

Abstract
Mutations in the methyl-CpG binding protein 2 gene, Mecp2, affect primarily the brain and lead to a wide range of neuropsychiatric disorders, most commonly Rett syndrome (RTT). Although the neuropathology of RTT is well understood, the cellular and molecular mechanism(s), which lead to the disease initiation and progression, has yet to be elucidated. RTT was initially attributed only to neuronal dysfunction, but our recent studies and those of others show that RTT is not exclusively neuronal but rather also involves interactions between neurons and glia. Importantly, studies have shown that MeCP2-restored astrocytes and microglia are able to attenuate the disease progression in otherwise MeCP2-null mice. Here we show that another type of glia, oligodendrocytes, and their progenitors are also involved in manifestation of specific RTT symptoms. Mice that lost MeCP2 specifically in the oligodendrocyte lineage cells, although overall normal, were more active and developed severe hindlimb clasping phenotypes. Inversely, restoration of MeCP2 in oligodendrocyte lineage cells, in otherwise MeCP2-null mice, although only mildly prolonging their lifespan, significantly improved the locomotor deficits and hindlimb clasping phenotype, both in male and female mice, and fully restored the body weight in male mice. Finally, we found that the level of some myelin-related proteins was impaired in the MeCP2-null mice. Expression of MeCP2 in oligodendrocytes of these mice only partially restored their expression, suggesting that there is a non–cell-autonomous effect by other cell types in the brains on the expression of myelin-related proteins in oligodendrocytes.
Introduction
Rett syndrome (RTT) is an autism spectrum disorder caused by sporadic mutations in the X-chromosome-linked gene methyl-CpG binding protein 2 (Mecp2) (Amir et al., 1999). Girls born with RTT attain normal developmental milestones; however, at 12–18 months of age, they regress, losing speech and motor skills and suffering other severe problems, including mental retardation, respiratory abnormalities, epileptic seizures, and overall retarded growth (Hagberg, 2002). Several mouse models with different germline mutations in Mecp2 have been generated, each of which recapitulates many characteristic features of RTT (Chen et al., 2001; Guy et al., 2001; Shahbazian et al., 2002). Importantly, conditional knock-out (k/o) of Mecp2 specifically in the brain results in a similar RTT phenotype (Chen et al., 2001), suggesting that RTT is caused primarily by dysfunction of MeCP2 in cells of the CNS.
Previously, RTT was attributed solely to neuronal dysfunction. However, studies by ourselves and others have shown that mutant glia, including astrocytes and microglia, are an integral part of manifestation of RTT, affecting neuronal structure and function (Ballas et al., 2009; Maezawa and Jin, 2010; Lioy et al., 2011; Derecki et al., 2012). Our recent studies further show that, like neurons, astrocytes of MeCP2-null mice also exhibit reduced process complexity (Nguyen et al., 2012). Importantly, reexpression of MeCP2 specifically in astrocytes or microglia of RTT mice significantly improves motor activities and behaviors, as well as prolongs lifespan to nearly normal (Lioy et al., 2011; Derecki et al., 2012).
Among glial cells, oligodendrocyte lineage cells, including oligodendrocyte precursor cells (OPCs, also known as NG2 cells) and oligodendrocytes, are active participants in neural networks. In addition to serving as progenitors for oligodendrocytes, OPCs also receive synaptic inputs from neurons (Bergles et al., 2010), and oligodendrocytes, the myelinating cells in the brain, play critical roles in propagation of neuronal signaling (Baumann and Pham-Dinh, 2001). Despite their important role in brain function, the critical question of whether oligodendrocyte lineage cells contribute to RTT neuropathology has not been addressed.
Here we show that mice lacking MeCP2 only in oligodendrocyte lineage cells, although overtly normal, are more active compared with their control siblings and develop a severe hindlimb clasping phenotype, indicative of a general neurological malaise. In reciprocal studies, expression of MeCP2 solely in oligodendrocyte lineage cells resulted in significant improvement of certain RTT symptoms, including full restoration of body weight in male mice, and significant improvement of hindlimb clasping phenotype and motor activity deficits in both male and female mice. Lifespan, however, was only mildly prolonged, distinguishing the role of oligodendrocytes and astrocytes in this parameter. Finally, we identified some myelin-related proteins whose expression was impaired in brains of MeCP2-null mice. While the reduced expression of myelin binding protein (MBP) was only partially restored upon expression of MeCP2 in oligodendrocyte lineage cells, the impaired expression of the myelin proteolipid protein (PLP) remained unchanged, suggesting that there is a non–cell-autonomous effect by other cell types in the brain on the expression of these proteins in the oligodendrocytes of RTT mice.
Materials and Methods
Animals
All animal studies were approved by the Institutional Animal Care and Use Committees at Stony Brook University and were in line with the guidelines established by the National Institute of Health.
Mecp2Stop/y (Guy et al., 2007) (B6.129P2-Mecp2tm2Bird/J) transgenic mice were obtained from The Jackson Laboratory. The Mecp2loxJ/y (Chen et al., 2001) (B6;129S4-Mecp2tm1Jae/Mmcd) transgenic mice were obtained from MMRRC and NG2Cre (Ng2CreBAC) (Komitova et al., 2009) transgenic mice were provided by Dr. Colognato (Stony Brook University). To generate the double-transgenic Mecp2loxJ/y/NG2Cre males and all their control littermates (wild-type [WT], NG2Cre, and Mecp2loxJ/y), we crossed female mice heterozygous for the Mecp2-lox allele (Mecp2loxJ/+) with male mice heterozygous for the NG2Cre transgene. Similar strategy was used to generate the Mecp2Stop/y/NG2Cre or Mecp2Stop/+/NG2Cre and the necessary control mice (WT, NG2Cre, and Mecp2Stop/y or Mecp2Stop/+). Only F1 mouse generation was used in this study. Mice were genotyped by tail biopsies, and the flox and Cre sequences were identified by PCR using the following sets of primers and according to the recommendation from The Jackson Laboratory: for Mecp2loxJ allele, forward, 5′-CAC CAC AGA AGT ACT ATG ATC-3′; reverse, 5′-CTA GGT AAG AGC TCT TGT TGA-3′. For Mecp2Stop allele, forward, 5′-AAC AGT GCC AGC TGC TCT TC-3′; mutant reverse, 5′-GCC AGA GGC CAC TTG TGT AG-3′; WT reverse, 5′-CTG TAT CCT TGG GTC AAG CTG-3′. For the NG2Cre allele, forward, 5′-CCG TAC ACC AAA ATT TGC C-3′; reverse, 5′-ATC GCG AAC ATC TTC AGG-3′. When necessary, female and male were distinguished by using Y-chromosome primers designated ZFY, forward, 5′-AAG ATA AGC TTA CAT AAT CAC ATG GA-3′; reverse, 5′-CCT ATG AAA TCC TTT GCT GCA CAT GT-3′.
Phenotypic scoring
Male and female mice were scored weekly or biweekly, respectively. Mice were removed from their home cage and placed onto a bench, at the same time during the day when possible. The scoring was performed essentially as we previously described (Nguyen et al., 2012). Each of the five symptoms, including mobility, gait, hindlimb clasping, tremor, and general condition was scored as 0 (symptom absent), 1 (symptom present), or 2 (symptom severe).
