Barker RA, Brooks DJ, Piccini P. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 2006;66:1638–1643. doi: 10.1212/01.wnl.0000222734.56412.17. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2008. [Google Scholar]
- Peng Q, Masuda N, Jiang M, Li Q, Zhao M, Ross CA, Duan W. The antidepressant sertraline improves the phenotype, promotes neurogenesis and increases BDNF levels in the R6/2 Huntington's disease mouse model. Exp Neurol. 2008;210:154–163. doi: 10.1016/j.expneurol.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda JR, Canals JM, Bosch M, Adell A, Mengod G, Artigas F, Ernfors P, Alberch J. Brain-derived neurotrophic factor modulates dopaminergic deficits in a transgenic mouse model of Huntington's disease. J Neurochem. 2005;93:1057–1068. doi: 10.1111/j.1471-4159.2005.03047.x. [DOI] [PubMed] [Google Scholar]
- Pineda JR, Pardo R, Zala D, Yu H, Humbert S, Saudou F. Genetic and pharmacological inhibition of calcineurin corrects the BDNF transport defect in Huntington's disease. Mol Brain. 2009;2:33. doi: 10.1186/1756-6606-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouladi MA, Stanek LM, Xie Y, Franciosi S, Southwell AL, Deng Y, Butland S, Zhang W, Cheng SH, Shihabuddin LS, Hayden MR. Marked differences in neurochemistry and aggregates despite similar behavioural and neuropathological features of Huntington disease in the full-length BACHD and YAC128 mice. Hum Mol Genet. 2012;21:2219–2232. doi: 10.1093/hmg/dds037. [DOI] [PubMed] [Google Scholar]
- Rangone H, Pardo R, Colin E, Girault JA, Saudou F, Humbert S. Phosphorylation of arfaptin 2 at Ser260 by Akt Inhibits PolyQ-huntingtin-induced toxicity by rescuing proteasome impairment. J Biol Chem. 2005;280:22021–22028. doi: 10.1074/jbc.M407528200. [DOI] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Lin CY, Kramár EA, Chen LY, Gall CM, Lynch G. Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J Neurosci. 2007;27:3017–3029. doi: 10.1523/JNEUROSCI.4037-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Rigamonti D, Bolognini D, Mutti C, Zuccato C, Tartari M, Sola F, Valenza M, Kazantsev AG, Cattaneo E. Loss of huntingtin function complemented by small molecules acting as repressor element 1/neuron restrictive silencer element silencer modulators. J Biol Chem. 2007;282:24554–24562. doi: 10.1074/jbc.M609885200. [DOI] [PubMed] [Google Scholar]
- Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, Bhide PG, Vonsattel JP, DiFiglia M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol. 2001;60:161–172. doi: 10.1093/jnen/60.2.161. [DOI] [PubMed] [Google Scholar]
- Scharfman HE. Brain-derived neurotrophic factor and epilepsy–a missing link? Epilepsy Curr. 2005;5:83–88. doi: 10.1111/j.1535-7511.2005.05312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid DA, Yang T, Ogier M, Adams I, Mirakhur Y, Wang Q, Massa SM, Longo FM, Katz DM. A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett Syndrome. J Neurosci. 2012;32:1803–1810. doi: 10.1523/JNEUROSCI.0865-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- Silvestroni A, Faull RL, Strand AD, Möller T. Distinct neuroinflammatory profile in post-mortem human Huntington's disease. Neuroreport. 2009;20:1098–1103. doi: 10.1097/WNR.0b013e32832e34ee. [DOI] [PubMed] [Google Scholar]
- Simmons DA, Casale M, Alcon B, Pham N, Narayan N, Lynch G. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington's disease. Glia. 2007;55:1074–1084. doi: 10.1002/glia.20526. [DOI] [PubMed] [Google Scholar]
- Simmons DA, Rex CS, Palmer L, Pandyarajan V, Fedulov V, Gall CM, Lynch G. Up-regulating BDNF with an ampakine rescues synaptic plasticity and memory in Huntington's disease knockin mice. Proc Natl Acad Sci U S A. 2009;106:4906–4911. doi: 10.1073/pnas.0811228106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DA, Mehta RA, Lauterborn JC, Gall CM, Lynch G. Brief ampakine treatments slow the progression of Huntington's disease phenotypes in R6/2 mice. Neurobiol Dis. 2011;41:436–444. doi: 10.1016/j.nbd.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spampanato J, Gu X, Yang XW, Mody I. Progressive synaptic pathology of motor cortical neurons in a BAC transgenic mouse model of Huntington's disease. Neuroscience. 2008;157:606–620. doi: 10.1016/j.neuroscience.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Grote HE, Varshney NK, Cordery PM, van Dellen A, Blakemore C, Hannan AJ. Environmental enrichment rescues protein deficits in a mouse model of Huntington's disease, indicating a possible disease mechanism. J Neurosci. 2004a;24:2270–2276. doi: 10.1523/JNEUROSCI.1658-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Grote HE, Garry S, Cordery PM, Van Dellen A, Blakemore C, Hannan AJ. Dendritic spine pathology and deficits in experience-dependent dendritic plasticity in R6/1 Huntington's disease transgenic mice. Eur J Neurosci. 2004b;19:2799–2807. doi: 10.1111/j.0953-816X.2004.03374.x. [DOI] [PubMed] [Google Scholar]
