

Abstract
Prolonged calpain activation is widely recognized as a key component of neurodegeneration in a variety of pathological conditions. Numerous reports have also indicated that synaptic activation of NMDA receptors (NMDARs) provides neuroprotection against a variety of insults. Here, we report the paradoxical finding that such neuroprotection involves calpain activation. NMDAR activation in cultured rat cortical neurons was neuroprotective against starvation and oxidative stress-induced damage. It also resulted in the degradation of two splice variants of PH domain and Leucine-rich repeat Protein Phosphatase 1 (PHLPP1), PHLPP1α and PHLPP1β, which inhibit the Akt and ERK1/2 pathways. Synaptic NMDAR-induced neuroprotection and PHLPP1 degradation were blocked by calpain inhibition. Lentiviral knockdown of PHLPP1 mimicked the neuroprotective effects of synaptic NMDAR activation and occluded the effects of calpain inhibition on neuroprotection. In contrast to synaptic NMDAR activation, extrasynaptic NMDAR activation had no effect on PHLPP1 and the Akt and ERK1/2 pathways, but resulted in calpain-mediated degradation of striatal-enriched protein tyrosine phosphatase (STEP) and neuronal death. Using μ-calpain- and m-calpain-selective inhibitors and μ-calpain and m-calpain siRNAs, we found that μ-calpain-dependent PHLPP1 cleavage was involved in synaptic NMDAR-mediated neuroprotection, while m-calpain-mediated STEP degradation was associated with extrasynaptic NMDAR-induced neurotoxicity. Furthermore, m-calpain inhibition reduced while μ-calpain knockout exacerbated NMDA-induced neurotoxicity in acute mouse hippocampal slices. Thus, synaptic NMDAR-coupled μ-calpain activation is neuroprotective, while extrasynaptic NMDAR-coupled m-calpain activation is neurodegenerative. These results help to reconcile a number of contradictory results in the literature and have critical implications for the understanding and potential treatment of neurodegenerative diseases.
Introduction
Calpains are calcium-dependent proteases that play critical roles in both physiological and pathological conditions in CNS (Lynch and Baudry, 1984; Liu et al., 2008; Baudry and Bi, 2013). Two major calpain isoforms are present in brain: μ-calpain (aka, calpain-1) and m-calpain (aka, calpain-2). Recent studies have shown that m-calpain can also be activated by phosphorylation (Zadran et al., 2010). Overactivation of calpain has been implicated in a wide range of pathological states, including stroke, epilepsy, traumatic nerve injury, neurodegenerative disorders, and aging (Xu et al., 2007; Liu et al., 2008; Vosler et al., 2008). However, a number of studies have reported opposite findings, indicating that calpain activation could also provide neuroprotection under certain conditions (Wu and Lynch, 2006; Jourdi et al., 2009; Pannaccione et al., 2012).
NMDARs play critical roles in both physiological and pathological conditions, and several studies have shown that NMDA receptor localization imparts opposite functions to NMDA receptor stimulation, with synaptic NMDAR activation providing neuroprotection, while extrasynaptic NMDARs are linked to prodeath pathways (Hardingham and Bading, 2010). The Akt and MAP kinase/extracellular signal-regulated kinase (ERK1/2) pathways are two key prosurvival pathways downstream of synaptic NMDARs (Hardingham et al., 2001a; Papadia et al., 2005; Wang et al., 2012). Akt phosphorylates and inhibits various proapoptotic substrates, such as glycogen synthase kinase-3 (GSK3), forkhead box O (FOXO) (Soriano et al., 2006), apoptosis signal-regulating kinase 1 (ASK1) (Kim et al., 2001), p53 (Yamaguchi et al., 2001), and Bcl2-associated death promoter (BAD) (Downward, 1999), while ERK1/2 activates the nuclear transcription factor, cyclic-AMP response element-binding protein (CREB) (Hardingham et al., 2001b). Although some upstream kinases linking NMDARs with Akt and ERK have been found (Perkinton et al., 2002; Krapivinsky et al., 2003), it is still unclear how Akt and ERK1/2 are activated by synaptic but not extrasynaptic NMDARs.
PH domain and Leucine-rich repeat Protein Phosphatase 1 (PHLPP1) exhibits two splice variants, PHLPP1α and PHLPP1β, which share amino acid sequence similarity but have different sizes (140 kDa and 190 kDa, respectively). PHLPP1α dephosphorylates Akt at Ser473 in cancer cells (Gao et al., 2005) and neurons (Jackson et al., 2010) and its down-regulation is related to cell survival in CNS (Jackson et al., 2009; Saavedra et al., 2010; Chen et al., 2013). However, how PHLPP1α level is regulated in CNS is not clear. PHLPP1β inhibits ERK1/2 by binding and trapping its activator Ras in the inactive form (Shimizu et al., 2003). PHLPP1β is degraded by calpain in hippocampus, and its degradation contributes to novel object recognition memory (Shimizu et al., 2007). We also found that calpain-mediated regulation of PHLPP1β degradation and synthesis plays opposite functions in LTP induction and consolidation (Y. Wang, G. Zhu, V. Briz, Y.-T. Hsu, X. Bi, M. Baudry, unpublished observations).
In this study, we used preferential inhibitors for μ-calpain and m-calpain and isoform-specific siRNAs to evaluate the relative contributions of μ-calpain and m-calpain in synaptic and extrasynaptic NMDAR-mediated neuroprotection and neurodegeneration, respectively. Our results indicate that synaptic NMDAR-induced activation of μ-calpain degrades both PHLPP1α and PHLPP1β, leading to activation of the Akt and ERK pathways and neuroprotection. On the other hand, extrasynaptic NMDARs specifically activate m-calpain, which degrades striatal-enriched protein tyrosine phosphatase (STEP) resulting in neurotoxicity.
Materials and Methods
Animal experiments were conducted in accordance with the principles and procedures of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All protocols were approved by the Institutional Animal Care and Use Committee of Western University of Health Sciences.
Chemicals.
Tetrodotoxin (TTX), CNQX, d-AP5, bicuculline (Bic), and 4-aminopyridine (4-AP) were purchased from Tocris Bioscience. MK801, NMDA, and glycine were purchased from Sigma-Aldrich. Calpain inhibitor III (CI-III) and μ-calpain inhibitor 3-(5-fluoro-3-indolyl)-2-mercapto-(Z)-2-propenoic acid (μCalp-I, also known as PD151746) were purchased from EMD Millipore. The m-calpain inhibitor Z-Leu-Abu-CONH-CH2-C6H3 (3, 5-(OMe)2) (mCalp-I) was a generous gift from Dr. James Power (University of Georgia Tech, Atlanta, GA).
μ-calpain KO mice.
μ-calpain−/− mice on a C57BL/6 background were originally generated in the laboratory of Dr. Athar Chishti (Azam et al., 2001) and were backcrossed >20 times into the C57BL/6 background, and a colony was established at Western University of Health Sciences. Control mice were age-matched C57BL/6 mice purchased from Jackson Laboratory.
Primary neuronal cultures.
Cortical tissues were dissected from embryonic day 18 rats and digested with papain (2 mg/ml, Sigma) for 30 min at 37°C. Dissociated cells were plated onto poly-d-lysine-coated 35 mm dishes and coverslips at a density of 6–10 × 104 cells/cm2 in Neurobasal medium (Invitrogen) supplemented with 2% B27 (Invitrogen) and Penicillin/Streptomycin Glutamine (Invitrogen) and kept at 37°C <5% CO2. Half of the culture medium was replaced with fresh culture medium at DIV4 and then every 7 d.
Synaptic NMDAR-dependent neuroprotection.
For synaptic NMDAR activation, cultured cortical neurons (DIV12) were first incubated overnight with TTX (1 μm), CNQX (40 μm), and d-AP5 (100 μm). They were then washed and incubated in neurobasal medium for 30–60 min to remove inhibitors. Neurons were then treated with 20 μm Bic and 100 μm 4-AP for 30 min. Treated neurons were lysed with cell lysis buffer (Cell Signaling Technology) containing 1 mm phenylmethanesulfonylfluoride (PMSF), and cell lysates were processed for Western blot following protein analysis with the bicinchoninic acid (BCA) assay (Thermo Scientific). In some groups, calpain inhibitors were added 10 min before Bic and 4-AP treatment. For induction of neuronal death, cortical neurons were incubated in either a starvation medium containing 10% MEM and 90% salt–glucose–glycine medium (114 mm NaCl, 0.22% NaHCO3, 5.3 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 10 mm HEPES, 1 mm glycine, 30 mm glucose, and 0.5 mm sodium pyruvate) for 3 d or an oxidative stress medium, 20 μm H2O2 in Neurobasal, for 24 h. For neuroprotection induced by synaptic NMDAR activation, 20 μm Bic and 100 μm 4-AP were applied together with the starvation medium or the H2O2-containing medium. For neuroprotection induced by previous NMDAR activity, Bic and 4-AP were first applied to neurons for 12 h. NMDAR activity was terminated by treatment with MK801 and TTX for 5 min before starvation or H2O2 insult. To test the effects of calpain inhibitors, CI-III, μCalp-I or mCalp-I was applied together with Bic and 4-AP.
Extrasynaptic NMDAR-dependent neurotoxicity.
Cortical neurons (DIV14–16) were incubated overnight with TTX (1 μm), CNQX (40 μm), and d-AP5 (100 μm). They were then washed and incubated in neurobasal medium for 30–60 min to remove inhibitors. Neurons were treated with 10 μm Bic and 40 μm MK801 for 2 min to activate and block synaptic NMDA receptors, then washed 3 times, before treating them with 100 μm NMDA and 10 μm glycine for 1 h. To test the effects of calpain inhibitors, CI-III, μCalp-I or mCalp-I was applied 10 min before addition of NMDA and glycine. For neuronal death assay, neurons were incubated in normal culture medium for another 24 h following NMDA and glycine treatment.