Behavioral assays
For all behavioral studies, the observer was blinded to genotype.
Digiscan activity monitor.
The mouse was placed in the center of an empty rat cage (44 cm × 21 cm) situated inside an Opto-Varimex-Minor animal activity meter (Columbus Instruments). The 15 × 15 infrared beams running in the x-y coordinates and 2 cm above the base of the machine recorded beam breaks associated with ambulatory activity (consecutive beam breaks) and total activity (all beam interruptions) during the 20 min observation period. Supported motor movements or full rearing (vertical activity) were defined as an animal using its upper and lower limbs to reach up the side of a cage and hold its body up.
Rotarod.
Mice were tested on a Medical Associates (model ENV-575M) Rotorod and ran by the “rotarod” controlled by a Dell Optiplex with a Windows 98 operating platform. The apparatus was a mechanized 30-cm-long rod divided into five 6 cm sections by 4 plastic white discs. The speed of rotation increased from 4 to 40 rpm through the course of 5 min session. The latency in seconds to drop off the rotarod was the main dependent measure of motor competence. Each mouse was tested for three consecutive trials with a 10 min rest between each session.
Wire hang test.
The mice were placed on a 44-cm-long stainless steel wire suspended 54 cm off the ground by two wooden posts so that only their front paws contact the wire. The latency to fall onto a foam pad below the apparatus was recorded. The maximum hang time was 60 s before removal. Each mouse was tested on three consecutive trials without resting.
Open-field activity.
Mecp2loxJ/y/NG2Cre and Mecp2loxJ/y mice were tested in this apparatus, which was square and made of wood covered with vinyl (60 × 60 cm), and the walls surrounding it were 25 cm high. The arena was divided in 16 squares (10 × 10 cm). The mice were left to explore the open arena for 10 min. The trial was videotaped. The number of times the mice crossed a line was counted.
Light-dark box test.
The light/dark box (43 × 21 × 21 cm) was made from a translucent Plexiglas box and consisted of two chambers connected by an opening (5 × 5 cm) located at the floor level in the center of the dividing wall. The small chamber (dark compartment) (14 × 21 cm) was painted black, and the large chamber (light compartment) (29 × 21 cm) was left translucent. Mice were placed in the dark chamber facing the opening immediately covered by a dark lid and were allowed to explore the apparatus for 5 min. The latency to exit the dark chamber, the length of time spent in the light side of the box, and the number of transition (number of time the animal passed in and out the light and dark chambers) were recorded.
Western blot analysis
Whole-brain tissues were homogenized in a Dounce homogenizer with the loose pestle in high-salt lysis buffer containing 20 mm HEPES, pH 7.9, 1.5 mm MgCl2, 0.2 mm EDTA, 420 mm NaCl, 0.5 mm DTT, 20% glycerol and protease inhibitors (Roche). Western blotting was performed as described previously (Ballas et al., 2009). The primary antibodies used are as follows: chicken anti-MeCP2 antibody (a generous gift from Dr. J.M. LaSalle, University of California Davis) (1:10,000) or a mouse anti-MeCP2 (Sigma) (1:2000), mouse anti-MBP (Covance) (1:1000), rabbit anti-PLP (Santa Cruz Biotechnology) (1:5000), mouse anti-CNPase (Sigma) (1:5000), and rabbit anti-myelin oligodendrocyte glycoprotein (MOG; Epitomics) (1:1000). Rabbit anti-β actin (Abcam) (1:5000) was used for loading control. The secondary antibodies used were IRDye800 or IRDye700-conjugated (LI-COR Biosciences) (1:10,000). Membranes were scanned using the Odyssey-Infrared Imaging System (LI-COR Biosciences), and the intensities of the bands of interest were determined from the captured images using the Odyssey imaging software.
Immunohistochemistry
Mice were fixed by transcardial perfusion with PBS-buffered 4% paraformaldehyde as described previously (Ballas et al., 2009). Brains were removed and postfixed overnight with PBS-buffered 4% paraformaldehyde at 4°C. The fixed tissue was cryoprotected with 30% sucrose in PBS buffer and frozen in optimal cutting medium (Neg50, Richard-Allan Scientific). Coronal sections were obtained by sectioning the tissue on a HM505E Microm Cryostat (Cryostat Industries) at 40 μm thickness and preserved at −20°C in PBS-buffered 50% glycerol until analyzed. The sections were immunostained using rabbit anti-MeCP2 antibody (a generous gift from Dr. M.E. Greenberg, Harvard Medical School) (1:500), chicken anti-GFAP (Millipore Bioscience Research Reagents) (1:500), mouse anti-NeuN (Millipore Bioscience Research Reagents) (1:150), guinea pig anti-NG2 (a generous gift from Dr. W.B. Stallcup, Burnham Institute), mouse anti-CC1 (Calbiochem) (1:100), mouse anti-S100β (Sigma) (1:2000), and mouse anti-MBP (Covance) (1:250), followed by incubation with the appropriate secondary antibodies conjugated to cyanine (Jackson ImmunoResearch Laboratories). MeCP2 immunostaining was enhanced using biotin streptavidin–conjugated secondary antibodies. Images were collected on a Zeiss confocal laser scanning LSM 510 microscope.
Statistics
To compare the two genotypes within each treatment or time point, t test or Mann–Whitney test was used. One-way ANOVA or two-way repeated-measures ANOVA was used for multiple-group comparisons and were followed by Holm-Sidak post hoc testing in all cases. The significance level of the main effect and interaction are presented following the description of each particular test in the figure legends. A minimum p value of 0.05 was accepted as statistically significant throughout. Data are presented as mean ± SEM.
Results
MeCP2 is depleted specifically in the oligodendrocyte lineage cells of the Mecp2loxJ/y/NG2Cre mouse brain
Previously, we showed that MeCP2 is present in all WT glia, including OPCs and their differentiated oligodendrocytes, in primary cultures as well as in postnatal mouse brain (Ballas et al., 2009). We confirmed the presence of MeCP2 in OPCs (NG2 cells) of the WT brain and showed that it is absent in OPCs of the RTT mouse brain (Fig. 1A). To analyze the role that MeCP2 might play in oligodendrocyte lineage cells, we generated an NG2-inducible Mecp2 k/o mouse model by crossing the transgenic mouse line NG2Cre (Komitova et al., 2009) with a mouse line carrying floxed Mecp2 allele (Mecp2loxJ) (Chen et al., 2001). We examined male progenies harboring the NG2Cre and the Mecp2loxJ alleles (Mecp2loxJ/y/NG2Cre) for the specificity of Mecp2-lox excision by the Cre recombinase in the OPCs and the differentiated oligodendrocytes. Immunostaining for MeCP2 and cell-specific markers confirmed the presence of MeCP2 in all cell types in the brain of the control NG2Cre mice and its specific absence in OPCs and oligodendrocytes in the brain of Mecp2loxJ/y/NG2Cre mice (Fig. 1B). Specifically, MeCP2 was undetectable in most OPCs in different brain areas (3–11% NG2+/MeCP2+ cells) of Mecp2loxJ/y/NG2Cre mice but remained present in neurons (93–98% NeuN+/MeCP2+) and astrocytes (90–96% GFAP+/Mecp2+ cells) (Fig. 1B; Table 1). Furthermore, only 8% of mature oligodendrocytes (CC1+) expressed MeCP2 in the corpus callosum of these mice (Fig. 1B; Table 1). The specific excision of the Mecp2-lox cassette by NG2Cre in oligodendrocyte lineage cells coincides with the published fate analysis of NG2 cells in the NG2Cre/ROSA26R double-transgenic mice (Komitova et al., 2009).
