

Abstract
Brief sounds produce a period of suppressed responsiveness in the auditory cortex (ACx). This forward suppression can last for hundreds of milliseconds and might contribute to mechanisms of temporal separation of sounds and stimulus-specific adaptation. However, the mechanisms of forward suppression remain unknown. We used in vivo recordings of sound-evoked responses in the mouse ACx and whole-cell recordings, two-photon calcium imaging in presynaptic terminals, and two-photon glutamate uncaging in dendritic spines performed in brain slices to show that synaptic depression at thalamocortical (TC) projections contributes to forward suppression in the ACx. Paired-pulse synaptic depression at TC projections lasts for hundreds of milliseconds and is attributable to a switch between firing modes in thalamic neurons. Thalamic neurons respond to a brief depolarizing pulse with a burst of action potentials; however, within hundreds of milliseconds, the same pulse repeated again produces only a single action potential. This switch between firing modes depends on Cav3.1 T-type calcium channels enriched in thalamic relay neurons. Pharmacologic inhibition or knockdown of Cav3.1 T-type calcium channels in the auditory thalamus substantially reduces synaptic depression at TC projections and forward suppression in the ACx. These data suggest that Cav3.1-dependent synaptic depression at TC projections contributes to mechanisms of forward suppression in the ACx.
Introduction
Sensory neurons possess the ability to selectively suppress responses to repetitive stimuli. For instance, a brief sound can render auditory cortex (ACx) neurons less responsive or unresponsive to a successive sound, and this decreased responsiveness or forward suppression can last for several hundred milliseconds (Calford and Semple, 1995; Brosch and Schreiner, 1997; Wehr and Zador, 2005).
The mechanisms of forward suppression are unclear. For instance, it is not known which neuronal circuits are responsible and how they suppress the second cortical response for so long. Several lines of evidence indicate that subcortical and cortical neurons in the auditory system may contribute to forward suppression in the ACx. Because thalamic neurons, but not ACx neurons, can follow trains of sounds at high frequency (Creutzfeldt et al., 1980; Miller et al., 2002), it has been proposed that neuronal circuits within the ACx contribute to mechanisms of forward suppression (Wehr and Zador, 2005). However, these circuits have not yet been clearly identified. Thus, thalamocortical (TC), corticocortical (CC), or cortical GABAergic inhibitory circuits may be involved in mechanisms of forward suppression in the ACx. It has been previously shown that cortical GABAergic inhibition only partially underlies the mechanisms of forward suppression (Calford and Semple, 1995; Brosch and Schreiner, 1997; Tan et al., 2004). Wehr and Zador (2005) showed that GABAergic mechanisms are responsible for forward suppression only for the first 100 ms after the first stimulus. Therefore, forward suppression at longer intervals must involve other mechanisms (Wehr and Zador, 2005).
We hypothesized that short-term plasticity at TC excitatory synapses underlies forward suppression in the ACx. These synapses are among a group of glutamatergic synapses that in response to a pair of stimuli applied to a presynaptic neuron elicit paired-pulse depression (PPD), wherein the second postsynaptic response is weaker than the first (Gil et al., 1999; Bartlett and Smith, 2002; Rose and Metherate, 2005; Blundon et al., 2011; Viaene et al., 2011; Chun et al., 2013). In contrast, most CC or other glutamatergic synapses are more prone to paired-pulse facilitation (PPF), wherein the second response is stronger than the first (Stratford et al., 1996; Beierlein and Connors, 2002; Beierlein et al., 2002).
In this study, we show that, similar to forward suppression, TC PPD can last for hundreds of milliseconds, and this prolonged phenomenon depends on slowly reactivating Cav3.1 T-type calcium channels that are enriched in thalamic relay neurons (Talley et al., 1999; Anderson et al., 2005). A switch between Cav3.1-dependent burst firing of action potentials (APs; bursting mode) and firing of a single AP (tonic mode) in thalamic neurons underlines TC PPD. We also show that pharmacologic inhibition or short interfering RNA (siRNA)-mediated knockdown of Cav3.1 T-type calcium channels in the thalamic neurons substantially diminishes forward suppression at longer interstimulus intervals (ISIs). Thus, Cav3.1-dependent synaptic depression at TC projections may contribute to the mechanisms of forward suppression in the ACx.
Materials and Methods
Animals.
Eight- to 12-week-old C57BL/6J mice (The Jackson Laboratory) of both sexes were used. The care and use of animals was reviewed and approved by the St. Jude Children's Research Hospital Institutional Animal Care and Use Committee.
In vivo recordings in the ACx.
Mice were anesthetized with Avertin (0.2 ml/kg, i.p.), and their heads were fixed in a stereotaxic apparatus by using a custom-made platform. Craniotomy was performed over the ACx, and a dental cement well was constructed around the ACx. The well was filled with artificial CSF (ACSF) containing the following (in mm): 136 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, and 10 glucose, pH 7.4 (300–310 mOsm). The surgical procedure, which on average lasted 30–40 min, was followed by in vivo recordings. During surgery and recordings, mice were maintained in the anesthetized state by supplementing Avertin every 20–30 min based on appearance of the positive tail-pinch reflex. Local field potential (LFP) responses to sound stimulation were recorded from the primary ACx (A1) at 250–400 μm from the pial surface, which corresponds to layer 3/4 (L3/4), with a glass pipette (open pipette resistance, 3.5–5 MΩ) filled with ACSF. Pairs of white-noise clicks [20 ms duration, 70 dB sound pressure level (SPL), cos2-gated 5 ms rise and fall time, interpulse intervals of 100–1000 ms] were generated using a RZ6 signal processor (processing rate, 100 kHz) and OpenEx software [Tucker-Davis Technologies (TDT)], with signal output to an electrostatic speaker (TDT) that was placed 10 cm from the contralateral ear of the mouse. Click parameters (SPL, duration, and rise and fall times) were calibrated and verified at a wide frequency range, using a free-field microphone, preamplifier, and signal conditioner (PS9200; ACO Pacific) that was digitized (100 kHz; DigiData 1440A; Molecular Devices) and recorded with the ClampEx 10.0 software (sampling frequency, 100 kHz; Molecular Devices) or the BioSigRZ software (TDT). LFPs evoked by clicks were recorded in current-clamp mode and amplified using a Multiclamp 700B (Molecular Devices), digitized (10 kHz; DigiData 1440A; Molecular Devices), and recorded with the pClamp 10.0 software (Molecular Devices). Pairs of clicks at different ISIs were delivered every 10–30 s.