Hoechst staining.
Treated neurons were fixed in 4% paraformaldehyde for 10 min, permeabilized with 0.05% Triton X-100 in PBS for 10 min, and then stained with 1 μg/ml Hoechst 33258 (Sigma) in PBS for 5 min. After washing with PBS for 2 times, neurons were mounted with antifade-mounting medium (Vector Laboratories). Apoptotic and healthy neurons were counted under confocal microscope by a blind observer to avoid bias.
Treatment of P2 membrane fractions with purified calpains.
Treatment of P2 fractions with purified calpains was performed as previously described (Briz et al., 2013). Briefly, brains from Sprague Dawley adult rats were placed in ice-cold homogenization buffer containing the following: 320 mm sucrose, 10 mm HEPES, pH 7.4, 2 mm EDTA, 2 mm EGTA, 0.1 mm PMSF, and protease and phosphatase inhibitor mixture (Thermo Scientific). Brain tissue was homogenized with 10 strokes using a Wheaton glass-Teflon homogenizer. The homogenate was centrifuged at 1000 × g for 10 min, and the supernatant collected and centrifuged at 14,000 × g for 20 min. The resultant pellet (P2 membrane fraction) was centrifuged again at 14,000 × g for 20 min to eliminate protease inhibitors. P2 pellets were then resuspended in Tris-acetate buffer (100 mm Tris-acetate, pH 7.4, 50 μm EGTA), and the protein concentration was determined by the BCA assay. Equal amounts of proteins (4 μg/μl) were incubated with μ-calpain (Calbiochem) or m-calpain (Origene; both at 2.4 U/ml) in the absence or presence of 2 mm calcium for 30 min at 37°C. Calpain reaction was stopped by adding Laemmli's loading buffer (final concentrations, 62.5 mm Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 0.005% bromophenol, 5% β-mercaptoethanol) followed by boiling at 100°C for 5 min, and samples were then subjected to Western blot.
Acute rat hippocampal slice preparation.
Adult male rats (2∼4-month-old) were anesthetized with halothane and decapitated. Brains were quickly removed and transferred to oxygenated, ice-cold cutting medium (in mm): 124 NaCl, 26 NaHCO3, 10 glucose, 3 KCl, 1.25 KH2PO4, 5 MgSO4, and 3.4 CaCl2. Hippocampal transversal slices (400 μm thick) were prepared using a McIlwain-type tissue chopper and transferred to a recovery chamber with a modified artificial CSF (aCSF) medium, containing the following (in mm): 124 NaCl, 2.5 KCl, 2.5 CaCl2, 1.5 MgSO4, 1.25 NaH2PO4, 24 NaHCO3, 10 d-glucose, and saturated with 95%O2/5%CO2 for 1 h at 37°C.
shRNA lentiviral infection.
PHLPP1 shRNA (rat) lentivirus (sc-270416-V, Santa Cruz Biotechnology) contains three shRNA plasmids targeting three different sites of the rat PHLPP1 mRNA. The siRNA sequences (sense) were GUAGACCUCUCAUGUUGUAtt, CGAUAUCAGUGGGAAUAAGtt, and GUAUGGAGUCAUGGUUACAtt, respectively. The scrambled shRNA lentivirus was sc-108080 from Santa Cruz Biotechnology. Cultured cortical neurons were infected at DIV 6, and 24–36 h after infection, 2/3 medium was replaced with fresh medium. Five to six days after infection, neurons were subjected to various treatments.
siRNA transfection.
Cortical neurons at DIV7 were transfected with 100 nm siRNAs (Qiagen) using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions and incubated at 37°C for 2 d before treatments were performed. The following siRNA sequences were used: negative control, AATTCTCCGAACGTCTCACGT; μ-calpain, TACCTCTGTTCAATTGCTCTA; m-calpain, GCGGTCAGATACCTTCATCAA.
Ki calculation for calpain inhibitors.
The hydrolysis of the fluorogenic substrate Suc-Leu-Tyr-7-amino-4-methylcoumarin (AMC, Calbiochem) by μ-calpain and m-calpain was performed as previously described (Sasaki et al., 1984), with minor modifications. Briefly, purified μ-calpain from porcine erythrocytes (8 μg, Millipore) or rat recombinant m-calpain (8 μg, a generous gift from Dr. Peter Davis from Queen's University, Ontario, CA) was incubated with Suc-Leu-Tyr-AMC (0.5 mm) in 60 mm imidazole-HCl buffer, pH 7.3, containing 5 mm CaCl2, 5 mm cysteine, 2.5 mm β-mercaptoethanol, and different concentrations of μCalp-I (ranging from 0 to 60 μm) or mCalp-I (0–20 μm). The reaction was initiated by adding the enzyme and continued at 30°C for 15 min, while the fluorescence of AMC (Ex 380 nm/Em 450 nm) was monitored every 30 s in a POLARstar Omega fluorescence polarization microplate reader (BMG Labtech). The rate of hydrolysis (increase in fluorescence/min) was determined from the linear portion of the curve. The half-inhibitory concentration (IC50) values were obtained by adjusting data from each experiment into a sigmoidal dose–response curve. The Ki value for m-Calp-I was calculated from the average of the IC50 values and from a single substrate concentration by using a Ki calculator tool for fluorescence-based competitive binding assays (http://sw16.i.m..med.umich.edu/software/calc_ki/). The Ki value for μCalp-I was calculated by using a different Ki calculator tool (http://botdb.abcc.ncifcrf.gov/toxin/kiConverter.jsp), considering that it is an uncompetitive inhibitor, as previously described (Wang et al., 1996). The Km values for Suc-Leu-Tyr-AMC used for the Ki calculation were 4.74 mm and 2.21 mm for μ-calpain and m-calpain, respectively, as previously reported (Sasaki et al., 1984).
Coimmunoprecipitation.
Cultured cortical neurons (DIV12–16) were lysed with a lysis buffer containing the following: 25 mm Tris, 150 mm NaCl, 1 mm EDTA, 1% NP-40, 5% glycerol, 0.1 mm PMSF, and a protease and phosphatase inhibitor mixture and 10 μm CI-III. Coimmunoprecipitation (Co-IP) was then performed using Pierce Classic IP Kit (Thermo Scientific) following the manufacturer instructions.
Immunocytochemistry.
Cultured cortical neurons at DIV14–16 were fixed in 1/2 culture medium and 1/2 fixation solution containing 20% sucrose and 4% PFA for 20 min at 37°C. After washing once with ice-cold PBS, neurons were permeabilized with 0.05% Triton X-100 in PBS for 7 min and then with 0.02% Tween-20 in PBS for 2 min. After washing twice with ice-cold PBS, neurons were blocked with 3% BSA in PBS for 30 min and then incubated with primary antibodies in blocking solution at 4°C overnight. After washing twice with ice-cold PBS, neurons were incubated with secondary antibodies in blocking solution for 2 h at 37°C. After washing with ice-cold PBS four times, neurons were mounted with anti-fade mounting medium containing DAPI (Vector Laboratories) and examined under confocal microscope (Nikon).
Acute mouse hippocampal slice preparation.
Mice at postnatal day 15 (P15) were anesthetized with halothane and decapitated. Brains were quickly removed and transferred to oxygenated, ice-cold cutting medium (in mm): 124 NaCl, 26 NaHCO3, 10 glucose, 3 KCl, 1.25 KH2PO4, 5 MgSO4, and 3.4 CaCl2. Hippocampal transversal slices (400 μm thick) were prepared using a McIlwain-type tissue chopper and transferred to a recovery chamber with a modified artificial CSF (aCSF) medium containing the following (in mm): 124 NaCl, 2.5 KCl, 2.5 CaCl2, 1.5 MgSO4, 1.25 NaH2PO4, 24 NaHCO3, and 10 d-glucose, and saturated with 95%O2/5%CO2 for 1 h at 37°C. Slices were then treated with NMDA (100 μm) in the absence or presence of calpain inhibitors, for 1 h (Western blots) or 3 h [lactate dehydrogenase (LDH) release]. At the end of the treatment, slices were homogenized and aliquots of the homogenates were processed for Western blots; alternatively, the medium was collected and analyzed for LDH activity.
LDH assay in acute hippocampal slices.
Neuronal damage following NMDA-treatment of acute hippocampal slices was assessed by measurement of LDH released into the medium solution. During the treatment, each two slices were incubated in 1 ml of aCSF solution saturated with 95% O2/5% CO2 at 37°C. At the end of treatment, 50 μl of medium solution was transferred to a 96-well plate and the LDH reaction was performed using Pierce LDH Cytotoxicity Assay Kit (Thermo Scientific) following the manufacturer's instructions. To determine LDH activity, the absorbance at 680 nm (background signal) was subtracted from the absorbance at 490 nm. LDH activity was normalized to protein concentration and results are shown as fold of controls.
Antibodies.
The following primary antibodies were used: PHLPP1 (07-1341, Millipore), pAkt S473 (#4060, Cell Signaling Technology), Akt (#2920), pERK1/2 (Thr202/Tyr204, #9101), ERK1/2 (#9107), PTEN (#9556), STEP (NB300-202, Novus), μ-calpain (monoclonal, MA3-940, Thermo Scientific), μ-calpain (polyclonal, #2556), m-calpain (PA5-17494, Thermo Scientific), NR2A (07-632, Millipore), NR2B (05-920, Millipore), and PSD95 (MA1-045, Thermo Scientific). The following secondary antibodies were used for Western blot: goat anti-rabbit IRDye 680RD and goat anti-mouse IRDye 800CW (LI-COR). The following secondary antibodies for immunocytochemistry were used: Alexa Fluor 594 goat anti-rabbit and Alexa Fluor 488 goat anti-mouse (Invitrogen).