Figure 1.

MeCP2 protein is specifically depleted from the oligodendrocyte lineage cells in brains of Mecp2loxJ/y/NG2Cre mice. A, Representative images of brain sections of WT and RTT (Mecp2J.-/y) (Chen et al., 2001) male mice coimmunolabeled for MeCP2 (green) and NG2 (red). DAPI (blue) represents nuclear staining. MeCP2 (green) is expressed in OPCs (NG2+, red) in the hippocampus of WT mice and absent in RTT mice. Scale bars, 20 μm. B, Representative images of brain sections showing the presence of MeCP2 in all cell types in the brain of NG2Cre male mice and its specific absence in the oligodendrocyte lineage cells (NG2+ and CC1+ cells) in the brain of MeCP2loxJ/y/NG2Cre male mice. Sections were coimmunolabeled for MeCP2 (green) and neuronal marker (NeuN, red) in the dentate gyrus area, MeCP2, and astrocytic (GFAP, red) or OPCs (NG2, red) markers in the hippocampus, and MeCP2 and mature oligodendrocytic marker (CC1, red) in the corpus callosum. DAPI (blue) represents nuclear staining. Scale bars, 20 μm. Arrows indicate colocalization or absence of MeCP2 in GFAP, NG2, or CC1-positive cells.
Table 1.
Specificity and recombination efficiency in different areas of Mecp2loxJ/y/NG2Cre mouse braina
|
MeCP2loxJ/y/NG2Cre |
||||
|---|---|---|---|---|
| % MeCP2+/NG2+ | % MeCP2+/CC1+ | % MeCP2+/NeuN+ | % MeCP2+/GFAP+ | |
| Cortex | 9.6 (99) | 7.7 (246) | 93.3 (932) | 96.4 (102) |
| Hippocampus | 11.3 (123) | 6.5 (170) | 94.7 (894) | 92.8 (98) |
| Thalamus | 6.6 (98) | 6.7 (388) | 97.4 (894) | 89.9 (335) |
| Hypothalamus | 9.3 (113) | 2.9 (614) | 97.7 (891) | 91.1 (417) |
| Cerebellum | 10.2 (108) | 4.9 (618) | 98.1 (915) | 96.4 (259) |
| Hindbrain | 3.5 (114) | 9.1 (460) | 95.9 (821) | 92.6 (286) |
| Corpus callosum | — | 8.3 (215) | — | — |
aValues in parentheses indicate total number of cells.
The loss of MeCP2 in oligodendrocyte lineage cells results in selective RTT-like behavioral deficits
We examined whether the Mecp2loxJ/y/NG2Cre mice develop any overt RTT-like phenotypes compared with MeCP2-null mice bearing the same mutation (Mecp2J.-/y) (Chen et al., 2001). For this, animals were weekly scored following the observational phenotypic scoring system (0–10) described previously (Guy et al., 2007; Nguyen et al., 2012), for five typical RTT phenotypes: mobility, gait, hindlimb clasping, tremors, and general condition. As previously published (Chen et al., 2001), the MeCP2-null mice developed overt RTT phenotypes beginning at 4–5 weeks of age, which worsen with time, reaching a score of 6–10 between 9 and 15 weeks of age (Fig. 2A), and died prematurely (Fig. 2B). In contrast, Mecp2loxJ/y/NG2Cre male mice had a normal lifespan; however, they developed a mild RTT-like phenotype starting at ∼10 weeks of age and progressing to an aggregate score of 2 at 20 weeks of age (Fig. 2C). The scores of all control littermate mice, including WT, NG2Cre, and Mecp2loxJ male mice, were 0 or close to 0 at all time points (Fig. 2C). Specifically, the Mecp2loxJ/y/NG2Cre mice exhibited dull fur, and some of them presented piloerection. A low percentage (∼20%) of the Mecp2loxJ/y control mice showed a mild hindlimb clasping phenotype (Fig. 2D) likely because of the hypomorphic Mecp2-lox allele. Indeed, quantitative Western blot analysis and immunostaining of different brain areas showed slightly reduced MeCP2 expression in Mecp2loxJ/y mice relative to their WT littermates (Fig. 3A,B). Nonetheless, a significantly higher percentage (∼60%) of the Mecp2loxJ/y/NG2Cre mice had hindlimb clasping phenotype compared with their control Mecp2loxJ/y littermates, which was also more severe and involved more frequent clasping of both hindlimbs (Fig. 2D,E).
Figure 2.

The loss of MeCP2 expression only in the oligodendrocyte lineage cells results in hindlimb clasping and increased motor activity phenotypes. A, B, Phenotypic scores (A) and Kaplan–Meier survival time course (B) of the MeCP2-null mice (Mecp2J.-/y) compared with WT control mice. C, D, Phenotypic scores and percentage of hindlimb clasping, respectively, of the Mecp2loxJ/y/NG2Cre and the indicated littermate control male mice. E, Images illustrating the hindlimb clasping phenotype of the Mecp2loxJ/y/NG2Cre mouse compared with the control Mecp2loxJ/y mouse. F, Rotarod test. G, Wire-hang test. H, Light/dark box test. I, J, Total and vertical supported activity test in digiscan activity monitor, respectively. K, Total activity in the open-field test. The number of mice in B is similar to that in A; the number of mice in D is similar to that in C; the number of mice in F, G is similar to that in H. The colors of the graphs and bar graphs correspond to the indicated genotypes (top/right). Two-way repeated-measures ANOVA (genotype and time) followed by Holm-Sidak post hoc for multiple-comparisons test was used to analyze the data in C and F–G. One-way ANOVA followed by Holm-Sidak post hoc for multiple-comparisons test was used to determine differences between indicated groups in H–K. Error bars indicate mean ± SEM. *p < 0.05, significant differences between Mecp2loxJ/y/NG2Cre mouse compared with other control littermates (WT, Mecp2loxJ/y, NG2Cre) in C; between the first and the third trial in F, G; and between the indicated mouse groups in H–K.
Figure 3.

The expression of MeCP2 is reduced in Mecp2loxJ/y mouse brain compared with that of the WT mouse brain. A, Quantitative Western blot showing a decrease in the levels of MeCP2 in the indicated brain areas of Mecp2loxJ/y mice versus control WT mice at 32 weeks of age. Actin served as loading control. B, Representative images of immunostaining of cortex (top) and hippocampus (bottom) areas of Mecp2loxJ/y and control WT mice immunolabeled for MeCP2 (green). DAPI (blue) represents nuclear staining. so, Stratum oriens; py, pyramidal layer; sr, stratum radiatum. Scale bars: bottom and left top panels, 100 μm; right top panel, 50 μm.