Brain slice preparation.
Acute primary TC slices (400 μm thick) containing the ACx and portions of the medial geniculate nucleus (MGv) of the thalamus were prepared as described previously (Cruikshank et al., 2002; Blundon et al., 2011). Briefly, mouse brains were quickly removed and placed in cold (4°C) dissecting ACSF containing 125 mm choline-Cl, 2.5 mm KCl, 0.4 mm CaCl2, 6 mm MgCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3, and 20 mm glucose (300–310 mOsm) at pH 7.4 with 95% O2/5% CO2. Primary TC slices were obtained from the left hemisphere by using a slicing angle of 15°. After a 1 h incubation in ACSF [125 mm NaCl, 2.5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3, and 10 mm glucose (300–310 mOsm) at pH 7.4, with 95% O2/5% CO2] at room temperature, slices were transferred into the recording chamber and superfused (2–3 ml/min) with warm (30–32°C) ACSF.
Brain slice electrophysiology.
Whole-cell recordings were obtained from cell bodies of L3/4 pyramidal thalamorecipient neurons in the ACx as described previously (Blundon et al., 2011; Chun et al., 2013). In the ACx of many mammalian species, pyramidal (not stellate) neurons are the principal cells in the thalamorecipient layers (Lund, 1973; Feldman and Peters, 1978; Smith and Populin, 2001; Richardson et al., 2009). Briefly, for voltage-clamp recordings, patch pipettes (open pipette resistance, 3.5–5 MΩ) were filled with an internal solution containing 125 mm CsMeSO3, 2 mm CsCl, 10 mm HEPES, 0.1 mm EGTA, 4 mm MgATP, 0.3 mm NaGTP, 10 mm Na2 creatine phosphate, 5 mm QX-314, 5 mm tetraethylammonium Cl, and 10–25 μm Alexa Fluor 594, pH 7.4, adjusted with CsOH (290–295 mOsm). Cell bodies of these neurons were found within the first 30–50% of the slice from the pial surface. All recorded neurons were visualized by two-photon imaging to ensure that the neurons were pyramidal and their dendritic tree was intact (see below). Voltage-clamp recordings were made by using a Multiclamp 700B (Molecular Devices), digitized (10 kHz; DigiData 1322A; Molecular Devices), and recorded using the pClamp 10.0 software (Molecular Devices). EPSCs were recorded at holding membrane potentials of −70 mV. In all experiments, membrane potentials were corrected for a liquid junction potential of −10 mV. Pairs of TC EPSCs were evoked by pairs of brief (0.2 ms) pulses at different ISIs delivered to the thalamic radiation (TR) via tungsten bipolar electrodes (FHC) placed in the white matter, midway between the MGv and the ACx (rostral to the hippocampus). CC EPSCs were evoked via stimulating electrodes (0.2 ms pulse) placed in L3/4, ∼200 μm lateral to the recording pipette. The monosynaptic component was measured using the initial (2 ms) slopes of EPSCs throughout experiments. The amplitude of stimulation was chosen to evoke 100–200 pA currents. To ensure consistent access resistance of the recording electrode during EPSC recordings, the peak amplitude of a 10 ms hyperpolarizing test pulse (−5 mV) was monitored. Access resistance in recorded neurons was typically 10–25 mΩ. Recordings were discarded if the access resistance was >25 mΩ or if the access resistance changed >15% during the course of the whole-cell recording.
Current-clamp recordings in MGv or ACx neurons were done by using an internal solution containing 115 mm KMeSO4, 20 mm KCl, 10 mm HEPES, 4 mm MgCl2·6H2O, 0.1 mm EGTA, 4 mm Na2ATP, 0.4 mm NaGTP, 10 mm Na2 creatine phosphate, and 10–25 μm Alexa Fluor 594, pH 7.3–7.4 (290–295 mOsm). Slow current (τ = 5 s) was injected through the patch pipette to maintain the resting membrane potential at −70 mV to reduce variability in the resting membrane potentials between recorded neurons. In current-clamp experiments, cell bodies of MGv or L3/4 neurons were targeted. In all experiments, Alexa Fluor 594 was added to the internal solution to visualize cell morphology. A pair of depolarizing pulses (0.2 ms, 3–4 nA) was delivered every 10–30 s at different ISIs (except for experiments shown in the last figure wherein pairs of stimuli were delivered every 500 ms, 970 ms, 1030 ms, 3 s, and 10 s) to generate APs in recorded neurons.
Cell-attached recordings in MGv neurons were done as described previously (Perkins, 2006). Briefly, the recording pipette (4–6 MΩ) was filled with NaCl (150 mm) and Alexa Fluor 594, and recordings were made in the voltage-clamp mode, which maintained an average current of 0 pA and is better suited for recording cell firing activity (Perkins, 2006). In some experiments, cell-attached recordings were performed in the current-clamp mode as described previously (Wehr and Zador, 2005), and similar results were obtained. The cell-attached recording was performed in the same cell after establishing the whole-cell recording (see above). Cell targeting and simultaneous cell-attached and whole-cell recordings were done using two-photon imaging.
Two-photon imaging and glutamate uncaging.
Two-photon laser-scanning microscopy was performed with an Ultima imaging system (Prairie Technologies), a titanium:sapphire Chameleon Ultra femtosecond-pulsed laser (Coherent), and 60× (0.9 numerical aperture) water-immersion infrared objectives (Olympus) as described previously (Blundon et al., 2011). To visualize dendrites, axons, dendritic spines, and presynaptic terminals, Alexa Fluor 594 (820 nm excitation wavelength) was included in intracellular solutions (see above). Dendritic spines were identified as protrusions emanating from a dendritic shaft of L3/4 neurons in the ACx. On the basis of previous data (Richardson et al., 2009), dendritic spines on L3/4 pyramidal neurons that were within 100 μm of the soma on basal or oblique dendrites were chosen as putative sites of thalamic inputs. Presynaptic terminals were identified as varicosities along TC projections emanating from thalamic neurons toward the TR. Calcium transients in presynaptic boutons were recorded in line-scan mode (500 Hz) and evoked by pairs of brief depolarizing pulses (see above) in current-clamp mode. Changes in fluorescence of Fluo 5F calcium indicator (green channel, G) added to intracellular solutions were measured. Line scans were analyzed as changes in Fluo 5F fluorescence relative to Alexa Fluor 594 fluorescence (red channel, R) as described previously (Yasuda et al., 2004).