Results
Calpain activity is required for synaptic NMDAR-dependent neuroprotection
We first determined whether calpain activity was involved in synaptic NMDAR-dependent neuroprotection against neurodegeneration elicited by starvation or oxidative stress. Cortical neurons cultured for 3 d under starvation conditions exhibited significant cell death, as indicated by Hoechst staining (Fig. 1A). Application of a combination of a GABAA receptor antagonist, bicuculline (Bic, 20 μm), and a K+ channel blocker, 4-aminopyridine (4-AP, 100 μm), which has been shown to specifically activate synaptic NMDARs (Hardingham et al., 2001a), significantly increased neuronal survival. Both ongoing synaptic NMDAR activity (see Materials and Methods, Bic + 4-AP in Fig. 1A) and previous synaptic NMDAR activity (Bic + 4-AP, followed by MK801 + TTX in Fig. 1A) reduced cell death, a result in good agreement with a previous report (Papadia et al., 2005). To evaluate the role of calpain in synaptic NMDAR-dependent neuroprotection against starvation, a broad spectrum calpain inhibitor, CI-III (10 μm), was applied together with Bic + 4-AP. Application of CI-III completely blocked the neuroprotection induced by both ongoing synaptic NDMAR activity and previous synaptic NMDAR activity (Fig. 1A). To rule out the possibility that CI-III treatment alone had neurotoxic effects, neurons in either normal growth medium or starvation medium were treated with 10 μm CI-III alone for 3 d. CI-III treatment did not cause cell death under these conditions (Fig. 1A). Synaptic NMDAR activation has also been shown to enhance neuronal antioxidant defense (Papadia et al., 2008). Both ongoing synaptic NMDAR activity (see Materials and Methods, Bic + 4-AP in Fig. 1B) and previous synaptic NMDAR activity (Bic + 4-AP, followed by MK801 + TTX in Fig. 1B) reduced neuronal death induced by a 24 h H2O2 treatment, a result in good agreement with previous results (Papadia et al., 2008). Application of CI-III (10 μm) blocked the neuroprotective effect induced by both ongoing synaptic NDMAR activity and previous synaptic NMDAR activity (Fig. 1B,C), indicating that calpain activity is also required for synaptic NMDAR-mediated neuroprotection against oxidative stress. Application of CI-III alone had no effect on neuronal death elicited by H2O2-treatment (Fig. 1B).
Figure 1.

Calpain activity is required for synaptic NMDAR-dependent neuroprotection against starvation and oxidative stress. A, Trophic deprivation for 3 d induced cultured cortical neuron death. Incubation with 20 μm Bic and 100 μm 4-AP during starvation period (Bic + 4-AP) or before starvation (Bic + 4-AP, then MK801 + TTX) reduced neuronal death. Coapplication of 10 μm CI-III with Bic and 4-AP blocked the neuroprotective effect of Bic and 4-AP. Incubation of neurons under either normal condition or starvation with 10 μm CI-III alone for 3 d did not cause cell death. Neuronal death was observed and quantified by Hoechst staining; 300–500 cells were counted for each group in each independent experiment. *p < 0.05; ns, not significantly different; one-way ANOVA followed by Bonferroni test. n = 3. Error bar indicates SEM. B, Similar experimental design as in A except that neuronal death was induced by a 24 h-H2O2 insult (20 μm). In all cases, results are means ± SEM of three independent experiments with 300–500 cells for each group in each independent experiment. *p < 0.05; ns, not significantly different; one-way ANOVA followed by Bonferroni test. C, Representative photos of Hoechst staining. Hoechst brightly stains condensed and/or fragmented nuclei of apoptotic neurons but only dimly stains normal nuclei of healthy neurons. Scale bar, 10 μm.
Synaptic NMDAR activation results in calpain-mediated degradation of PHLPP1 and activation of the Akt and ERK1/2 pathways
Synaptic NMDARs exert their neuroprotective effect by activating the Akt and ERK1/2 prosurvival pathways (Hardingham and Bading, 2010). The PHLPP1 splice variants, PHLPPα and PHLPP1β (SCOP) inhibit Akt and ERK1/2 pathways, respectively (Shimizu et al., 2003; Gao et al., 2005). PHLPP1β has previously been shown to be a calpain substrate in hippocampal neurons (Shimizu et al., 2007). As PHLPPα and PHLPP1β have similar amino acid sequences, we postulated that they might also share the same calpain cleavage site(s). To assess whether both PHLPP1 splice variants were calpain substrates, aliquots of P2 fractions from rat brain homogenates (4 μg/μl) were treated with purified μ-calpain or m-calpain (both at 2.4 U/ml) with or without Ca2+ (2 mm) for 30 min, and the levels of PHLPPα and PHLPP1β were determined by Western blots, using a pan-PHLPP1 antibody. Treatment with both μ-calpain and m-calpain in the presence of calcium resulted in PHLPPα and PHLPP1β degradation, as compared with treatment without Ca2+ (Fig. 2A), indicating that both PHLPPα and PHLPP1β are calpain substrates. We then tested whether synaptic NMDAR activation could produce PHLPP1 degradation. Acute rat hippocampal slices were treated with Bic (20 μm) and 4-AP (100 μm) for 30 min to activate synaptic NMDARs. Bic + 4-AP treatment significantly decreased the levels of both PHLPPα and PHLPP1β and increased the levels of phospho-Akt S473 (pAkt) and phospho-ERK1/2 (pERK1/2) (Fig. 2B,C). When CI-III (10 μm) was applied 15 min before Bic and 4-AP treatment, Bic and 4-AP treatment no longer produced PHLPP1 degradation nor activation of Akt and ERK (Fig. 2B,C, CI-III). Similar results were obtained with cultured cortical neurons. Bic (20 μm) and 4-AP (100 μm) treatment of cortical neurons for 30 min significantly decreased PHLPPα and PHLPP1β levels and increased p-Akt and p-ERK1/2 levels. Preapplication of CI-III (10 μm) before Bic and 4-AP treatment completely blocked PHLPP1 degradation and Akt activation, and partially blocked ERK activation (Fig. 2D,E). Preapplication of the NMDAR antagonist MK801 (50 μm) 10 min before Bic and 4-AP treatment completely blocked Bic and 4-AP-induced changes, indicating those changes are NMDAR dependent (Fig. 2E). These results indicate that calpain mediates PHLPP1 degradation and Akt and ERK activation following synaptic NMDAR activation.
Figure 2.

Synaptic NMDAR-dependent calpain activation degrades PHLPP1 and activates Akt and ERK1/2 pathway. A, Treatment of rat brain P2 fraction aliquots with purified μ-calpain (4 μg/ml) in the presence of 200 μm Ca2+ or purified m-calpain (4 μg/ml) in the presence of 2 mm Ca2+ for 1 h reduced the levels of full-length PHLPP1α and PHLPP1β, as compared with treatment with μ-calpain or m-calpain alone. B, Western blots for rat acute hippocampal slices treated with 20 μm Bic and 100 μm 4-AP for 30 min. In another group, 10 μm CI-III was applied 20 min before Bic and 4-AP treatment. PHLPP1α, PHLPP1β, phospho-Akt S473 (pAkt), Akt, phospho-ERK 1/2 (pERK 1/2), and ERK1/2 were detected by Western blot. C, Quantitative analysis of Western blots similar to those shown in B. The ratios of PHLPP1 to Akt (loading control), p-Akt to Akt, and p-ERK 1/2 to ERK 1/2 were compared. All ratios were normalized to control values before analysis. Bic and 4-AP treatment decreased PHLPP1/Akt and increased p-Akt/ Akt and p-ERK/ ERK. Preapplication of CI-III blocked those changes. *p < 0.05; ns, not significantly different; one-way ANOVA followed by Bonferroni test; n = 3. D, Cultured cortical neurons (DIV12) were treated with 20 μm Bic and 100 μm 4-AP for 30 min. In another group, 10 μm CI-III was applied 10 min before Bic and 4-AP treatment. The levels of PHLPP1, p-Akt, Akt, p-ERK1/2, ERK1/2, and PTEN were detected by Western blot. E, Quantitative analysis of Western blots similar to those shown in D. Bic and 4-AP treatment decreased PHLPP1/Akt and increased p-Akt/Akt and p-ERK/ERK, effects which were blocked by CI-III. PTEN levels were not affected by treatments. *p < 0.05; ns, not significantly different; one-way ANOVA followed by Bonferroni test; n = 3. Error bars indicate SEM.
PHLPP1 is required for calpain-mediated Akt and ERK regulation and neuroprotection
The above results showed that PHLPP1 degradation was associated with Akt and ERK activation. To further confirm that PHLPP1 degradation was necessary for Akt and ERK activation and neuroprotection, we infected cortical neurons with PHLPP1 RNAi lentivirus, resulting in down-regulation of PHLPP1α and PHLPP1β (Fig. 3A,C; p < 0.05; Bonferroni's test between scrambled RNAi and PHLPP1 RNAi). Knockdown of PHLPP1 significantly elevated p-Akt levels (Fig. 3A,C; p < 0.05 between scrambled RNAi and PHLPP1 RNAi) and slightly but not significantly elevated p-ERK levels (Fig. 3A,C; not significantly different between scrambled RNAi and PHLPP1 RNAi). In addition, downregulation of PHLPP1 significantly decreased starvation-induced neuronal death (Fig. 3D; p < 0.05 between scrambled RNAi and PHLPP1 RNAi).
Figure 3.