We then examined the behavioral performance of the Mecp2loxJ/y/NG2Cre mice at 32 weeks of age, when their aggregate symptom score had reached a plateau of ∼2 (Fig. 2C). Our data show that these mice had no deficits in motor coordination, grip strength, or anxiety-like behaviors compared with their control littermates, as assessed by the rotarod, wire hang, and light/dark box tests, respectively (Fig. 2F–H). The motor skill learning of the Mecp2loxJ/y/NG2Cre mice was not affected either as their performance in the rotarod significantly increased between the first and the third trials (Fig. 2F). As expected, because of the hypomorphic allele (Fig. 3), in the digiscan test, Mecp2loxJ/y mice were less active than the WT or NG2Cre control littermates (Fig. 2I,J). Surprisingly, however, Mecp2loxJ/y/NG2Cre mice showed an increase in total and vertical activity compared with the Mecp2loxJ/y mice, reaching a level close to that of WT and NG2Cre mice (Fig. 2I,J). This relative greater activity level was confirmed in an open field test in which the Mecp2loxJ/y/NG2Cre mice crossed significantly more lines (70% and 100% more lines in day 1 and day 2 trials) than the Mecp2loxJ/y mice in a 10 min session (Fig. 2K).
MeCP2 expression is restored specifically in the oligodendrocyte lineage cells of the Mecp2Stop/y/NG2Cre mouse brain
As the absence of MeCP2 just in oligodendrocyte lineage cells led to partial RTT phenotypes, we asked whether normal MeCP2 expression in oligodendrocyte lineage cells in otherwise MeCP2-null mice, could rescue, at least in part, RTT-like symptoms. For this, we crossed heterozygous NG2Cre male mice with heterozygous female mice containing a Cre-excisable transcriptional Stop sequence in the endogenous Mecp2 gene (Mecp2Stop/+) (Guy et al., 2007), to generate male progeny harboring the NG2Cre transgene and the Mecp2Stop allele (Mecp2Stop/y/NG2Cre), and the control littermates, including WT, NG2Cre, and Mecp2Stop/y male mice. We first confirmed by immunostaining for MeCP2 and cell-specific markers that neurons (NeuN+), astrocytes (GFAP+), OPCs (NG2+), and mature oligodendrocytes (CC1+) in the brain of Mecp2Stop/y mice were deficient for MeCP2 (Fig. 4A). We then examined whether MeCP2 was specifically expressed in the oligodendrocyte lineage cells in the brain of Mecp2Stop/y/NG2Cre mice. Our data show that 75–85% of OPCs (NG2+) in different areas of the brain, including cortex, hippocampus, and striatum, and 90% of the mature oligodendrocytes (CC1+) in the corpus callosum express MeCP2 (Fig. 4B; Table 2). The expression of MeCP2 protein is specific to the oligodendrocyte lineage cells as indicated by the absence of MeCP2 in the majority of the neurons (0.5%–4.3% NeuN+/MeCP2+) and astrocytes (1%–2.2% GFAP+/MeCP2+) of different brain areas (Fig. 4B; Table 2). We were cognizant of previous observation that a subset of protoplasmic astrocytes, which were GFAP-negative but S100β-positive in the ventral area of the cortex, were derived from cells expressing NG2Cre (Zhu et al., 2008). Therefore, we also counted cells that are S100β+/MeCP2+ in this specific area; whereas most S100β cells expressed MeCP2 in the WT mice, only 11–12% reexpressed MeCP2 in the Mecp2Stop/y/NG2Cre mice (Fig. 4B, bottom; Table 2).
Figure 4.

MeCP2 protein is restored specifically in the oligodendrocyte lineage cells in brains of Mecp2Stop/y/NG2Cre mice. A, Representative images of brain sections of Mecp2Stop/y male mice showing global absence of MeCP2 in all cell types. Sections were coimmunostained with MeCP2 (green) and neuronal marker (NeuN, red) in the dentate gyrus area, MeCP2 with astrocytic (GFAP, red), and OPCs (NG2, red) markers in the hippocampus, and MeCP2 with mature oligodendrocyte marker (CC1, red) in the corpus callosum. DAPI (blue) represents nuclear staining. B, Representative images of immunostaining of brain sections of Mecp2Stop/y/NG2Cre male mice showing specific expression of MeCP2 in OPCs (NG2+) and oligodendrocytes (CC1+) but not in S100β+ or GFAP+ astrocytes and NeuN+ neurons. Cell-specific markers and brain areas are as indicated in A. Scale bars: 20 μm. Arrows indicate colocalization or absence of MeCP2 in GFAP, S100β, NG2, or CC1-positive cells.
Table 2.
Specificity and recombination efficiency in different areas of Mecp2Stop/y/NG2Cre mouse braina
|
MeCP2Stop/y/NG2Cre |
|||
|---|---|---|---|
| % MeCP2+/NeuN+ | % MeCP2+/GFAP+ | % MeCP2+/NG2+ | |
| Cortex | 1.9 (1936) | 1.6 (321) | 81.1 (285) |
| 0.5 (1752) | 1.0 (387) | 75.6 (245) | |
| Striatum | 4.3 (2398) | 2.2 (273) | 85.6 (401) |
| % MeCP2+/S100β+ | % MeCP2+/CC1+ | ||
| Corpus callosum | — | — | 89.7 (871) |
| Ventral cortex | — | 11.7 (817) | — |
aValues in parentheses indicate total number of cells.
The expression of MeCP2 solely in oligodendrocyte lineage cells partially restores some of the overt RTT-like phenotypes in male and female RTT mice
We first analyzed whether normal MeCP2 expression in oligodendrocyte lineage cells could rescue some of the classical RTT-like phenotypes previously described for the Mecp2Stop/y mice (Guy et al., 2007) as well as other MeCP2-null mice (Chen et al., 2001; Guy et al., 2001) (Fig. 2A,B) or delayed their onset. For this, Mecp2Stop/y/NG2Cre mice and age-matched control littermates were weekly weighted and scored following the observational phenotypic scoring system (0–10) described above. Although WT and NG2Cre mice had no symptoms, as expected, Mecp2Stop/y mice showed severe progression of aggregate symptom scores followed by lethality, with 50% survival at 13 weeks of age (Fig. 5A,C). Mecp2Stop/y/NG2Cre progeny showed similar kinetics of symptom progression as the Mecp2Stop/y mice (Fig. 5A); however, a significantly lower percentage of these mice developed the hindlimb clasping phenotype (∼40%) compared with the Mecp2Stop/y mice (∼65%) (Fig. 5B). In addition, the lifespan of Mecp2Stop/y/NG2Cre mice was prolonged by ∼3 weeks compared with the Mecp2Stop/y mice with a 50% survival at 16 weeks of age (Fig. 5C). Furthermore, whereas the Mecp2Stop/y mice were underweight, the body weight of the Mecp2Stop/y/NG2Cre mice was fully restored to the level of the control WT and NG2Cre littermate male mice (Fig. 5D). These data show that the expression of MeCP2 just in oligodendrocyte lineage cells can partially or fully restore some of the overt RTT-like phenotypes, although the shortened lifespan was only mildly improved.
Figure 5.