For two-photon glutamate uncaging (TGU), MNI-glutamate [(S)-α-amino-2,3-dihydro-4-methoxy-7-nitro-δ-oxo-1H-indole-1-pentanoic acid; 2.5 mm] was added to the recording ACSF. The timing and intensity of glutamate uncaging were controlled by TriggerSync (Prairie Technologies). In typical experiments, 0.2 ms pulses from a second titanium:sapphire Chameleon Ultra laser (720 nm) were delivered to a targeted dendritic spine, and TGU-evoked EPSCs (uEPSCs) were recorded. The duration and intensity of illumination of the uncaging laser were then adjusted to induce responses that mimic spontaneous miniature EPSCs, which were recorded in L3/4 neurons and averaged 10–15 pA (Richardson et al., 2009). These uncaging parameters (site, laser duration, and laser intensity) were used to conduct the TGU experiments.
In vivo injections of virus or drugs.
Lentivirus vector siRNA plasmids (control, 5′-TACGTCCAAGGTCGGGCAGGAAGA-3′; and Cav3.1 shRNA, 5′-CTTCTCTTCATGCTATTATTTTTCATCTT-3′) were generated by Applied Biological Materials, and viruses (1.8 × 108 to 1 × 109 particles/ml) were produced by either the St. Jude Vector Laboratory or University of Tennessee Health Sciences Center Viral Vector Core. Anesthesia was induced with 2–2.5% isoflurane (in 100% O2) and maintained during surgery by using 1.5% isoflurane. Mouse heads were fixed in a stereotaxic device. A 28 gauge cannula (Plastics One) was inserted into the brain to deliver 0.4 μl of lentiviruses at two locations in the MGv [anteroposterior (AP), −3.0 mm; mediolateral (ML), ±2.0 mm; dorsoventral (DV), −3.1 mm; and AP, −3.5 mm; ML, ±2.28 mm; DV, −3.2 mm) at 0.5–2 μl/h, using a standard procedure (Zakharenko et al., 2003; Chun et al., 2013). The cannula was left in place for 15–20 min before being slowly retracted. After viral injections, incisions were sutured and mice were allowed to recover and then returned to holding cages. Electrophysiological experiments in vivo or in slices from these mice were performed 1–3 weeks after injections.
For drug injections in vivo, mice were anesthetized with Avertin, and their heads were fixed in a stereotaxic apparatus as described above. A cannula was inserted into the MGv at the following stereotaxic coordinates: AP, −3.0 mm; ML, ±2.0 mm; DV, −3.1 mm. A craniotomy was then performed over the ACx as described above. Drugs were slowly injected into the MGv or ACx at 5–10 μl/h. In vivo paired-pulse recordings of LFPs were taken after 40–60 min of drug perfusion.
Quantitative real-time PCR.
RNA was isolated from the thalamus containing the MGv 5–7 d after viral injection by using the mirVana RNA isolation kit (Applied Biosystems). cDNA was synthesized from 300 ng of total RNA, using the SuperScript III reverse transcriptase kit (Invitrogen). Quantitative real-time PCR (qPCR) was performed by using SYBR Green (Applied Biosystems) with the following primers: Gapdh, 5′-GTCGGTGTGAACGGATTTG-3′ and 5′-TAGACTCCACGACATACTCAGCA-3′; Cacna1g, 5′-TGTCTCCGCACGGTCTGTAA-3′ and 5′-AGATACCCAAAGCGACCATCTT-3′; and Cacna1c, 5′-CCTGCTGGTGGTTAGCGTG and 5′-TCTGCCTCCGTCTGTTTAGAA-3′.
Drugs.
Drugs were purchased from Sigma-Aldrich or Tocris Bioscience. Mibefradil and NNC 55-0396 were dissolved in ACSF. ACSF was used as a vehicle in control experiments.
Statistical analyses.
All data are represented as mean ± SEM. Statistics for all experiments were computed by using nonparametric Mann–Whitney rank-sum, Wilcoxon's signed-rank, or t tests measured in Sigma Stat (Systat Software).
Results
Forward suppression of sound-evoked responses in the mouse ACx and PPD of TC synaptic transmission and firing in thalamic neurons
We characterized forward suppression by using extracellular recordings and measuring sound-evoked responses in A1 of anesthetized mice in response to pairs of identical brief (20 ms) white noise sound clicks presented at different ISIs (Fig. 1a). Sound-evoked LFPs were measured 250–400 μm from the pial surface, which corresponds to L3/4, the major thalamorecipient layer of the ACx. Consistent with previous observations made using a loose cell-attached electrophysiological technique (Wehr and Zador, 2005), the second sound-evoked response was substantially depressed compared with the first response, and this forward suppression lasted for hundreds of milliseconds (Fig. 1b,c). Thus, the paired-pulse ratio (PPR) of the second LFP to the first LFP (PPR of LFPs) was <1 for all tested ISIs (p < 0.01, n = 15 mice; Fig. 1c). Forward suppression in the ACx at longer (200–1000 ms) intervals was similar (64–73% of the first response, n = 15 mice), suggesting similar mechanisms at those intervals. However, at the 100 ms interval, forward suppression was substantially stronger (25.7 ± 2.6% of the first response, p = 0.006, n = 15 mice), suggesting the contribution of an additional mechanism such as GABAergic inhibition that is effective only during the first 100 ms (Wehr and Zador, 2005).
Figure 1.