Lentiviral shRNA knockdown of PHLPP1 mimics the neuroprotective effect of synaptic NMDAR activation and eliminates the effect of calpain inhibition on neuroprotection. A, Cultured cortical neurons were infected by PHLPP1 shRNA lentivirus or scrambled shRNA lentivirus. The levels of indicated proteins in infected neurons were assessed by Western blot. B, shRNA lentivirus-infected neurons were treated with Bic and 4-AP or with CI-III, Bic, and 4-AP for 30 min. The levels of indicated proteins were assessed by Western blot. C, Quantitative analysis of Western blots similar to those shown in A and B. Bic and 4-AP treatment reduced PHLPP1α and PHLPP1β levels and increased p-Akt/Akt levels in scrambled RNAi-infected neurons but not in PHLPP1 RNAi-infected neurons. CI-III blocked Bic and 4-AP-induced changes in PHLPP1, p-Akt/Akt, and p-ERK/ERK levels in scrambled RNAi-infected neurons but not in PHLPP1 RNAi-infected neurons. *p < 0.05; ns, not significantly different; one-way ANOVA followed by Bonferroni test; n = 3. Error bars indicate SEM. D, shRNA lentivirus-infected neurons were subjected to starvation for 3 d. PHLPP1 RNAi-infected neurons showed decreased neuronal death compared with scrambled RNAi-infected neurons. Bic and 4-AP incubation along with starvation decreased neuronal death in scrambled RNAi-infected neurons but not in PHLPP1 RNAi-infected neurons. CI-III blocked Bic and 4-AP induced reduction of neuronal death in scrambled RNAi-infected neurons but not in PHLPP1 RNAi-infected neurons. Approximately 300–500 cells were counted for each group in each independent experiment. *p < 0.05; ns, not significantly different; one-way ANOVA followed by Bonferroni test. n = 3. Error bar indicates SEM.
Cortical neurons were transfected with PHLPP1 RNAi lentivirus before treatment with Bic and 4-AP to activate synaptic NMDARs. Bic and 4-AP treatment caused no further PHLPP1 degradation or Akt activation under these conditions (Fig. 3B,C), suggesting that PHLLP1 knockdown occluded synaptic NMDAR activation-induced PHLPP1 degradation. Interestingly, p-ERK was still elevated following Bic and 4-AP treatment in PHLPP1-depleted neurons, which is consistent with the mild elevation of pERK1/2 by PHLPP1-knockdown and suggests that at least another pathway also contributes to ERK activation. Unlike in scrambled RNAi neurons, calpain inhibition had no effect on PHLPP1, p-Akt, or p-ERK levels following Bic and 4-AP treatment in PHLPP1-depleted neurons (Fig. 3B,C), suggesting that PHLPP1 is required for calpain-dependent regulation of the Akt and ERK pathways. In the neuroprotection model, PHLPP1 knockdown by itself reduced starvation-elicited neuronal death, and Bic and 4-AP treatment did not further reduce neuronal death. More importantly, calpain inhibition did not block synaptic NMDAR activation-mediated neuroprotection in PHLPP1-depleted neurons, as it did in scrambled RNAi neurons (Fig. 3D). The above results indicate that PHLPP1 is necessary for calpain-mediated regulation of the Akt and ERK pathways and neuroprotection.
Extrasynaptic NMDAR stimulation-induced calpain activation does not cleave PHLPP1 but cleaves STEP and causes neurotoxicity
It has previously been reported that calpain is activated by extrasynaptic NMDAR stimulation, resulting in STEP cleavage, p38 activation, and neuronal death (Xu et al., 2009). We first replicated these results; synaptic NMDARs were first activated by Bic (10 μm) and irreversibly blocked by MK801 (40 μm) application; NMDA (100 μm) was then added to activate extrasynaptic NMDARs (see Materials and Methods). Extrasynaptic NMDAR stimulation resulted in STEP61 degradation, which generates a cleavage product with a molecular weight of 33 kDa (STEP33), and in neuronal death, as previously reported (Xu et al., 2009). Pretreatment with CI-III (10 μm) before extrasynaptic NMDAR stimulation blocked STEP cleavage and reduced neuronal death (Fig. 4). Notably, PHLPP1 was not cleaved under these conditions, nor was Akt or ERK activated (Fig. 4A,B). These results indicate that calpain activation following synaptic or extrasynaptic NMDAR stimulation results in the degradation of different substrates and produces opposite effects on neuronal survival.
Figure 4.

Extrasynaptic NMDAR-induced calpain activation degrades STEP but not PHLPP1 and mediates neurotoxicity. A, Cortical neurons were treated with 40 μm MK801 and 10 μm Bic for 2 min, washed three times, then treated with 100 μm NMDA and 10 μm glycine for 1 h to induce extrasynaptic NMDAR activation. In another group, 10 μm CI-III was applied 10 min before addition of NMDA and glycine. The levels of indicated proteins were assessed by Western blot. B, Quantitative analysis of Western blots similar to those shown in A. Extrasynaptic NMDAR activation decreased STEP61 levels and increased STEP33 levels. CI-III blocked extrasynaptic activation-induced STEP cleavage. PHLPP1, pAkt/Akt, and pERK/ERK levels were not affected by extrasynaptic NMDAR activation. *p < 0.05; ns, not significantly different; one-way ANOVA followed by Bonferroni test; n = 3. Error bars indicate SEM. C, Extrasynaptic NMDAR activation for 2 h induced neuronal death. Addition of 10 μm CI-III along with extrasynaptic activation reduced neuronal death. At least 300–500 Hoechst-stained cells were counted for each group in each independent experiment. *p < 0.05; one-way ANOVA followed by Bonferroni test. n = 3. Error bar indicates SEM.
μ-calpain is preferentially coupled to synaptic NMDAR-dependent prosurvival pathways and m-calpain to extrasynaptic NMDAR-dependent prodeath pathway
Since synaptic NMDAR activation did not degrade PTEN (Fig. 2D), an m-calpain-specific substrate we recently identified (Briz et al., 2013), we hypothesized that synaptic NMDAR activation results in μ-calpain but not m-calpain stimulation. To test this hypothesis, we used an m-calpain preferential inhibitor, Z-Leu-Abu-CONH-CH2-C6H3 (3, 5-(OMe)2) (mCalp-I), which has been reported to have a Ki of 22 nm against m-calpain versus a Ki of 2.3 μm against μ-calpain (Li et al., 1996). We verified these Ki values by testing its inhibition against purified μ-calpain or m-calpain cleavage of Suc-Leu-Tyr-AMC, a fluorescent calpain substrate, and we obtained Ki values of 25 nm and 1.3 μm against m-calpain and μ-calpain, respectively (Wang et al., unpublished observations), in close agreement with the reported values. To determine the role of m-calpain in synaptic NMDAR activation-mediated effects, 200 nm mCalp-I was applied to neurons 10 min before Bic and 4-AP treatment. Application of mCalp-I did not affect PHLPP1 degradation or Akt and ERK activation following Bic and 4-AP treatment (Fig. 5A,B). Consistently, application of mCalp-I (200 nm) together with Bic and 4-AP did not affect the neuroprotective effect of Bic and 4-AP against starvation or hydrogen peroxide (Fig. 5C,D). However, when mCalp-I concentration was increased to 5 μm, which should inhibit both μ-calpain and m-calpain according to the calculated Ki values, it blocked the neuroprotective effect elicited by Bic and 4-AP (Fig. 5C,D). To assess the role of m-calpain in extrasynaptic NMDAR activation-mediated effects, mCalp-I was added to neurons 10 min before extrasynaptic NMDAR stimulation. Under these conditions, mCalp-I (200 nm) significantly reduced STEP degradation. Extrasynaptic NMDAR stimulation in the absence or presence of mCalp-I (200 nm) had no effect on PHLPP1, p-Akt, and p-ERK levels (Fig. 5E,F). mCalp-I at both 200 nm and 5 μm significantly reduced neuronal death caused by extrasynaptic NMDAR activation (Fig. 5G).
Figure 5.

An m-calpain-specific inhibitor blocks extrasynaptic NMDAR-induced neurotoxicity but not synaptic NMDAR-induced neuroprotection. A, Cortical neurons were treated with 20 μm Bic and 100 μm 4-AP for 30 min and lysed for Western blot. In another group, 200 nm mCalp-I was applied 10 min before Bic and 4-AP treatment. The levels of indicated proteins were assessed by Western blot. B, Quantitative analysis of Western blots similar to those shown in A. mCalp-I (200 nm) did not affect Bic- and 4-AP-induced changes in PHLPP1, pAkt/Akt, and pERK/ERK levels. *p < 0.05; ns, not significantly different; one-way ANOVA followed by Bonferroni test; n = 3. Error bars indicate SEM. C, mCalp-I at 5 μm but not 200 nm blocked Bic- and 4-AP-induced neuroprotection against starvation in cortical neurons. At least 300–500 Hoechst-stained cells were counted for each group in each independent experiment. *p < 0.05; ns, not significantly different; one-way ANOVA followed by Bonferroni test. n = 3–6. Error bar indicates SEM. D, 5 μm but not 200 nm mCalp-I blocked Bic- and 4-A-induced neuroprotection against H2O2 insult. n = 3. E, Cortical neurons were treated with the protocol of extrasynaptic NMDAR activation and then lysed for Western blot. In another group, 200 nm mCalp-I was applied 10 min before extrasynaptic activation. F, Quantitative analysis of Western blots similar to those shown in E. mCalp-I (200 nm) reduced the cleavage of STEP induced by extrasynaptic NMDAR activation. *p < 0.05; one-way ANOVA followed by Bonferroni test; n = 3–5. Error bars indicate SEM. G, Pretreatment with 200 nm or 5 μm mCalp-I reduced neuronal death induced by extrasynaptic NMDAR activation. Approximately 300–500 Hoechst-stained cells were counted for each group in each independent experiment. *p < 0.05; one-way ANOVA followed by Bonferroni test. n = 4. Error bar indicates SEM.