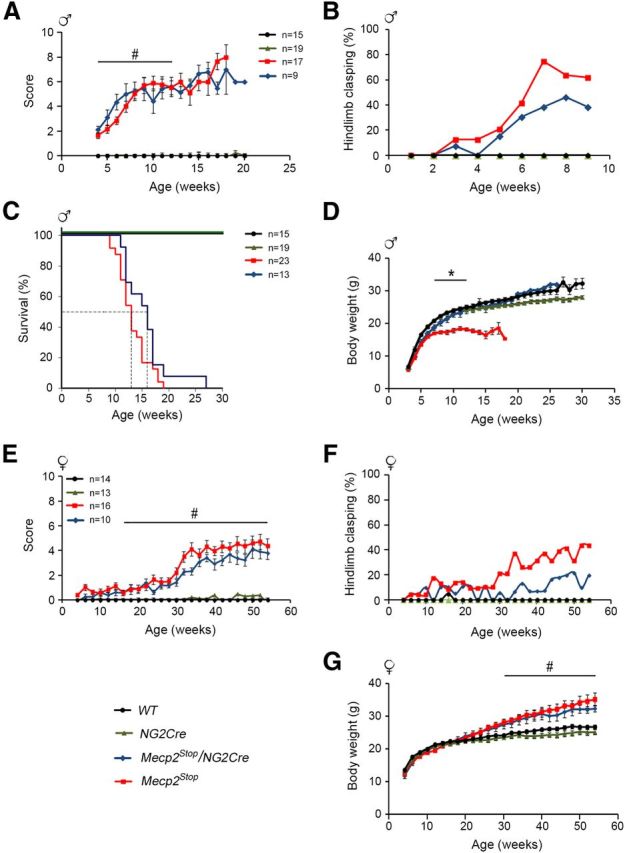
Expression of MeCP2 in oligodendrocyte lineage cells partially rescues some RTT-like features in Mecp2Stop/y/NG2Cre male and Mecp2Stop/+/NG2Cre female mice. A–D, Phenotypic scores, percentage of hindlimb clasping, Kaplan–Meier survival, and body weight time course, respectively, of Mecp2Stop/y/NG2Cre and the indicated littermate control male mice. B, D, The number of mice is similar to that indicated in A. E–G, Phenotypic scores, percentage of hindlimb clasping, and body weight time course, respectively, of Mecp2Stop/+/NG2Cre and the indicated littermate control female mice. F, G, The number of mice is similar to the indicated in E. Male and female symbols were used in the figure to distinguish the sex of each animal's set. Two-way repeated-measures ANOVA (genotype and time) followed by Holm-Sidak post hoc for multiple-comparisons test was used to analyze the data in A, D, E, and G. Error bars indicate mean ± SEM. #p < 0.05, significant differences between the controls WT, NG2Cre male mice, and Mecp2Stop/y and Mecp2Stop/y/NG2Cre male mice in A; between the controls WT, NG2Cre female mice, and the Mecp2Stop/+ and Mecp2Stop/+/NG2Cre female mice in E, G. *p < 0.05, significant differences between Mecp2Stop/y mice and the WT, NG2Cre, and Mecp2Stop/y/NG2Cre littermate male mice in D. Statistical calculation could not be performed for the age more than 12 weeks because of the increase number of mortality in the Mecp2Stop/y and the Mecp2Stop/y/NG2Cre mouse group.
In humans, RTT manifests predominantly in girls. Therefore, we also analyzed the effect of the restoration of MeCP2 expression in oligodendrocyte lineage cells on the RTT-like phenotype in heterozygous female mice. To this end, Mecp2Stop/+/NG2Cre female mice and their control WT, NG2Cre, and Mecp2Stop/+ littermate female mice were monitored biweekly for their body weight and overt RTT-like symptoms. Like the Mecp2Stop/y/NG2Cre males, the Mecp2Stop/+/NG2Cre females showed similar aggregate symptom scores to the Mecp2Stop/+ females (Fig. 5E). Although a trend of improvement in aggregate symptom scores was noticeable in Mecp2Stop/+/NG2Cre female mice, it was statistically nonsignificant (Fig. 5E). However, as in Mecp2Stop/y/NG2Cre male mice, a significantly lower percentage of Mecp2Stop/+/NG2Cre females (∼20%) developed the hindlimb clasping phenotype compared with the Mecp2Stop/+ mice (∼40%) (Fig. 5F). Consistent with previous studies (Guy et al., 2007), our data show that, after a normal period of development, female Mecp2Stop/+ mice became obese starting at ∼26 weeks of age; and by 38 weeks of age, they weighed 20% more than the WT or NG2Cre control female littermates (Fig. 5G). The weight gain coincides with the initiation of symptoms in the Mecp2Stop/+ female mice, reaching a plateau corresponding to an aggregate score of 4 beginning at 36 weeks of age (Fig. 5E). Interestingly, whereas the underweight phenotype of the Mecp2Stop/y male mice was fully rescued by restoring MeCP2 in oligodendrocyte lineage cells in the Mecp2Stop/y/NG2Cre mice (Fig. 5D), the overweight phenotype observed in Mecp2Stop/+ mice, which is typical for female RTT mice (Guy et al., 2007), was not rescued in the Mecp2Stop/+/NG2Cre female mice (Fig. 5G).
Restoration of MeCP2 in oligodendrocyte lineage cells significantly improves specific RTT-like behavioral deficits in male and female RTT mice
Mice deficient for MeCP2 demonstrate motor activity, grip strength, and anxiety defaults (Guy et al., 2001; McGill et al., 2006; Santos et al., 2007; Stearns et al., 2007). We examined whether some of the behavioral deficits of MeCP2-null mice are improved in the Mecp2Stop/NG2Cre mice. We subjected the male Mecp2Stop/y/NG2Cre mice and their control littermates for behavioral tests at 9–10 weeks of age, a stage when the Mecp2Stop/y and Mecp2Stop/y/NG2Cre mice were not severely symptomatic, reaching a median score of 5–6 (Fig. 5A). As expected, the motor coordination and forelimb strength of the Mecp2Stop/y male mice were significantly impaired as they fell off faster than their WT and NG2Cre control male littermates in the rotarod and wire hang tests (Fig. 6A,C). Interestingly, the Mecp2Stop/y/NG2Cre mice performed significantly better than the Mecp2Stop/y mice and stayed two times longer at trials 2 and 3 in the wire hang test (Fig. 6C). Indeed, in trail 2, there were no statistical significant differences between their performance and the performance of the WT and NG2Cre male littermates. There was also a trend of improvement in the performance of Mecp2Stop/y/NG2Cre mice compared with Mecp2Stop/y mice in the rotarod test, although it was statistically nonsignificant (Fig. 6A). Similar strength improvements were noticeable in the digiscan monitoring experiment; whereas the Mecp2Stop/y supported vertical activity was only ∼30% of the WT and NG2Cre littermate activity, the Mecp2Stop/y/NG2Cre mice improved up to ∼65% of the activity level of the WT and NG2Cre male mice (Fig. 6E), indicating a significant restoration of the hindlimb strength. In the digiscan tests, Mecp2Stop/y showed a dramatic decrease in motor activity, reaching only 35% of their control littermate activity (Fig. 6G). Importantly, the motor activity of Mecp2Stop/y/NG2Cre mice was improved up to ∼70% of the activity level of the control WT and NG2Cre male mice (Fig. 6G). Furthermore, motor activity enhancement was also observed in the light/dark box experiments in which the number of total transitions between the dark and light compartments of the Mecp2Stop/y/NG2Cre mice was fully restored to the level of the WT and NG2Cre control male mice, whereas the number of transitions of Mecp2Stop/y mice was only ∼40% the level of the WT male mice (Fig. 6I). The total time spent in the light or dark compartment and the latency to exit the dark were the same for all genotypes (data not shown), indicating no differences in the anxiety-like behavior between these mice.