Forward suppression of sound-evoked responses in the ACx and PPD at TC synapses. a, Illustration of the experimental setup in which a pair of clicks (20 ms, 70 dB) was delivered to the contralateral ear of a mouse. LFPs were recorded from L3/4 of the ACx. The MGv is shown in blue, and the ACx is shown in red. b, LFP traces (average of 10–15 sweeps) in response to pairs of clicks delivered at 200, 400, and 700 ms ISIs. c, Mean PPR of LFPs (peak LFP2/peak LFP1) as a function of the ISI. d, A diagram of the TC slice containing portions of the MGv, hippocampus, and the ACx. L3/4 neurons in the ACx are shown in green. Stimulation electrodes (black) placed into the TR to stimulate TC projections and in the ACx to stimulate CC projections. e, Representative traces of TC PPD and CC PPF. f, Mean TC and CC PPR of EPSCs (EPSC2/EPSC1) as a function of ISI. g, Depiction of whole-cell recordings from thalamic relay neurons in the MGv and L3/4 pyramidal neurons in the ACx. Pairs of brief depolarization pulses (0.2 ms, 3–4 nA) are delivered to evoke APs in these cells. h, Representative voltage traces in MGv and ACx neurons in response to a pair of depolarization pulses. i, Mean PPR of APs (AP2/AP1) at different ISIs. Dots correspond to the delivery of clicks in vivo (b), stimulation of TC or CC projections (e), or depolarizing pulses (h) in slice experiments.
Thalamorecipient L3/4 pyramidal neurons receive excitatory inputs from the MGv of the thalamus through TC projections and from cortical sources through CC projections. If forward suppression observed in vivo is encoded in TC or CC synapses, those synapses should display a similarly reduced second response, and this reduction should last as long as forward suppression. To test this hypothesis, we measured the PPR of EPSCs at TC and CC synapses by using whole-cell voltage-clamp recordings in TC slices containing parts of the MGv and ACx (Cruikshank et al., 2002). Recording from L3/4 pyramidal neurons, the PPR of EPSCs was measured in response to pairs of brief (0.2 ms) electrical stimuli applied either to the TR or ACx to test the PPR at TC or CC synapses, respectively (Fig. 1d). Consistent with previous findings in the mouse ACx (Blundon et al., 2011; Chun et al., 2013), CC synapses responded with PPF, and TC synapses responded with PPD (Fig. 1e). The PPF at CC synapses was highest at shorter ISIs (<200 ms), with a substantial decline or even disappearance at longer ISIs (≥500 ms; Fig. 1f). In contrast, TC synapses displayed robust PPD at all intervals tested, which was similar to forward suppression in vivo. Thus, only TC projections display properties of forward suppression in the ACx, that is, reduction of the second response that can last for hundreds of milliseconds.
To determine the mechanisms of TC PPD, we tested excitability of presynaptic MGv thalamic relay neurons in response to a pair of stimulations. We tested generation of APs in response to a pair of stimuli by using whole-cell current-clamp recordings. The resting membrane potential in these neurons was −68.9 ± 1.4 mV (n = 15 cells). We injected a pair of brief depolarizing pulses (0.2 ms) at the same ISIs as in previous experiments (Fig. 1g). Interestingly, thalamic relay neurons typically responded with a burst of several APs in response to the first depolarization pulse and with only one AP in response to the second depolarization pulse (Fig. 1h). In contrast, L3/4 cortical pyramidal neurons faithfully responded to each brief depolarization pulse with a single AP (Fig. 1h). The resting membrane potential of cortical neurons was −69.8 ± 1.7 mV, which was not significantly different from that of thalamic neurons (p > 0.05, n = 9 cells). However, the PPR of the number of APs generated by the second pulse to the number of APs generated by the first pulse (PPR of APs) was 1.0 for cortical neurons (nine cells) and significantly less than 1.0 for thalamic neurons (p < 0.01, n = 15 cells) at all tested ISIs (Fig. 1i).
Although these data suggest that presynaptic bursting activity underlies TC PPD, postsynaptic mechanisms may also be involved in this process. We tested postsynaptic mechanisms by the TGU assay, which allows for the release of exogenous glutamate in a time- and synapse-controlled manner, bypassing the release of endogenous glutamate from presynaptic terminals (Fig. 2). We previously identified dendritic spines on L3/4 pyramidal neurons that are the inputs of TC projections (Richardson et al., 2009). The extracellular solution used in these experiments contained MNI-glutamate (2.5 mm) as the source of caged glutamate and TTX (1 μm) to block spontaneous APs. We released exogenous glutamate in pairs of brief (0.2 ms) pulses of a two-photon laser (720 nm wavelength) at thalamic inputs at different ISIs and measured uEPSCs from the soma of L3/4 pyramidal neurons (Fig. 2a,b). At all tested intervals, uEPSCs elicited by the second pulse were similar to those elicited by the first pulse (p > 0.05, n = 5; Fig. 2b,c). These data rule out the contribution of the postsynaptic locus (e.g., saturation or desensitization of postsynaptic glutamatergic receptors; Trussell et al., 1993) and suggest that bursting activity in presynaptic thalamic neurons is responsible for TC PPD.
Figure 2.

TC PPD is not expressed postsynaptically. a, Diagram showing TGU at thalamic inputs on L3/4 pyramidal neurons in the ACx. b, Representative traces of uEPSCs in response to a pair of TGU pulses (0.2 ms) at thalamic inputs at 100, 200, or 500 ms ISIs. Red dots indicate delivery of TGU pulses to individual dendritic spines. c, The average PPR of uEPSCs (uEPSC2/uEPSC1) as a function of ISI.
T-type calcium channel underlies PPD of APs in thalamic neurons
Several groups have reported that bursting activity of thalamic relay neurons is caused by T-type calcium channels (Llinás and Jahnsen, 1982; Coulter et al., 1989; Crunelli et al., 1989). We confirmed that the T-type calcium channel blockers mibefradil (10 μm) and NNC 55-0396 (15 μm) eliminate bursting activity in MGv neurons. After 60 min in the extracellular solution, both blockers reduced the number of APs resulting from the first depolarizing pulse to 1 without affecting AP generation during the second depolarizing pulse (Fig. 3a). The second depolarizing pulse also produced a single AP in the presence of these blockers, similar to what was observed in the absence of blockers. During the 60 min drug application, the PPR of APs changed from 0.37 ± 0.02 to 0.89 ± 0.06 (p < 0.01, n = 5 cells) in neurons treated with mibefradil and from 0.44 ± 0.02 to 1.0 ± 0.0 (p < 0.01, n = 7 cells) in those treated with NNC 55-0396, whereas the PPR of APs in cells treated with the vehicle control did not change significantly (0.46 ± 0.02 before and 0.43 ± 0.03 after 60 min; p > 0.05, n = 12 cells; Fig. 3b).