We then analyzed the effects of a μ-calpain preferential inhibitor, 3-(5-fluoro-3-indolyl)-2-mercapto-(Z)-2-propenoic acid (μCalp-I), which has been reported to display >20-fold greater selectivity for μ-calpain-1 over m-calpain (Wang et al., 1996). We first verified the specificity of μCalp-I by determining its IC50 against purified μ-calpain and m-calpain cleavage of fluorescent calpain substrate Suc-Leu-Tyr-AMC. The IC50 values obtained against μ-calpain and m-calpain were 2.88 ± 0.74 μm and 9.26 ± 2.06 μm, respectively (p = 0.002, N = 4, two-tailed t test). Using a variant of the Cheng–Prusoff equation (Cer et al., 2009), and considering that μCalp-I is an uncompetitive inhibitor (Wang et al., 1996), the calculated Ki values were 0.27 and 1.71 μm for μCalp-I against μ-calpain and m-calpain, respectively, which are close to those reported previously (0.26 and 5.33 μm, respectively). To assess the role of μ-calpain in synaptic NMDAR activation-mediated effects, 2 μm μCalp-I was applied 10 min before Bic and 4-AP treatment. Application of μCalp-I blocked PHLPP1 degradation and Akt and ERK activation following Bic and 4-AP treatment (Fig. 6A,B). We then determined the effects of μCalp-I on synaptic NMDAR-dependent neuroprotection. Application of 2 μm μCalp-I together with Bic and 4-AP blocked the neuroprotective effects of Bic and 4-AP against starvation or oxidative stress (Fig. 6C,D). Application of 2 μm μCalp-I alone to neurons for 3 d did not induce cell death (Fig. 6C). To assess the role of μ-calpain in extrasynaptic NMDAR activation-mediated effects, 2 μm μCalp-I was applied 10 min before extrasynaptic NMDAR stimulation. μCalp-I did not affect STEP degradation (Fig. 6E,F) or neuronal death (Fig. 6G) elicited by extrasynaptic NMDAR activation.
Figure 6.

A μ-calpain-specific inhibitor blocks synaptic NMDAR-induced neuroprotection but not extrasynaptic NMDAR-induced neurotoxicity. A, Cortical neurons were treated with 20 μm Bic and 100 μm 4-AP for 30 min and lysed for Western blot. In another group, 2 μm μCalp-I was applied 10 min before Bic and 4-AP treatment. The levels of indicated proteins were assessed by Western blot. B, Quantitative analysis of Western blots similar to those shown in A. μCalp-I (2 μm) blocked Bic- and 4-AP-induced changes in PHLPP1, pAkt/Akt, and pERK/ERK levels. *p < 0.05; one-way ANOVA followed by Bonferroni test; n = 3. Error bars indicate SEM. C, μCalp-I (2 μm) blocked Bic- and 4-AP-induced neuroprotection against starvation in cortical neurons. Incubation of neurons with 2 μm μCalp-I alone for 3 d did not cause cell death. At least 300–500 Hoechst-stained cells were counted for each group in each independent experiment. *p < 0.05; ns, not significantly different; one-way ANOVA followed by Bonferroni test. n = 3–5. Error bar indicates SEM. D, μCalp-I (2 μm) blocked Bic- and 4-AP-induced neuroprotection against H2O2. n = 3. E, Cortical neurons were treated with the protocol for extrasynaptic NMDAR activation and then lysed for Western blot. In another group, 2 μm μCalp-I was applied 10 min before extrasynaptic NMDAR activation. F, Quantitative analysis of Western blots similar to those shown in E. μCalp-I (2 μm) did not affect STEP degradation induced by extrasynaptic NMDAR activation (ns, not significantly different; two-tailed t test; n = 3). Error bars indicate SEM. G, μCalp-I (2 μm) did not affect neuronal death induced by extrasynaptic NMDAR activation. Approximately 300–500 Hoechst-stained cells were counted for each group in each independent experiment. ns, not significantly different; two-tailed t test; n = 4–5. Error bar indicates SEM.
To further verify the specific role of μ-calpain in the signaling pathways downstream of synaptic NMDARs, we downregulated μ-calpain or m-calpain in cultured cortical neurons using specific siRNAs (Fig. 7A,B) before synaptic NMDAR stimulation. Neither μ-calpain or m-calpain downregulation affected PHLPP1, p-Akt, and p-ERK levels in neurons under basal conditions (Fig. 7C). However, downregulation of μ-calpain but not m-calpain prevented PHLPP1 degradation and significantly reduced Akt and ERK activation following Bic and 4-AP treatment (Fig. 7C,D). Altogether, these results indicate that μ-calpain is preferentially coupled to synaptic NMDAR-PHLPP1-Akt/ERK prosurvival pathway and m-calpain to extrasynaptic NMDAR-STEP prodeath pathway.
Figure 7.

μ-calpain but not m-calpain knockdown blocks PHLPP1 degradation and Akt and ERK activation induced by synaptic NMDAR activation. A, Cultured cortical neurons at DIV 7 were infected with scrambled siRNA, μ-calpain siRNA, or m-calpain siRNA separately. Two days after infection, the levels of indicated proteins were assessed by Western blot. B, Quantitative analysis of Western blots similar to those shown in A. μ-calpain and m-calpain siRNA significantly reduced μ-calpain and m-calpain levels, respectively. *p < 0.05; one-way ANOVA followed by Bonferroni test. n = 3. C, siRNA-infected neurons were treated with Bic and 4-AP for 30 min. The levels of indicated proteins in infected neurons before or after the treatment were assessed by Western blot. D, Quantitative analysis of Western blots similar to those shown in C. The levels of PHLPP1α and PHLPP1β were significantly higher and the ratios of pAkt/Akt and pERK/ERK were significantly lower in μ-calpain siRNA-infected neurons than that in scrambled siRNA-infected neurons. *p < 0.05; ns, not significantly different; one-way ANOVA followed by Bonferroni test; n = 3.
μ-calpain is recruited to synaptic NMDAR complex following synaptic NMDAR activation
To further explore the mechanism underlying μ-calpain activation following synaptic NMDAR stimulation in cultured cortical neurons, we performed coimmunoprecipitation (co-IP) using an antibody against the NR2A subunit of NMDAR, which is preferentially localized in postsynaptic density (Steigerwald et al., 2000) and mediates prosurvival signaling (Liu et al., 2007). Under basal conditions, μ-calpain was weakly associated with NR2A, whereas m-calpain was not associated at all with NR2A (Fig. 8A,B). We then treated neurons with Bic, 4-AP, and CI-III for 1 h before co-IP, to prevent calpain activation, without affecting its translocation. μ-calpain, but not m-calpain, was significantly recruited to NR2A-containing NMDARs following Bic, 4-AP, and CI-III treatment. As a positive control, we verified that PSD95 and NR2B coimmunoprecipitated with NR2A (Fig. 8A,B). On the other hand, PHLPP1 was not detected in the immunoprecipitation with the NR2A antibody we used (data not shown). We then performed co-IP using a PHLPP1 antibody. Under basal conditions, μ-calpain was weakly associated with PHLPP1, but their interaction was increased following Bic, 4-AP, and CI-III treatment (Fig. 8C,D). In contrast, m-calpain showed no interaction or recruitment with PHLPP1 under the same conditions. NR2A was also immunoprecipitated by PHLPP1 antibody under basal conditions, but there was no further recruitment following synaptic NMDA receptor activation. Together, these co-IP results suggest that synaptic NMDAR activation recruits μ-calpain but not m-calpain to NMDAR and PHLPP1 complex.
Figure 8.
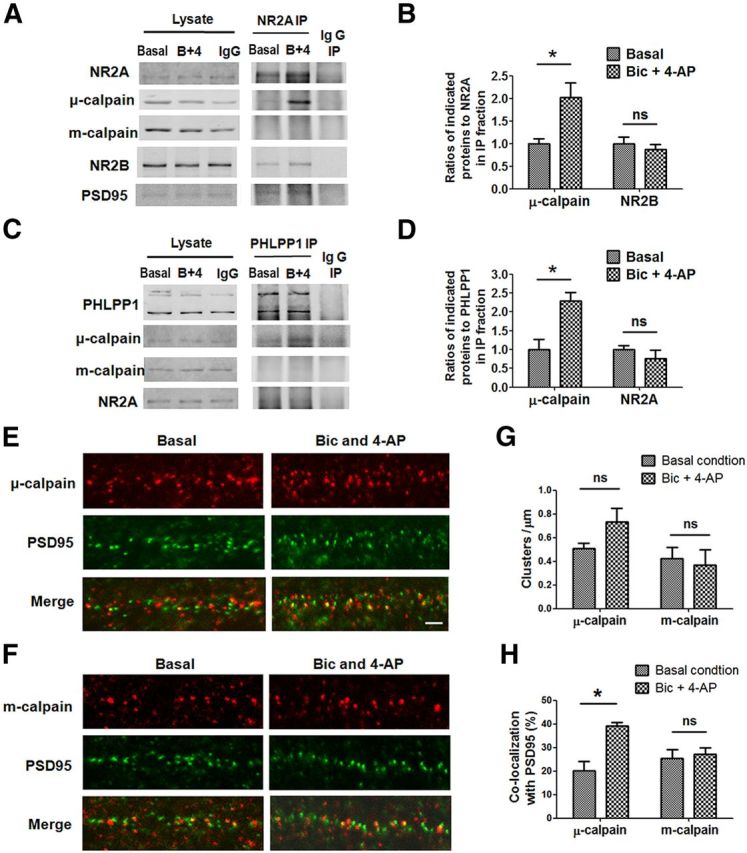
μ-calpain but not m-calpain is recruited to synaptic NMDAR complex following synaptic NMDAR activation. A, NR2A coprecipitated μ-calpain,NR2B, and PSD95 but not m-calpain from lysates of cultured cortical neurons. Treatment of neurons with 20 μm Bic, 100 μm 4-AP, and 10 μm CI-III for 1 h increased the association of μ-calpain with NR2A. IP with nonimmune rabbit IgG was included as a negative control. B, Statistical analysis of three independent co-IP experiments. The ratios of μ-calpain or NR2B to NR2A in IP fraction were compared. All ratios were normalized to the average ratio of basal group before t test (*p < 0.05; ns, no significant difference; two-tailed t test; n = 3). C, PHLPP1 coprecipitated μ-calpain and NR2A but not m-calpain from lysates of cultured cortical neurons. Treatment of neurons with Bic, 4-AP, and CI-III for 1 h increased the association of μ-calpain with PHLPP1. D, Statistical analysis of three independent co-IP experiments. The ratios of μ-calpain or NR2A to PHLPP1 in IP fraction were compared (*p < 0.05; ns, no significant difference; two-tailed t test; n = 3). E, F, Representative images of μ-calpain/m-calpain and PSD95 costaining in dendrites of cultured cortical neurons before and after 1 h of Bic (20 μm) and 4-AP (100 μm) treatment. Scale bar, 2 μm. G, Quantitative analysis of cluster density in images similar to those shown in E and F. Bic and 4-AP treatment did not affect cluster densities of μ-calpain and m-calpain. H, Quantitative analysis of percentage of clusters immunopositive for μ-calpain and m-calpain and PSD95 in images similar to those shown in E and F. Bic and 4-AP treatment increased the colocalization of PSD95 with μ-calpain but not with m-calpain. Approximately 800 clusters of each protein from three separate experiments were quantified; *p < 0.05; ns, not significantly different; two-tailed t test. Error bars indicate SEM.