Figure 6.
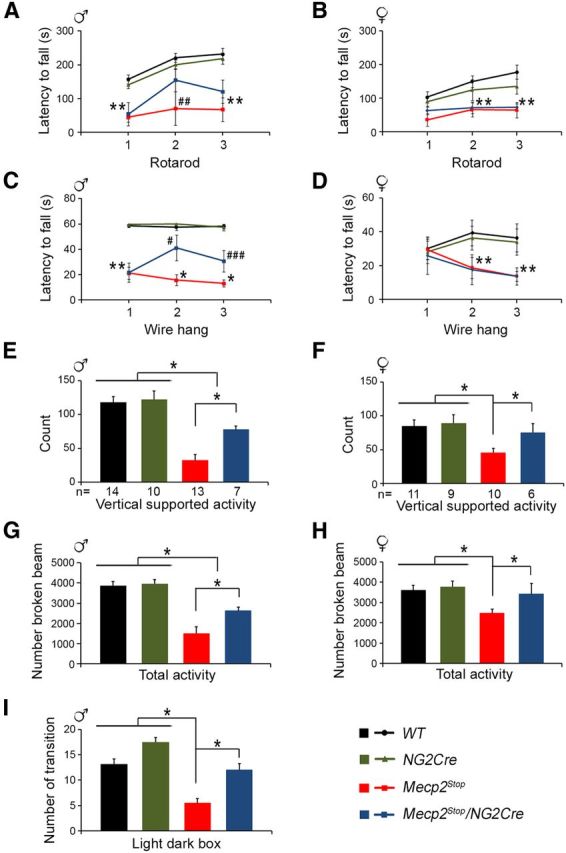
Expression of MeCP2 in oligodendrocyte lineage cells in Mecp2Stop/y/NG2Cre male and Mecp2Stop/+/NG2Cre female mice partially rescues some of the RTT behavioral deficits. Male and female mice were subjected to the indicated behavioral tests at 9–10 weeks or 1 year of age, respectively. A, B, Rotarod test for male (A) and female (B) mice. C, D, Wire hang test for male (C) and female mice (D). E–H, Vertical supported activity (E, F) and total activity (G, H) tests in digiscan activity monitor. E, G, Males. F, H, Females. I, Light/dark box test for male mice. The colors of the graphs and bar graphs correspond to the indicated genotypes in the right bottom corner. Male and female symbols were used in the figure to distinguish the sex of each animal's set. Two-way repeated-measures ANOVA (genotype and trial) followed by Holm-Sidak post hoc for multiple-comparisons test was used to analyze the data in A–D. One-way ANOVA followed by Holm-Sidak post hoc for multiple-comparisons test was used to determine differences between groups in E–I. Error bars indicate mean ± SEM. A–D, *p < 0.05, significant differences between the Mecp2Stop mouse group versus all other groups. **p < 0.05, significant differences between the both Mecp2Stop and Mecp2Stop/NG2Cre mouse groups versus the WT and NG2Cre littermate groups. #p < 0.05, significant differences between the Mecp2Stop/NG2Cre mouse group versus the Mecp2Stop littermate group. ##p < 0.05, significant differences between the Mecp2Stop mouse group versus the WT and NG2Cre littermate groups. ###p < 0.05, significant differences between the Mecp2Stop/NG2Cre mouse group versus all other groups. A, C, G, I, The number of mice is similar to that indicated in E. B, D, H, The number of mice is similar to that indicated in F.
Next, we evaluated the behavior of the female Mecp2Stop/+/NG2Cre mice when they were 12 months old, a stage when their symptoms stabilized and reached a mean score of 4–5 (Fig. 5E). Like the male Mecp2Stop/y/NG2Cre mice, we found that the motor activity of the female Mecp2Stop/+/NG2Cre mice measured in the digiscan test was improved up to 90% of the activity levels of WT or NG2Cre female mice, whereas the activity of Mecp2Stop/+ mice was only 60% of their control littermate female mice (Fig. 6H). In the rotarod and wire hang tests, the female Mecp2Stop/+ mice demonstrated deficits in motor coordination and forelimb strength compared with the control littermates; however, no improvement was noticeable in these two parameters for the Mecp2Stop/+/NG2Cre mice (Fig. 6B,D). On the other hand, the supported rearing activity of the Mecp2Stop/+/NG2Cre mice was restored to ∼90% of the level of the WT control mice (Fig. 6F), indicating a significant hindlimb strength improvement, which allowed the animals to hold up their body transitorily, unlike the Mecp2Stop/+ mice (Fig. 6F). It is likely that the underperformance of Mecp2Stop/+/NG2Cre mice in the rotarod and in the wire hang tests, which require whole-body support and coordination, is due to their obese phenotype (Fig. 5G). Together, our data indicate that MeCP2 expression in oligodendrocyte lineage cells in otherwise MeCP2-null brain ameliorates some of the behavioral deficits observed in male and female RTT mice.
The expression of proteins involved in myelin production is altered in Mecp2Stop/y mice but is only partially restored in the Mecp2Stop/y/NG2Cre mice
We first examined whether the absence of MeCP2 just in the oligodendrocyte lineage cells affects the expression of key proteins involved in myelin production. We analyzed the protein expression of the myelin-related genes at postnatal day 16 (P16), a time when myelination is actively occurring in the forebrain. Quantitative Western blot analysis showed no differences in the protein expression of CNPase (2′,3′-cyclic-nucleotide-3′-phosphodiesterase), MOG (myelin oligodendrocyte glycoprotein), MBP, or PLP (myelin proteolipid protein) between WT, Mecp2loxJ/y, and Mecp2loxJ/y/NG2Cre mice (Fig. 7A).
Figure 7.

MeCP2 expression in the oligodendrocyte lineage cells only partially rescues the aberrant expression pattern of MBP protein in RTT mice. A, The loss of MeCP2 only in the oligodendrocyte lineage does not affect the expression pattern of proteins involved in myelination. Quantitative Western blot analysis showing the expression levels of proteins involved in myelin formation in whole-brain extracts of P16 Mecp2loxJ/y/NG2Cre and the indicated control mice. B, Expression of MeCP2 in Mecp2Stop/y/NG2Cre mice only partially restored the reduced expression of MBP observed in Mecp2Stop/y mice. Quantitative Western blot analysis showing aberrant expression of MBP and PLP in whole-brain extracts of P16 Mecp2Stop/y mice and partial restoration of MBP expression in Mecp2Stop/y/NG2Cre age-matched littermate mice. C, Reconstruction of MBP confocal images of the corpus callosum (CC) and the striatum (ST) areas of the WT and Mecp2Stop/y brains. CTX, Cortex; LV, lateral ventricle. Scale bars, 200 μm. D, High magnification of the MBP immunostaining in the corpus callosum areas. Arrows indicate the thickness of MBP staining in the corpus callosum area calculated in E. E, Bar graphs representing the thickness of MBP staining in the corpus callosum area of the WT, Mecp2Stop/y, and Mecp2Stop/y/NG2Cre brains. An average of three measurements along the corpus callosum for each image was used for the thickness calculation. One-way ANOVA followed by Holm-Sidak post hoc for multiple-comparisons test was used to determine differences between groups in A, B and E. Error bars indicate mean ± SEM. *p < 0.05. n = 3 mice per genotype.