Figure 3.

T-type calcium channel blockers eliminate the bursting pattern of firing in MGv relay neurons. a, Representative voltage traces in MGv relay neurons in response to a pair of depolarization pulses delivered through a whole-cell pipette in the absence of drug (left) or 60 min after adding the vehicle, mibefradil, or NNC 55-0396. b, Average PPR of APs as a function of time in the presence of vehicle, mibefradil, or NNC 55-0396. *p < 0.01.
Presynaptic calcium at TC projections undergoes T-type calcium channel-dependent PPD
Bursting activity of thalamic neurons during the first pulse presumably causes more APs to arrive at presynaptic terminals and produces larger depolarization than it does during the second pulse. This should cause a stronger calcium influx and subsequently more glutamate release during the first pulse than the second pulse. To test this, we measured calcium transients in presynaptic terminals of TC projections. In a whole-cell current-clamp mode, we filled a thalamic neuron with the fluorescent calcium indicator Fluo 5F (green channel, G) and the fluorescent cytoplasmic dye Alexa Fluor 594 (red channel, R; Fig. 4a). By two-photon imaging, we visualized an axon that emanated from the recorded MGv neuron and propagated toward the ACx through the TR. Along these long axons, we observed varicosities that are the sites of presynaptic boutons (Fig. 4b). A pair of brief depolarizing pulses delivered to the soma of the thalamic neuron resulted in PPD of APs as reported above and in a pair of calcium transients in TC presynaptic boutons that were measured by changes in Fluo 5F fluorescence (Fig. 4c). The amplitude of calcium transients was quantified as the increase in Fluo 5F fluorescence normalized to Alexa Fluor 594 fluorescence (ΔG/R; Yasuda et al., 2004). Pairs of depolarizing pulses delivered to the soma of thalamic neurons produced a pair of calcium transients in which the second calcium transient was substantially smaller than the first (Fig. 4d). Thus, similar to the PPR of TC EPSCs and LFPs in vivo, the PPR of ΔG/R values in TC presynaptic terminals was <1.0 at all measured ISIs (p < 0.01, n = 13 boutons; Fig. 4f). However, in the presence of T-type calcium channel blockers, the PPR of ΔG/R values significantly changed (p < 0.05, n = 6–10 boutons). At short ISIs (100–500 ms), the PPR of ΔG/R values reached even higher than 1.0 in the presence of either mibefradil or NNC 55-0396. This is likely attributable to a slow decay of calcium transient traces, which depends on the slow dissociation of calcium from Fluo 5F. At shorter ISIs, the second calcium transient started before the first calcium transient reached the baseline (Fig. 4d), and this contributes to overestimation of PPR of ΔG/R. At a longer ISI (1000 ms), when both first and second calcium transients started from the same baseline (data not shown), the PPR of ΔG/R values was not significantly different from 1.0 (p > 0.05, n = 6–10 boutons) in the presence of channel blockers (Fig. 4f). Note that, under the control condition, PPD values of calcium transients, although smaller than 1.0, did not reach PPD values of EPSCs. This result is expected because there is a nonlinear relationship between presynaptic calcium and neurotransmitter release at central synapses, which predicts that a small change in presynaptic calcium is translated into a larger change in neurotransmitter release (Mintz et al., 1995; Borst and Sakmann, 1996).
Figure 4.
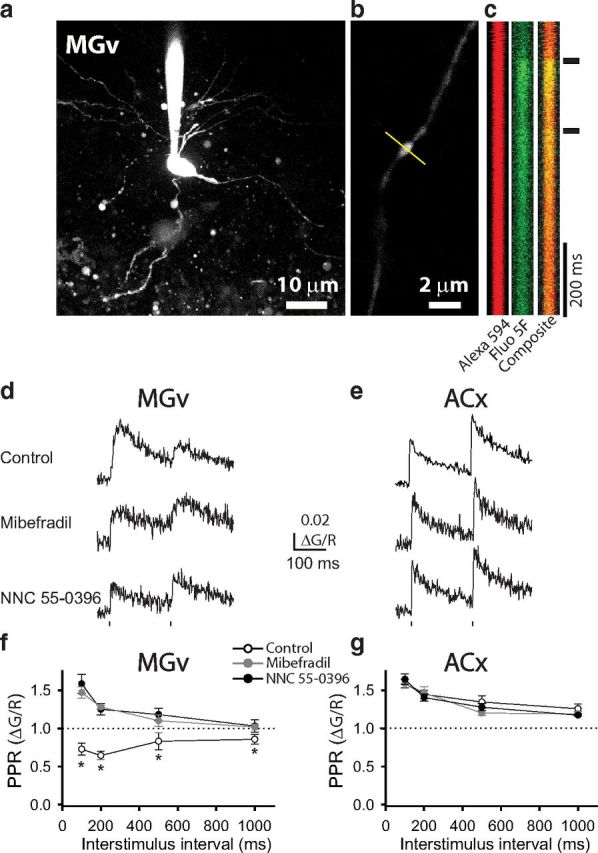
PPD of calcium transients in presynaptic terminals at TC projections. a, Image of an MGv thalamic relay neuron filled with fluorescent dyes Alexa Fluor 594 and Fluo 5F. b, Image of a TC projection with a presynaptic bouton. The yellow line represents the direction of line scans. c, Successive line scans of Alexa Fluor 594 (red), Fluo 5F (green), and composite fluorescence through a presynaptic bouton in response to a pair of depolarization pulses delivered to an MGv soma. d, e, Representative traces of calcium transients in TC presynaptic boutons from MGv neurons (d) and CC presynaptic boutons from L3/4 cortical neurons (e) in the presence of vehicle, mibefradil, or NNC 55-0396. Dots represent the delivery of depolarizing pulses. f, g, Average PPR of calcium transients (ΔG/R2/ΔG/R1) in the presence of vehicle, mibefradil, or NNC 55-0396 as a function of ISI in TC presynaptic boutons from MGv neurons (f) and CC presynaptic boutons from L3/4 cortical neurons (g). *p < 0.01.