- Stack EC, Kubilus JK, Smith K, Cormier K, Del Signore SJ, Guelin E, Ryu H, Hersch SM, Ferrante RJ. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington's disease transgenic mice. J Comp Neurol. 2005;490:354–370. doi: 10.1002/cne.20680. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand AD, Baquet ZC, Aragaki AK, Holmans P, Yang L, Cleren C, Beal MF, Jones L, Kooperberg C, Olson JM, Jones KR. Expression profiling of Huntington's disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J Neurosci. 2007;27:11758–11768. doi: 10.1523/JNEUROSCI.2461-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroppolo A, Guinea B, Tian C, Sommer J, Ehrlich ME. Role of phosphatidylinositide 3-kinase in brain-derived neurotrophic factor-induced DARPP-32 expression in medium size spiny neurons in vitro. J Neurochem. 2001;79:1027–1032. doi: 10.1046/j.1471-4159.2001.00651.x. [DOI] [PubMed] [Google Scholar]
- Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci U S A. 2000;97:8093–8097. doi: 10.1073/pnas.110078997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dellen A, Blakemore C, Deacon R, York D, Hannan AJ. Delaying the onset of Huntington's in mice. Nature. 2000a;404:721–722. doi: 10.1038/35008142. [DOI] [PubMed] [Google Scholar]
- van Dellen A, Welch J, Dixon RM, Cordery P, York D, Styles P, Blakemore C, Hannan AJ. N-Acetylaspartate and DARPP-32 levels decrease in the corpus striatum of Huntington's disease mice. Neuroreport. 2000b;11:3751–3757. doi: 10.1097/00001756-200011270-00032. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EJ., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Wacker JL, Huang SY, Steele AD, Aron R, Lotz GP, Nguyen Q, Giorgini F, Roberson ED, Lindquist S, Masliah E, Muchowski PJ. Loss of Hsp70 exacerbates pathogenesis but not levels of fibrillar aggregates in a mouse model of Huntington's disease. J Neurosci. 2009;29:9104–9114. doi: 10.1523/JNEUROSCI.2250-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CE, Tydlacka S, Orr AL, Yang SH, Graham RK, Hayden MR, Li S, Chan AW, Li XJ. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington's disease. Hum Mol Genet. 2008;17:2738–2751. doi: 10.1093/hmg/ddn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K, Xu JL, Suen PC, Levine E, Huang YY, Mount HT, Lin SY, Black IB. Functional trkB neurotrophin receptors are intrinsic components of the adult brain postsynaptic density. Brain Res Mol Brain Res. 1996;43:286–290. doi: 10.1016/S0169-328X(96)00211-2. [DOI] [PubMed] [Google Scholar]
- Xie Y, Hayden MR, Xu B. BDNF overexpression in the forebrain rescues Huntington's disease phenotypes in YAC128 mice. J Neurosci. 2010;30:14708–14718. doi: 10.1523/JNEUROSCI.1637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sobue G, Yamamoto K, Terao S, Mitsuma T. Expression of mRNAs for neurotrophic factors (NGF, BDNF, NT-3, and GDNF) and their receptors (p75NGFR, trkA, trkB, and trkC) in the adult human peripheral nervous system and non-neural tissues. Neurochem Res. 1996;21:929–938. doi: 10.1007/BF02532343. [DOI] [PubMed] [Google Scholar]
- Zhou L, Martinez SJ, Haber M, Jones EV, Bouvier D, Doucet G, Corera AT, Fon EA, Zisch AH, Murai KK. EphA4 signaling regulates phospholipase Cgamma1 activation, cofilin membrane association, and dendritic spine morphology. J Neurosci. 2007;27:5127–5138. doi: 10.1523/JNEUROSCI.1170-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Role of brain-derived neurotrophic factor in Huntington's disease. Prog Neurobiol. 2007;81:294–330. doi: 10.1016/j.pneurobio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington's disease. Physiol Rev. 2010;90:905–981. doi: 10.1152/physrev.00041.2009. [DOI] [PubMed] [Google Scholar]