We then analyzed the expression of the same proteins in whole-brain extracts of Mecp2Stop/y/NG2Cre mice at P16. Western blot analysis showed no changes in the expression of CNPase and MOG proteins between the different types of mice; however, the expression of MBP and PLP proteins was significantly altered in Mecp2Stop/y mice compared with WT or NG2Cre mice (Fig. 7B). Specifically, MBP expression was decreased to ∼40%, and the PLP protein level was increased up to ∼200% of the level in age-matched WT and NG2Cre control littermate mice (Fig. 7B). When we analyzed the expression of the same proteins in the Mecp2Stop/y/NG2Cre mouse brains, we found that, whereas the level of PLP remained aberrant, the level of MBP was only partially restored to ∼60% of the level of the control WT or NG2Cre mice (Fig. 7B). We further analyzed the expression pattern of MBP at P16 by immunostaining and found that there was reduced expression of MBP in the corpus callosum and striatum of Mecp2Stop/y mice compared with WT mice (Fig. 7C). We also evaluated the thickness of MBP in the corpus callosum and found a significant reduction in the Mecp2Stop/y brain compared with that of the WT littermate mice (Fig. 7D,E); however, there were no significant differences between the MBP thickness between Mecp2Stop/y mice and Mecp2Stop/y/NG2Cre mice (Fig. 7D,E). Together, these data show that the expression of MeCP2 in oligodendrocytes in otherwise MeCP2-null mice had no effect (PLP) or only mild effect (MBP) on restoration of proteins involved in myelination and suggest that there is likely a non–cell-autonomous effect on the expression of these proteins in oligodendrocytes.
Discussion
Increasing evidence supports the view that RTT is not exclusively a neuronal disorder but rather a disease that also involves interactions between neurons and glia. Our recent studies and those of others indicated that at least two glial types, astrocytes and microglia, negatively affected neurons in a non–cell-autonomous fashion and contribute to the disease progression (Ballas et al., 2009; Maezawa and Jin, 2010; Lioy et al., 2011; Derecki et al., 2012). Here, we tested for contributions to RTT of a third type of glia, oligodendrocytes and their progenitor cells, OPCs. We found that, although there was some overlap in contributions of oligodendrocytes and astrocytes to the initiation of RTT-like symptoms, and in their ability to rescue RTT phenotypes, some features were idiosyncratic to the oligodendrocytic lineage.
Loss of MeCP2 from oligodendrocyte lineage cells, as in the astrocyte k/o mouse model, resulted in an overall mild phenotype. A unique behavior mediated by loss of MeCP2 from oligodendrocyte lineage cells, however, was relatively increased activity, which was similar to the WT level of activity but elevated compared with the activity of the floxed Mecp2 male mice. This is likely an authentic phenotype because the oligodendrocyte lineage k/o mice contain the same floxed allele as the floxed Mecp2 mice, and the latter develops a weak RTT-like phenotype with time, including mild hypoactivity, resulting from the hypomorphic allele (Figs. 2 and 3). A certain degree of behavioral deficits resulting from hypomorphic allele was also reported for another Mecp2-lox mouse model (Mecp2B. lox/y) (Samaco et al., 2008); however, the reduced level of MeCP2 and behavioral deficits in the mouse model (Mecp2J. lox/y) we used in the present studies were milder. Although enhanced activity as observed in the oligodendrocyte k/o mice is not one of the classical RTT phenotypes, Mecp2S421A;S424A knock-in mice, in which MeCP2 phosphorylation is impaired, have increased locomotion (Tao et al., 2009). Furthermore, patients with a C-terminal mutation (R294X) are hyperactive (Chahrour and Zoghbi, 2007; Temudo et al., 2011). Thus, mutations in Mecp2 can lead to a hyperactivity phenotype, in mice and humans, perhaps reflecting specific functions mediated by different domains in MeCP2; however, further studies are warranted to fully understand this phenotype.
In the rescue paradigm, although the mice still died prematurely, several RTT-like symptoms were significantly improved in both male and female RTT mice by restoration of MeCP2 in oligodendrocyte lineage cells. Specifically, motor skills, including activity, strength, and coordination, were significantly improved. Thus, all glial types expressing MeCP2 seem able to restore, at least in part, the motor levels of male and female RTT mice (Lioy et al., 2011; Derecki et al., 2012; Lioy and Mandel, personal communication; Fig. 6). These results together suggest that the loss of MeCP2 from the different glial types of RTT mice and patients likely has additive effects on locomotion deficits.
Compared with astrocytes and microglia, oligodendrocyte lineage cells expressing MeCP2 had a relatively milder ability to prolong lifespan. This differential effect could be the result of the greater ability of MeCP2-restored astrocytes and microglia over MeCP2-restored oligodendrocyte lineage cells to reverse breathing defects in the MeCP2-null mice (Lioy et al., 2011; Derecki et al., 2012). Although there is not much information on the role of microglia in respiration, astrocytes in particular have been shown to play an important role in regulating chemosensitive neurons (Gourine et al., 2010). In addition, unlike astrocytes (Lioy et al., 2011), oligodendrocyte lineage cells expressing MeCP2 were unable to improve the anxiety-like behavior observed in RTT mice. Nonetheless, oligodendrocyte rescue mice showed an improvement in the hindlimb clasping phenotype that is a prominent RTT-like symptom in mice. This improvement was not apparent in mice with MeCP2-restored astrocytes or microglia (Lioy et al., 2011; Derecki et al., 2012; and data not shown). Interestingly, unlike the male mice whose underweight phenotype was fully restored, the obesity phenotype normally observed in female RTT mice but not in RTT patients was not rescued by restoration of MeCP2 in oligodendrocyte lineage cells. Together, the comparison of the phenotypes in the MeCP2-deficient and rescued mice of different glial lineages suggests that, like the different neuronal subtypes (Chen et al., 2001; Fyffe et al., 2008; Chao et al., 2010), there is also differential and specific contributions of the different glia to RTT neuropathology.