Measurements of PPR of calcium transients in the ACx revealed that, unlike presynaptic terminals in TC projections, those in L3/4 neurons underwent facilitation (Fig. 4e). PPF was present at all tested ISIs, and the strongest PPF was observed at shorter intervals. Again, these data may be an overestimation because of the slow decay rate of calcium transients, which causes the second calcium transients to start at higher baseline levels than the first calcium transients, especially at shorter ISIs. However, in contrast to the results obtained at thalamic presynaptic terminals, neither mibefradil nor NNC 55-0396 affected the PPR of ΔG/R values in cortical presynaptic terminals (Fig. 4e,g).
Thalamic T-type calcium channels are necessary for PPD of TC EPSCs and forward suppression of sound-evoked LFPs in the ACx
To determine whether T-type calcium channels underlie the PPD of synaptic transmission at TC projections, we measured TC PPR of EPSCs in TC slices in the presence or absence of T-type calcium channel blockers in the extracellular solution (Fig. 5a). Mibefradil or NNC 55-0396 significantly reduced PPD of EPSCs at TC synapses at all measured ISIs (p < 0.01, n = 6–8 neurons per group; Fig. 5b). In contrast, neither blocker significantly affected the PPF of EPSCs at CC synapses (p > 0.05, n = 5–6 neurons per group; Fig. 5c,d). Together, these data indicate that T-type calcium channels specifically regulate the PPD of synaptic transmission at TC projections. It is important to note that, although treatment of slices with T-type channel blockers substantially reduced TC PPR of EPSCs, they did not completely eliminate it. Thus, in the presence of mibefradil or NNC 55-0396, TC PPD of EPSCs was significantly different from 1.0 at all tested ISIs (Fig. 5b). These data suggest that other, T-type channel-independent mechanisms contribute to TC PPD of EPSCs. These mechanisms may include presynaptic mechanisms, such as changes in probability of glutamate release in synaptic vesicles containing VGluT2, a glutamate transporter enriched at TC synapses (Weston et al., 2011), or activity-dependent accumulation of adenosine, an inhibitory modulator of neurotransmitter release, in the synaptic cleft (Flagmeyer et al., 1997; Blundon and Zakharenko, 2013).
Figure 5.
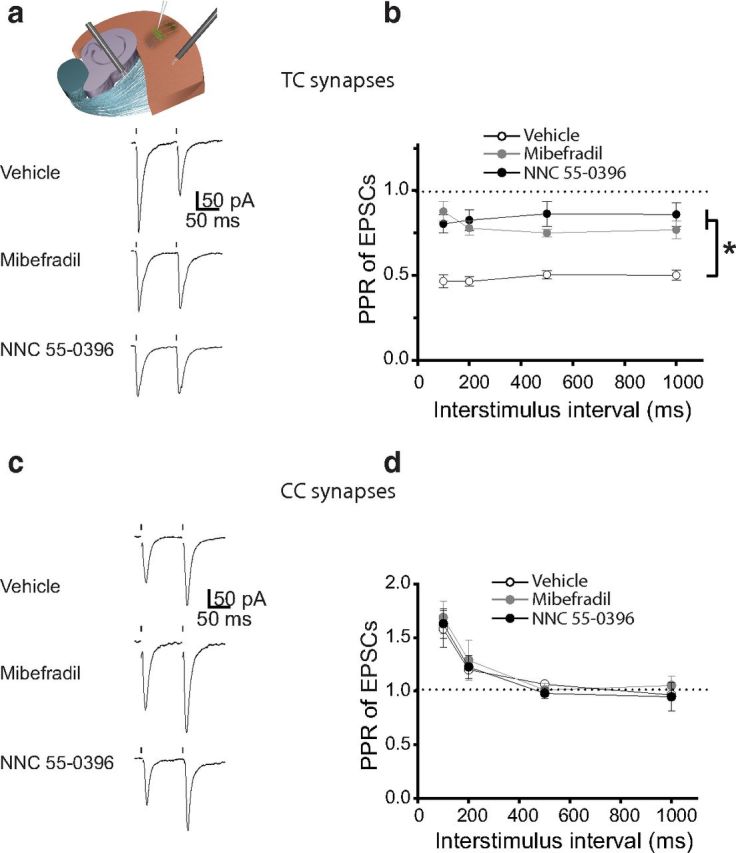
TC PPD of EPSCs is mediated by T-type calcium channels. a, Representative traces of TC EPSCs in response to pairs of stimuli delivered to the TR in TC slices in the presence of vehicle, mibefradil, or NNC 55-0396 (>60 min exposure). Dots represent the delivery of stimuli to the TR. b, Average PPR of EPSCs at different ISIs in the presence of vehicle, mibefradil, or NNC 55-0396. c, Representative traces of EPSCs recorded from L3/4 pyramidal neurons in the ACx in response to a pair of CC stimulations in the presence of vehicle, mibefradil, or NNC 55-0396. d, The average PPR of CC EPSCs as a function of ISI in the presence of vehicle, mibefradil, or NNC 55-0396. *p < 0.01.
If the PPD at TC projections is the mechanism underlying forward suppression in the ACx, then reducing TC PPD should inhibit sound-evoked forward suppression in vivo. To test this hypothesis, we slowly injected T-type channel blockers into the MGv of an anesthetized mouse and measured forward suppression of sound-evoked LFPs in the ACx by delivering pairs of clicks (Fig. 6a). When either mibefradil or NNC 55-0396 was delivered into the MGv, the PPR of LFPs at all measured intervals was significantly lower than that in mice injected with a vehicle control (p < 0.01, n = 6–8 mice per group; Fig. 6b). In contrast, the delivery of either mibefradil or NNC 55-0396 into the ACx did not significantly reduce the PPD of LFPs in vivo (p > 0.05; n = 5–10 mice per group; Fig. 6c,d). These findings indicate that T-type calcium channels in thalamic neurons contribute to mechanisms of forward suppression in the ACx.
Figure 6.

Inhibitors of T-type calcium channels delivered into the MGv but not ACx reduce forward suppression in the ACx. a, c, Illustrations of the experimental setup show the measurement of forward suppression in response to a pair of clicks (20 ms, 70 dB) delivered to the contralateral ear while locally delivering drugs into the MGv (a) or ACx (c). b, d, Average PPR of LFPs measured in the presence of vehicle, mibefradil, or NNC 55-0396 delivered into the MGv (b) or ACx (d). *p < 0.01.