We then examined the subcellular localization of the two calpain isoforms in cortical neurons by double-immunostaining for μ-calpain or m-calpain and PSD95 before and after Bic and 4-AP treatment. Both μ-calpain and m-calpain staining exhibited puncta along dendrites, which were partially colocalized with PSD95 puncta under basal conditions (20% of μ-calpain and 25% of m-calpain; Fig. 8E–H). Noteworthy, colocalization of μ-calpain with PSD95 was significantly increased (from 20 to 39%) following Bic and 4-AP treatment for 1 h. Furthermore, μ-calpain cluster density was slightly, although not significantly, increased after Bic and 4-AP treatment. In contrast, neither m-calpain cluster density nor colocalization with PSD95 was affected by Bic and 4-AP treatment (Fig. 8E–H). Together, these results suggest that following synaptic NMDAR activation, μ-calpain, but not m-calpain, is recruited to synaptic NMDAR complexes containing NR2A, PSD95, and PHLPP1, which most likely facilitates the cleavage of PHLPP1 by μ-calpain.
m-calpain activity exacerbates while μ-calpain activity suppresses NMDA-induced neurotoxicity in acute hippocampal slices
Our previous study showed that NMDA treatment of acute hippocampal slices from young rats (1–3 weeks) caused neurotoxicity, which was reduced by calpain inhibition (Zhou and Baudry, 2006). Here, we further explored the specific role of μ-calpain and m-calpain in this process. Acute hippocampal slices from wild-type mice at postnatal day 15 were incubated with NMDA (100 μm) for 1 h. NMDA treatment, which activates both synaptic and extrasynaptic NMDARs, caused the degradation of both STEP and PHLPP1 (Fig. 9A,B). PHLPP1β was significantly decreased and PHLPP1α was slightly but not significantly decreased. Correspondingly, ERK but not Akt was activated. Coapplication of CI-III (10 μm) completely blocked NMDA-induced degradation of both STEP and PHLPP1 and ERK activation. However, coapplication of mCalp-I (200 nm) only blocked the degradation of STEP but not of PHLPP1, which supports the idea that STEP is degraded by m-calpain but not μ-calpain under these conditions. We then used μ-calpain KO mice to further test the role of μ-calpain in NMDA-induced neurotoxicity in hippocampal slices. μ-calpain was completely absent, while m-calpain levels were not affected in both cortex and hippocampus of μ-calpain KO mice (Fig. 9C). NMDA treatment of hippocampal slices from μ-calpain KO mice caused the degradation of STEP but not of PHLPP1 (Fig. 9D,E), indicating that μ-calpain is required for the cleavage of PHLPP1 but not of STEP. The ERK pathway was not activated, probably because PHLPP1β was unchanged following NMDA treatment in slices from μ-calpain KO mice. Coapplication of CI-III in these slices, which under this condition only targets m-calpain, blocked NMDA-induced degradation of STEP (Fig. 9D,E), further supporting the idea that STEP is cleaved by m-calpain. Finally, we tested LDH release, an index of excitotoxicity, from slices prepared from P15 wild-type and μ-calpain KO mice treated with NMDA in the presence or absence of CI-III for 3 h. NMDA treatment of slices from P15 wild-type mice caused a significant LDH release (approximately fourfold increase over control), which was partially blocked by CI-III (Fig. 9F), a result in good agreement with our previous results in young rat slices (Zhou and Baudry, 2006). NMDA treatment also induced a large LDH release in μ-calpain KO slices (Fig. 9F). LDH release was also partially blocked by CI-III. Importantly, NMDA treatment caused more LDH release in slices from μ-calpain KO than wild-type mice (Fig. 9G), supporting the notion that μ-calpain reduces neurotoxicity following NMDA treatment.
Figure 9.

m-calpain inhibition decreases while μ-calpain knockdown increases NMDA-induced LDH release in acute hippocampal slices. A, Acute hippocampal slices from wild-type mice on postnatal day 15 (P15) were treated with NMDA (100 μm) ± CI-III (10 μm) or mCalp-I (200 nm) for 1 h. The levels of indicated proteins were detected by Western blot. B, Quantitative analysis of Western blots similar to those shown in A. NMDA treatment significantly increased the levels of STEP33 (calpain-mediated STEP breakdown product) and p-ERK and decreased the level of full-length PHLPP1β. CI-III blocked all those changes. mCalp-I blocked the increase of STEP33 but not the decrease in PHLPP1β. *p < 0.05; one-way ANOVA followed by Bonferroni test. n = 3–5. C, Levels of μ-calpain and m-calpain in cortical and hippocampal homogenates from wild-type and μ-calpain KO mice. D, Acute hippocampal slices from P15 μ-calpain KO mice were treated with NMDA (100 μm) ± CI-III (10 μm) for 1 h. The levels of indicated proteins were detected by Western blot. E, Quantitative analysis of Western blots similar to those shown in D. NMDA treatment of slices from μ-calpain KO mice significantly increased STEP33 but did not affect PHLPP1 or p-ERK. *p < 0.05; one-way ANOVA followed by Bonferroni test. n = 3. F, NMDA-induced LDH release in acute hippocampal slices from P15 wild-type and μ-calpain KO mice. Slices were treated with NMDA (100 μm) in the absence or presence of CI-III (10 μm) for 3 h. LDH release was normalized to protein concentration and results are shown as fold of the average value of the control group. *p < 0.05; one-way ANOVA followed by Bonferroni test. n = 4 (16–24 slices each treatment from 4 independent experiments). Error bar indicates SEM. G, NMDA-induced LDH release from slices of P15 wild-type and μ-calpain KO mice was expressed as fold of the respective control and compared. *p < 0.05; two-tailed t test. n = 4. Error bars indicate SEM.
Discussion
Our results clearly indicate that μ-calpain and m-calpain are activated by different NMDAR populations (synaptic vs extrasynaptic NMDARs) and regulate different substrates (PHLPP1 and STEP) to produce opposite effects on neuronal fate (neuroprotection and neurodegeneration; Fig. 10). These findings challenge the prevalent theory that the duration of calpain activation determines whether calpain plays physiological or pathological roles. Instead, our results support the notion that calpain function is determined by local scaffolding with upstream receptors and downstream substrates. In particular, our study indicates that μ-calpain and m-calpain are preferentially coupled to synaptic and extrasynaptic NMDARs, respectively, underlying the importance of considering specific calpain isoforms as potential clinical targets for the treatment of NMDAR-related neurodegenerative diseases.
Figure 10.

Schematic illustration of the separate pathways downstream of synaptic and extrasynaptic NMDARs. Left, Synaptic NMDAR activation stimulates μ-calpain, resulting in PHLPP1degradation and in the activation of Akt and ERK1/2 and their downstream prosurvival pathways. Right, Extrasynaptic NMDAR activation induces m-calpain activation and the resulting STEP degradation and in the activation of p38 prodeath pathway, leading to neurotoxicity. There are no cross-talks between those two pathways.
μ-calpain-dependent cleavage of PHLPP1 downstream of synaptic NMDARs regulates both Akt and ERK prosurvival cascades
Calpain-induced cleavage of PHLPP1β and the resulting ERK activation have been shown to regulate synaptic plasticity (Shimizu et al., 2007). Here, we report that μ-calpain-mediated PHLPP1β degradation is specifically triggered by synaptic but not extrasynaptic NMDAR activation and contributes to the neuroprotective effects of synaptic NMDARs. In addition, we found that PHLPP1α, which dephosphorylates and inhibits Akt, is also cleaved by μ-calpain following synaptic NMDAR activation. This result supports and further refines the hypothesis proposed by Jackson and Foster (2009). Calpain-mediated cleavage of PHLPP1 1α and β is necessary and sufficient for synaptic NMDAR-induced activation of the Akt and ERK pathways since calpain inhibition blocked, while PHLPP1 knockdown mimicked, the effects of synaptic NMDAR activation on Akt and ERK pathways. PHLPP1 suppresses Akt and ERK pathways under basal conditions; following synaptic NMDAR activation, calpain cleaves PHLPP1α and β, thus releasing the inhibition of these two major prosurvival signaling cascades in neurons. Consistently, calpain cleavage of PHLPP1 was required for the neuroprotective effects of synaptic NMDARs, as calpain inhibition blocked the neuroprotection elicited by synaptic NMDAR activation. This result was further confirmed by PHLPP1 knockdown experiments, as downregulation of PHLPP1 not only suppressed the blockade of neuroprotection caused by calpain inhibition but also induced neuroprotection without synaptic NMDAR activation. Consistent with our results, a recent study reported that PHLPP1 knock-out mice are more resistant to ischemic brain injury (Chen et al., 2013). Thus, PHLPP1 should be considered as a novel potential target for the treatment of neurodegenerative diseases.