The loss of MeCP2 specifically in OPCs and differentiated oligodendrocytes had no effect on the expression level of several proteins involved in myelin formation, a primary function of this lineage. Interestingly, the MeCP2-null male mice used in this study did display aberrant expression of at least two abundant myelin-related proteins in the mammalian CNS; MBP and PLP (Jahn et al., 2009). Nonetheless, restoration of MeCP2 in oligodendrocyte lineage cells in these mice resulted in only partial restoration of the MBP level, whereas the level of PLP still remained higher than normal. These results infer that MeCP2 expression in oligodendrocytes has little effect on the expression of myelin-related proteins and further suggest that there is likely a non–cell-autonomous influence on the expression of these genes from other cell types in the brain, lacking MeCP2. Such non–cell-autonomous effects could be mediated by neurons and/or astrocytes, which are known to influence myelination by secretion of signaling molecules. For example, the leukemia growth factor, LIF, which promotes myelination by mature oligodendrocytes, is released by astrocytes in response to ATP liberated from axons firing action potentials (Stevens et al., 2002). Another example is the release of glutamate from synaptic vesicles along axons, which promotes myelin induction by stimulating the formation of cholesterol-rich signaling domains between oligodendrocytes and axons, and increasing local synthesis of the major protein in the myelin sheath, MBP (Wake et al., 2011).
Collectively, our studies provide further support for the idea that different cell types in the brain contribute differentially to RTT neuropathology, in a cell-autonomous and non–cell-autonomous manner, through dynamic interactions among the different neurons and glia. As such, although ubiquitous removal or reexpression of MeCP2 can recapitulate or reverse, respectively, the full spectrum of RTT symptoms (Chen et al., 2001; Guy et al., 2007; McGraw et al., 2011; Cheval et al., 2012; Nguyen et al., 2012; Robinson et al., 2012), the loss or restoration of MeCP2 in specific cell types only partially initiates or rescues specific RTT symptoms.
Footnotes
This work was supported by the National Institutes of Health (N.B. and G.M.) and International Rett Syndrome Foundation (N.B.). G.M. is an investigator of the Howard Hughes Medical Institute. We thank Drs. Aguirre and Colognato for their advice in the analysis of oligodendrocytic genes, Dr. Stallcup for providing anti-NG2 antibodies, Drs. Greenberg and LaSalle for anti-MeCP2 antibodies, Dr. Lioy for sharing unpublished data, Dr. Trocme for helping in statistical analysis, and Ms. Picot for technical assistance.
The authors declare no competing financial interests.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Ballas N, Lioy DT, Grunseich C, Mandel G. Non–cell-autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12:311–317. doi: 10.1038/nn.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jabs R, Steinhäuser C. Neuron-glia synapses in the brain. Brain Res Rev. 2010;63:130–137. doi: 10.1016/j.brainresrev.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JL, Noebels JL, Rosenmund C, Zoghbi HY. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Cheval H, Guy J, Merusi C, De Sousa D, Selfridge J, Bird A. Postnatal inactivation reveals enhanced requirement for MeCP2 at distinct age windows. Hum Mol Genet. 2012;21:3806–3814. doi: 10.1093/hmg/dds208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG, Kipnis J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyffe SL, Neul JL, Samaco RC, Chao HT, Ben-Shachar S, Moretti P, McGill BE, Goulding EH, Sullivan E, Tecott LH, Zoghbi HY. Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron. 2008;59:947–958. doi: 10.1016/j.neuron.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Kasymov V, Marina N, Tang F, Figueiredo MF, Lane S, Teschemacher AG, Spyer KM, Deisseroth K, Kasparov S. Astrocytes control breathing through pH-dependent release of ATP. Science. 2010;329:571–575. doi: 10.1126/science.1190721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg B. Clinical manifestations and stages of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8:61–65. doi: 10.1002/mrdd.10020. [DOI] [PubMed] [Google Scholar]
- Jahn O, Tenzer S, Werner HB. Myelin proteomics: molecular anatomy of an insulating sheath. Mol Neurobiol. 2009;40:55–72. doi: 10.1007/s12035-009-8071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komitova M, Zhu X, Serwanski DR, Nishiyama A. NG2 cells are distinct from neurogenic cells in the postnatal mouse subventricular zone. J Comp Neurol. 2009;512:702–716. doi: 10.1002/cne.21917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, Hirrlinger PG, Kirchhoff F, Bissonnette JM, Ballas N, Mandel G. A role for glia in the progression of Rett's syndrome. Nature. 2011;475:497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J Neurosci. 2010;30:5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill BE, Bundle SF, Yaylaoglu MB, Carson JP, Thaller C, Zoghbi HY. Enhanced anxiety and stress-induced corticosterone release are associated with increased Crh expression in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2006;103:18267–18272. doi: 10.1073/pnas.0608702103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw CM, Samaco RC, Zoghbi HY. Adult neural function requires MeCP2. Science. 2011;333:186. doi: 10.1126/science.1206593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MV, Du F, Felice CA, Shan X, Nigam A, Mandel G, Robinson JK, Ballas N. MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J Neurosci. 2012;32:10021–10034. doi: 10.1523/JNEUROSCI.1316-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson L, Guy J, McKay L, Brockett E, Spike RC, Selfridge J, De Sousa D, Merusi C, Riedel G, Bird A, Cobb SR. Morphological and functional reversal of phenotypes in a mouse model of Rett syndrome. Brain. 2012;135:2699–2710. doi: 10.1093/brain/aws096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaco RC, Fryer JD, Ren J, Fyffe S, Chao HT, Sun Y, Greer JJ, Zoghbi HY, Neul JL. A partial loss of function allele of methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndrome. Hum Mol Genet. 2008;17:1718–1727. doi: 10.1093/hmg/ddn062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos M, Silva-Fernandes A, Oliveira P, Sousa N, Maciel P. Evidence for abnormal early development in a mouse model of Rett syndrome. Genes Brain Behav. 2007;6:277–286. doi: 10.1111/j.1601-183X.2006.00258.x. [DOI] [PubMed] [Google Scholar]
- Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/S0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- Stearns NA, Schaevitz LR, Bowling H, Nag N, Berger UV, Berger-Sweeney J. Behavioral and anatomical abnormalities in Mecp2 mutant mice: a model for Rett syndrome. Neuroscience. 2007;146:907–921. doi: 10.1016/j.neuroscience.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Stevens B, Porta S, Haak LL, Gallo V, Fields RD. Adenosine: a neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron. 2002;36:855–868. doi: 10.1016/S0896-6273(02)01067-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Hu K, Chang Q, Wu H, Sherman NE, Martinowich K, Klose RJ, Schanen C, Jaenisch R, Wang W, Sun YE. Phosphorylation of MeCP2 at Serine 80 regulates its chromatin association and neurological function. Proc Natl Acad Sci U S A. 2009;106:4882–4887. doi: 10.1073/pnas.0811648106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temudo T, Santos M, Ramos E, Dias K, Vieira JP, Moreira A, Calado E, Carrilho I, Oliveira G, Levy A, Barbot C, Fonseca M, Cabral A, Cabral P, Monteiro J, Borges L, Gomes R, Mira G, Pereira SA, Santos M, et al. Rett syndrome with and without detected MECP2 mutations: an attempt to redefine phenotypes. Brain Dev. 2011;33:69–76. doi: 10.1016/j.braindev.2010.01.004. [DOI] [PubMed] [Google Scholar]
- Wake H, Lee PR, Fields RD. Control of local protein synthesis and initial events in myelination by action potentials. Science. 2011;333:1647–1651. doi: 10.1126/science.1206998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development. 2008;135:145–157. doi: 10.1242/dev.004895. [DOI] [PubMed] [Google Scholar]