Thalamic Cav3.1 T-type calcium channels underlie forward suppression in the ACx
Cav3.1 T-type calcium channels are predominantly expressed in excitatory thalamic relay neurons (Talley et al., 1999; Anderson et al., 2005). Therefore, these channels may underlie the T-type calcium channel-mediated PPD of TC EPSCs and forward suppression of sound-evoked responses in the ACx. To test this, we used an siRNA approach to specifically knockdown the expression of Cav3.1 channels in the MGv. We injected lentiviruses encoding either siRNA against Cacna1g, which encodes Cav3.1 (Cav3.1 siRNA), or a scrambled control siRNA into the MGv (Fig. 7a). Both siRNAs were coexpressed with GFP. GFP expression in MGv neurons was verified by two-photon imaging in TC slices (Fig. 7a,b). Also, Cav3.1 siRNA reduced the levels of Cav3.1 mRNA in the thalamus. qPCR studies showed that, after injection of Cav3.1 siRNA, Cav3.1 was reduced to 74.4 ± 5.5% (p < 0.05, n = 5 mice) of that in mice injected with control siRNA (n = 5 mice), which was similar to that from mice injected with ACSF (p > 0.05, n = 5 mice). To prove the selectivity of Cav3.1 siRNA in these experiments, we also measured levels of Cav1.2 encoded by Cacna1c. The Cav1.2 level was not reduced in mice injected with either Cav3.1 siRNA (p > 0.05, n = 5 mice) or control siRNA (p > 0.05, n = 5 mice) relative to that in ACSF-injected mice. Cav3.1 siRNA-driven knockdown appeared modest, mainly because RNA levels were measured in the thalamus containing MGv and other nuclei after injecting viruses in the MGv only. Therefore, the RNA changes measured in the thalamus in these experiments are probably an underestimation of RNA knockdown in the MGv.
Figure 7.
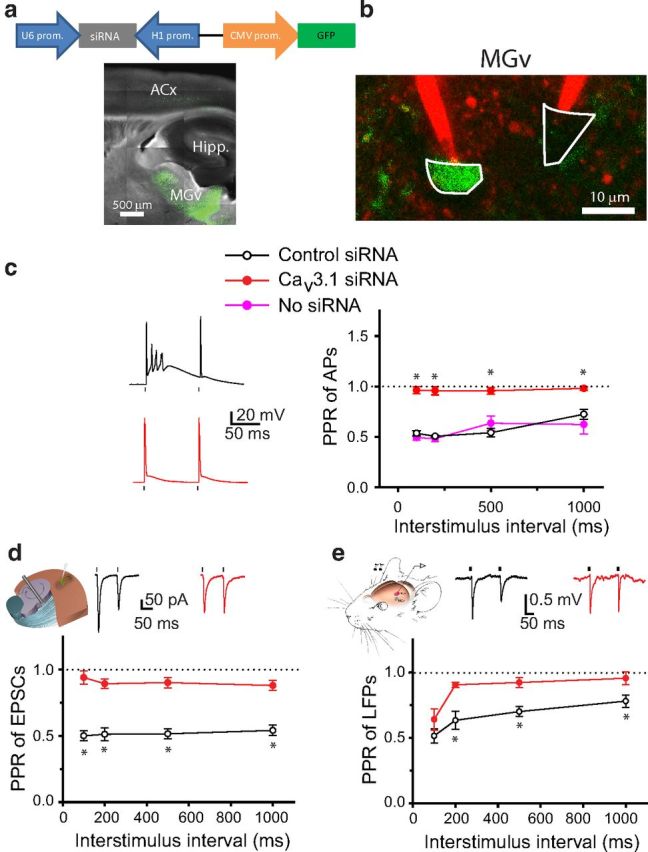
Knockdown of Cav3.1 T-type calcium channels in MGv neurons reduces TC PPD and forward suppression of sound-evoked responses in the ACx. a, Diagram of a lentivirus encoding Cav3.1 siRNA and GFP (top). Representative image of a TC slice from a mouse injected with Cav3.1 siRNA into the MGv (bottom). b, Image of GFP-positive (infected with siRNA lentivirus, green) and GFP-negative (not infected, black) MGv neurons that are targeted with two glass pipettes filled with Alexa Fluor 594 (red). c, Representative voltage traces (left) and average PPR of APs as a function of ISI recorded in MGv neurons either not infected or infected with viruses containing control siRNA or Cav3.1 siRNA (right). d, Mean PPR of EPSCs recorded in slices from mice injected with control siRNA or Cav3.1 siRNA into the MGv. Inset shows representative EPSP traces in response to a pair of stimuli. e, Mean PPR of LFPs recorded in the ACx in vivo in mice injected with control siRNA or Cav3.1 siRNA into the MGv. Inset shows representative LFP traces in response to a pair of clicks. c–e, Dots above or below the traces represent the delivery of depolarizing pulses to cell bodies of MGv neurons (c), stimuli to the TR (d), or clicks to the contralateral ear (e). *p < 0.01.
At the cellular level, the effect of Cav3.1 siRNA-mediated knockdown was more profound. Whole-cell recordings from thalamic neurons revealed that GFP-expressing neurons injected with the control siRNA exhibited the same bursting activity as did the GFP-negative neurons (p > 0.05, n = 5–8 neurons per group; Fig. 7c). In contrast, all but one GFP neuron infected with Cav3.1 siRNA responded with a single AP to the first depolarizing pulse. As a result, the PPR of APs was substantially lower in neurons infected with Cav3.1 siRNA than in neurons infected with control siRNA or in non-infected neurons (p < 0.01, n = 5–8 neurons per group; Fig. 7c).