Opposite roles of synaptic versus extrasynaptic calpain
In our study, calpain activated by extrasynaptic NMDAR stimulation cleaved STEP and caused neuronal death, a result in good agreement with previous results (Xu et al., 2009). The opposite effects of PHLPP1 and STEP on neuroprotection/neurodegeneration account for the opposite effects of synaptic versus extrasynaptic calpain activity on neuronal fate. The results presented here indicate that selective activation of synaptic calpain induces neuroprotection, while extrasynaptic calpain stimulation contributes to neurotoxicity. These findings challenge the general view that prolonged or excessive activation of calpain is responsible for calpain-mediated neurotoxicity. In our study, prolonged activation of synaptic calpain (by Bic and 4-AP treatment) for as long as 3 d did not result in STEP cleavage (data not shown), nor in neuronal damage, but produced neuroprotection against starvation and oxidative stress. On the other hand, activation of extrasynaptic NMDARs with 100 μm NMDA, a concentration that would cause calcium overload and calpain overactivation, did not affect PHLPP1 or its downstream pathways, suggesting that there are two separate pools of calpain downstream of synaptic and extrasynaptic NMDARs, which regulate different substrates and therefore exert separate functions.
μ-calpain and m-calpain are downstream of synaptic and extrasynaptic NMDARs, respectively
The possibility that μ-calpain and m-calpain exert different roles in CNS has not been extensively discussed. However, the discovery that m-calpain can be activated by phosphorylation (Glading et al., 2004; Zadran et al., 2010), coupled with the identification of PTEN as a specific m-calpain substrate (Briz et al., 2013), supported the notion that μ-calpain and m-calpain could play distinct functions. In the present study, we observed that synaptic NMDAR activation by Bic and 4-AP treatment did not result in the degradation of PTEN, a specific m-calpain substrate, suggesting that synaptic NMDAR activation does not activate m-calpain. The use of μ-calpain- and m-calpain-specific inhibitors confirmed this idea: an m-calpain-specific inhibitor did not affect synaptic NMDAR-dependent PHLPP1 cleavage and neuroprotection but blocked extrasynaptic NMDAR-dependent STEP cleavage and neurotoxicity. In contrast, a μ-calpain-specific inhibitor blocked synaptic NMDAR-mediated effects but not extrasynaptic NMDAR-mediated neurotoxicity. We verified these results by using μ-calpain and m-calpain-specific siRNAs in cultured neurons and found that only μ-calpain knockdown blocked synaptic NMDAR-mediated neuroprotective pathways. As for the effect of calpain knockdown on NMDAR-mediated neurotoxicity, it has been previously reported that knockdown of m-calpain, but not μ-calpain, by AAV-shRNA transfection increases survival of primary hippocampal neurons following NMDA treatment (Bevers et al., 2009). To further verify the role of μ-calpain and m-calpain in neuronal survival beyond cultured neurons, we used NMDA-induced neurotoxicity in acute hippocampal slices from young mice. We use this model because our previous study showed that NMDA treatment of acute hippocampal slices caused neurotoxicity in young but not adult rats (Zhou and Baudry, 2006), probably because young rats have more NR2B-containing NMDARs, which preferentially localize at extrasynaptic sites (Tovar and Westbrook, 1999). We found that μ-calpain knock-out eliminated NMDA-induced degradation of PHLPP1 but not of STEP and ERK activation, and exacerbated neurotoxicity. On the other hand, m-calpain-specific inhibition by applying either mCalp-I to slices from wild-type mice or CI-III to slices from μ-calpain KO mice blocked NMDA-induced degradation of STEP and reduced neurotoxicity. Together, these results strongly suggest that μ-calpain is preferentially activated by synaptic NMDAR stimulation, whereas m-calpain is preferentially activated by extrasynaptic NMDAR stimulation. In support of this idea, μ-calpain is present in synaptic compartments (Perlmutter et al., 1988), where it could regulate synaptic function through its action on synaptic elements such as cytoskeletal and scaffolding proteins as well as glutamate receptors (Liu et al., 2008). In contrast, little is known regarding the ultrastructural localization of m-calpain in neurons. One of the newly discovered physiological roles of m-calpain is to regulate activity-dependent local protein synthesis (Wang and Huang, 2012; Briz et al., 2013), which takes place not in synapses but in nearby extrasynaptic areas (Steward and Wallace, 1995; Frey and Morris, 1998). In addition, m-calpain has been reported to control synaptogenesis in dendritic shafts through constitutive proteolysis of the cytoskeletal protein, cortactin (Mingorance-Le Meur and O'Connor, 2009). The discovery of new and specific substrates for both μ-calpain or m-calpain should help differentiating the physiological functions of these two major calpain isoforms in the CNS.
Different scaffolding of μ-calpain and m-calpain determines their separate roles in neurons
The existence of separate signaling pathways for μ-calpain and m-calpain reported here suggests that these two calpain isoforms belong to different protein scaffolds, which could segregate them in different compartments in neurons. PHLPP1 can be cleaved by both purified μ-calpain and m-calpain in membrane fractions (Fig. 2A), yet it was cleaved only by synaptic μ-calpain activity in hippocampal slices, suggesting that substrate specificity for calpains depends not only on amino acid sequences within substrates, but also on localization and scaffolding of both substrates and calpains in neurons. The co-IP results presented here also suggest that NR2A-containing NMDARs, PSD95, μ-calpain, and PHLPP1 form a complex in neurons. Furthermore, synaptic NMDAR activity recruits more μ-calpain to this NMDAR multiprotein complex, as shown both by co-IP and immunostaining experiments; such recruitment could facilitate the proteolysis of PHLPP1 and possibly of other μ-calpain substrates in the complex. In contrast, m-calpain was not associated with this complex under basal conditions or recruited by activity, consistent with the absence of m-calpain activation following synaptic NMDAR activation. It is conceivable that an m-calpain-containing multiprotein complex is associated with extrasynaptic NMDARs. Besides the extrasynaptic NMDAR-STEP pathway, it has been reported that SAP102 mediates the movement of NR2B-containing NMDARs from synaptic to extrasynaptic membranes (Chen et al., 2012), where calpain has been shown to cleave NR2B and disrupt its interaction with SAP102 under neurotoxic conditions (Briz et al., 2012). Further studies are needed to examine scaffolding proteins that interact with μ-calpain or m-calpain and specifically associate with synaptic or extrasynaptic NMDAR multiprotein complexes. The poorly characterized different types of PDZ-binding motifs present in μ-calpain and m-calpain C-terminals may be involved in the different scaffolding of μ-calpain and m-calpain.
Clinical implications of specific m-calpain inhibition and μ-calpain activation
Our results have important implications for the development of new approaches for treating diseases associated with excitotoxicity, such as epilepsy, stroke, Alzheimer's and Parkinson's disease, Huntington disease, and ischemia. In all these cases, it has been suggested that extrasynaptic NMDAR activation and STEP degradation are involved in neurodegeneration. Our results would therefore suggest that specific inhibition of m-calpain but not μ-calpain would have neuroprotective effects under these conditions. Conversely, overexpression or activation of μ-calpain, by cleaving PHLPP1 and stimulating prosurvival cascades, could also have beneficial effects. Interestingly, we recently discovered that m-calpain activation is also involved in regulating the magnitude of long-term potentiation (LTP) in hippocampus, due to the existence of a molecular brake consisting in m-calpain-mediated PTEN degradation and stimulation of m-TOR-dependent PHLPP1β synthesis (Y. Wang, G. Zhu, V. Briz, Y.-T. Hsu, X. Bi, M. Baudry, unpublished observations). In particular, specific inhibition of m-calpain resulted in an enhancement of LTP magnitude, without interfering with LTP induction. Together, these findings indicate that specific m-calpain inhibition could therefore be extremely beneficial for preventing neurodegeneration, while facilitating certain forms of learning and memory.
Footnotes
This work was supported by Grant P01NS045260–01 from NINDS (PI: Dr. C. M. Gall). We also thank Dr. Steve Standley for numerous stimulating discussions.
The authors declare no competing financial interests.
References
- Azam M, Andrabi SS, Sahr KE, Kamath L, Kuliopulos A, Chishti AH. Disruption of the mouse mu-calpain gene reveals an essential role in platelet function. Mol Cell Biol. 2001;21:2213–2220. doi: 10.1128/MCB.21.6.2213-2220.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry M, Bi X. Learning and memory: an emergent property of cell motility. Neurobiol Learn Mem. 2013;104:64–72. doi: 10.1016/j.nlm.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevers MB, Lawrence E, Maronski M, Starr N, Amesquita M, Neumar RW. Knockdown of m-calpain increases survival of primary hippocampal neurons following NMDA excitotoxicity. J Neurochem. 2009;108:1237–1250. doi: 10.1111/j.1471-4159.2008.05860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briz V, Parkash J, Sánchez-Redondo S, Prevot V, Suñol C. Allopregnanolone prevents dieldrin-induced NMDA receptor internalization and neurotoxicity by preserving GABA(A) receptor function. Endocrinology. 2012;153:847–860. doi: 10.1210/en.2011-1333. [DOI] [PubMed] [Google Scholar]
- Briz V, Hsu YT, Li Y, Lee E, Bi X, Baudry M. Calpain-2-mediated PTEN degradation contributes to BDNF-induced stimulation of dendritic protein synthesis. J Neurosci. 2013;33:4317–4328. doi: 10.1523/JNEUROSCI.4907-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cer RZ, Mudunuri U, Stephens R, Lebeda FJ. IC50-to-Ki: a web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 2009;37:W441–W445. doi: 10.1093/nar/gkp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BS, Gray JA, Sanz-Clemente A, Wei Z, Thomas EV, Nicoll RA, Roche KW. SAP102 mediates synaptic clearance of NMDA receptors. Cell Rep. 2012;2:1120–1128. doi: 10.1016/j.celrep.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Van Winkle JA, Lyden PD, Brown JH, Purcell NH. PHLPP1 gene deletion protects the brain from ischemic injury. J Cereb Blood Flow Metab. 2013;33:196–204. doi: 10.1038/jcbfm.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. How BAD phosphorylation is good for survival. Nat Cell Biol. 1999;1:E33–E35. doi: 10.1038/10026. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG. Synaptic tagging: implications for late maintenance of hippocampal long-term potentiation. Trends Neurosci. 1998;21:181–188. doi: 10.1016/S0166-2236(97)01189-2. [DOI] [PubMed] [Google Scholar]
- Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Glading A, Bodnar RJ, Reynolds IJ, Shiraha H, Satish L, Potter DA, Blair HC, Wells A. Epidermal growth factor activates m-calpain (calpain-II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol Cell Biol. 2004;24:2499–2512. doi: 10.1128/MCB.24.6.2499-2512.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H. A calcium microdomain near NMDA receptors: a switch for ERK-dependent synapse-to-nucleus communication. Nat Neurosci. 2001a;4:565–566. doi: 10.1038/88380. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci. 2001b;4:261–267. doi: 10.1038/85109. [DOI] [PubMed] [Google Scholar]
- Jackson TC, Foster TC. Regional health and function in the hippocampus: Evolutionary compromises for a critical brain region. Biosci Hypotheses. 2009;2:245–251. doi: 10.1016/j.bihy.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson TC, Rani A, Kumar A, Foster TC. Regional hippocampal differences in AKT survival signaling across the lifespan: implications for CA1 vulnerability with aging. Cell Death Differ. 2009;16:439–448. doi: 10.1038/cdd.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson TC, Verrier JD, Semple-Rowland S, Kumar A, Foster TC. PHLPP1 splice variants differentially regulate AKT and PKCalpha signaling in hippocampal neurons: characterization of PHLPP proteins in the adult hippocampus. J Neurochem. 2010;115:941–955. doi: 10.1111/j.1471-4159.2010.06984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdi H, Hamo L, Oka T, Seegan A, Baudry M. BDNF mediates the neuroprotective effects of positive AMPA receptor modulators against MPP+-induced toxicity in cultured hippocampal and mesencephalic slices. Neuropharmacology. 2009;56:876–885. doi: 10.1016/j.neuropharm.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001;21:893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, Ben-Ari Y, Clapham DE, Medina I. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron. 2003;40:775–784. doi: 10.1016/S0896-6273(03)00645-7. [DOI] [PubMed] [Google Scholar]
- Li Z, Ortega-Vilain AC, Patil GS, Chu DL, Foreman JE, Eveleth DD, Powers JC. Novel peptidyl alpha-keto amide inhibitors of calpains and other cysteine proteases. J Med Chem. 1996;39:4089–4098. doi: 10.1021/jm950541c. [DOI] [PubMed] [Google Scholar]
- Liu J, Liu MC, Wang KK. Calpain in the CNS: from synaptic function to neurotoxicity. Sci Signal. 2008;1:re1. doi: 10.1126/stke.114re1. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G, Baudry M. The biochemistry of memory: a new and specific hypothesis. Science. 1984;224:1057–1063. doi: 10.1126/science.6144182. [DOI] [PubMed] [Google Scholar]
- Mingorance-Le Meur A, O'Connor TP. Neurite consolidation is an active process requiring constant repression of protrusive activity. EMBO J. 2009;28:248–260. doi: 10.1038/emboj.2008.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannaccione A, Secondo A, Molinaro P, D'Avanzo C, Cantile M, Esposito A, Boscia F, Scorziello A, Sirabella R, Sokolow S, Herchuelz A, Di Renzo G, Annunziato L. A new concept: abeta1-42 generates a hyperfunctional proteolytic NCX3 fragment that delays caspase-12 activation and neuronal death. J Neurosci. 2012;32:10609–10617. doi: 10.1523/JNEUROSCI.6429-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Stevenson P, Hardingham NR, Bading H, Hardingham GE. Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. J Neurosci. 2005;25:4279–4287. doi: 10.1523/JNEUROSCI.5019-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Léveillé F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, McKenzie G, Craigon M, Corriveau R, Ghazal P, Horsburgh K, Yankner BA, Wyllie DJ, Ikonomidou C, Hardingham GE. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008;11:476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkinton MS, Ip JK, Wood GL, Crossthwaite AJ, Williams RJ. Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurones. J Neurochem. 2002;80:239–254. doi: 10.1046/j.0022-3042.2001.00699.x. [DOI] [PubMed] [Google Scholar]
- Perlmutter LS, Siman R, Gall C, Seubert P, Baudry M, Lynch G. The ultrastructural localization of calcium-activated protease “calpain” in rat brain. Synapse. 1988;2:79–88. doi: 10.1002/syn.890020111. [DOI] [PubMed] [Google Scholar]
- Saavedra A, García-Martínez JM, Xifró X, Giralt A, Torres-Peraza JF, *Canals JM, Díaz-Hernández M, Lucas JJ, Alberch J, Pérez-Navarro E. PH domain leucine-rich repeat protein phosphatase 1 contributes to maintain the activation of the PI3K/Akt pro-survival pathway in Huntington's disease striatum. Cell Death Differ. 2010;17:324–335. doi: 10.1038/cdd.2009.127. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Kikuchi T, Yumoto N, Yoshimura N, Murachi T. Comparative specificity and kinetic studies on porcine calpain I and calpain II with naturally occurring peptides and synthetic fluorogenic substrates. J Biol Chem. 1984;259:12489–12494. [PubMed] [Google Scholar]
- Shimizu K, Okada M, Nagai K, Fukada Y. Suprachiasmatic nucleus circadian oscillatory protein, a novel binding partner of K-Ras in the membrane rafts, negatively regulates MAPK pathway. J Biol Chem. 2003;278:14920–14925. doi: 10.1074/jbc.M213214200. [DOI] [PubMed] [Google Scholar]
- Shimizu K, Phan T, Mansuy IM, Storm DR. Proteolytic degradation of SCOP in the hippocampus contributes to activation of MAP kinase and memory. Cell. 2007;128:1219–1229. doi: 10.1016/j.cell.2006.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano FX, Papadia S, Hofmann F, Hardingham NR, Bading H, Hardingham GE. Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci. 2006;26:4509–4518. doi: 10.1523/JNEUROSCI.0455-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steigerwald F, Schulz TW, Schenker LT, Kennedy MB, Seeburg PH, Köhr G. C-Terminal truncation of NR2A subunits impairs synaptic but not extrasynaptic localization of NMDA receptors. J Neurosci. 2000;20:4573–4581. doi: 10.1523/JNEUROSCI.20-12-04573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Wallace CS. mRNA distribution within dendrites: relationship to afferent innervation. J Neurobiol. 1995;26:447–449. doi: 10.1002/neu.480260316. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CF, Huang YS. Calpain 2 activated through N-methyl-D-aspartic acid receptor signaling cleaves CPEB3 and abrogates CPEB3-repressed translation in neurons. Mol Cell Biol. 2012;32:3321–3332. doi: 10.1128/MCB.00296-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KK, Nath R, Posner A, Raser KJ, Buroker-Kilgore M, Hajimohammadreza I, Probert A W, Jr, Marcoux FW, Ye Q, Takano E, Hatanaka M, Maki M, Caner H, Collins JL, Fergus A, Lee KS, Lunney EA, Hays SJ, Yuen P. An alpha-mercaptoacrylic acid derivative is a selective nonpeptide cell-permeable calpain inhibitor and is neuroprotective. Proc Natl Acad Sci U S A. 1996;93:6687–6692. doi: 10.1073/pnas.93.13.6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YB, Wang JJ, Wang SH, Liu SS, Cao JY, Li XM, Qiu S, Luo JH. Adaptor protein APPL1 couples synaptic NMDA receptor with neuronal prosurvival phosphatidylinositol 3-kinase/Akt pathway. J Neurosci. 2012;32:11919–11929. doi: 10.1523/JNEUROSCI.3852-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Lynch DR. Calpain and synaptic function. Mol Neurobiol. 2006;33:215–236. doi: 10.1385/MN:33:3:215. [DOI] [PubMed] [Google Scholar]
- Xu J, Kurup P, Zhang Y, Goebel-Goody SM, Wu PH, Hawasli AH, Baum ML, Bibb JA, Lombroso PJ. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J Neurosci. 2009;29:9330–9343. doi: 10.1523/JNEUROSCI.2212-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Wong TP, Chery N, Gaertner T, Wang YT, Baudry M. Calpain-mediated mGluR1alpha truncation: a key step in excitotoxicity. Neuron. 2007;53:399–412. doi: 10.1016/j.neuron.2006.12.020. [DOI] [PubMed] [Google Scholar]
- Yamaguchi A, Tamatani M, Matsuzaki H, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J Biol Chem. 2001;276:5256–5264. doi: 10.1074/jbc.M008552200. [DOI] [PubMed] [Google Scholar]
- Zadran S, Jourdi H, Rostamiani K, Qin Q, Bi X, Baudry M. Brain-derived neurotrophic factor and epidermal growth factor activate neuronal m-calpain via mitogen-activated protein kinase-dependent phosphorylation. J Neurosci. 2010;30:1086–1095. doi: 10.1523/JNEUROSCI.5120-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Baudry M. Developmental changes in NMDA neurotoxicity reflect developmental changes in subunit composition of NMDA receptors. J Neurosci. 2006;26:2956–2963. doi: 10.1523/JNEUROSCI.4299-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]