TC PPD measured in slices and forward suppression of sound-evoked response in the ACx in vivo were also substantially reduced (Fig. 7d,e). Thus, Cav3.1 siRNA injected into the MGv significantly reduced the PPR of TC EPSCs in TC slices (p < 0.01, n = 14–19 neurons per group) and the PPR of LFPs in anesthetized mice (p < 0.05, n = 8–9 mice per group) compared with mice injected with control siRNA. Of note, PPD of TC EPSCs and LFPs was not completely eliminated in the presence of Cav3.1 siRNA, as seen for PPD of APs. These data suggest that either Cav3.1 siRNA-mediated knockdown did not affect all TC projections (only a fraction of thalamic neurons was infected with Cav3.1 siRNA in these experiments) or other synaptic mechanisms (see above) contribute to TC PPD. Nonetheless, these data indicate that there is substantial contribution of Cav3.1 T-type channels in PPD of TC EPSCs and LFPs.
Previous studies on forward suppression used different time intervals between pairs of stimulations, ranging from hundreds of milliseconds (Brosch and Schreiner, 1997) to several seconds (Wehr and Zador, 2005). Our data indicate that repetitive delivery of pairs of sounds with intervals between pairs <1 s can convert the bursting and a single AP (burst–1AP) pattern of firing in thalamic neurons to the 1AP–1AP pattern of firing. However, it was not clear whether this conversion also occurred when the interval between pairs of stimuli is larger than 1 s. We tested this by delivering pairs of brief depolarizing pulses to thalamic neurons at different intervals between pairs. We defined these intervals as the period from the offset of the second pulse in a preceding pair to the onset of the first pulse in a successive pair (Δt; Fig. 8a). We found that the burst–1AP pattern was disrupted at shorter Δt. For instance, at Δt = 500 ms, the burst–1AP pattern of firing either switched to a 1AP–1AP pattern or there was a reduction in the number of APs during bursts or a reduction in frequency of bursts (16 neurons; Fig. 8a). However, the burst–1AP pattern of firing was preserved at longer Δt, for example, Δt = 1030 ms (21 neurons; as in the study by Brosch and Schreiner, 1997), Δt = 3 s (15 neurons; as in the study by Wehr and Zador, 2005), and Δt = 10 s (10 neurons; Fig. 8a,b). Also, the magnitude of PPD of APs was similar at longer Δt (3–10 s) and decreased at shorter Δt (Fig. 8c). The transition point at which the switch from a burst–1AP pattern of firing to a 1AP–1AP pattern of firing occurred was ∼1 s, because it occurred more frequently at Δt = 970 than at Δt = 1030 ms (p < 0.05 at ISI = 200–1000 ms, 12–21 neurons; data not shown). As expected, when Δt was shorter than ISI, the PPR of APs became 1.0 (Fig. 8c). In these instances, a burst–1AP pattern of firing occasionally switched to a burst–1AP pattern of firing (Fig. 8c, inset). Smaller magnitudes of PPD of APs at a short Δt suggest incomplete recovery of Cav3.1 T-type calcium channels from inactivation at frequencies >1 Hz. These data also suggest that Cav3.1 T-type calcium channels have a greater contribution to the forward suppression at lower frequencies.
Figure 8.
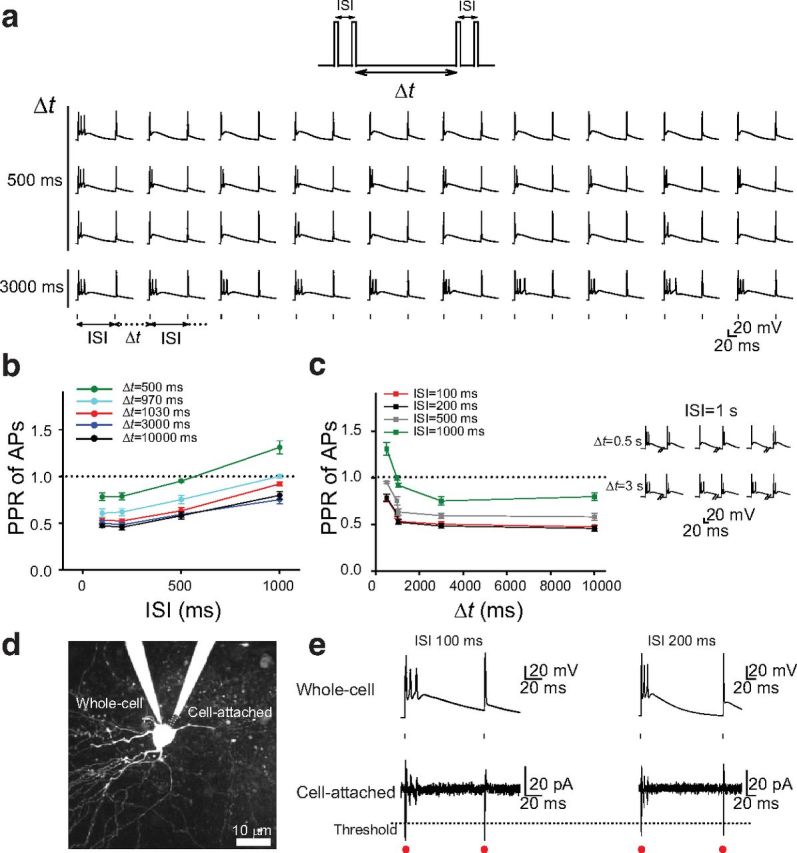
The burst–1AP pattern of firing in thalamic neurons at different time intervals between pairs of stimulation. a, Top, A train of pairs of depolarizing pulses with different ISIs was delivered at different interpair intervals (Δt) to the soma of thalamic neurons. Bottom, Examples of sequences of 10 whole-cell recordings with ISI = 100 ms delivered at Δt = 500 (from three different neurons) and 3000 ms. b, Mean PPR of APs recorded in thalamic neurons at five different Δt. PPR of APs from each cell was calculated as an average of 10 PPRs of APs in each sequence. c, Mean PPR of APs as a function of Δt from the data shown in b. Inset, Examples of sequences consisting of three whole-cell recordings with ISI = 1 s at different Δt. Note the occurrence of bursts during the second pulse at shorter but not longer Δt. d, Image of a thalamic neuron with recording pipettes at whole-cell and cell-attached configurations. Dotted lines outline the tip of a cell-attached pipette. e, Representative whole-cell and cell-attached traces at two different ISIs. Dotted line represents an arbitrary threshold for cell-attached recordings. Red dots show the number of detected APs using this threshold. Small dots show the delivery of depolarizing pulses.
However, this conclusion contradicts previous findings, which show that firing in thalamic neurons recovers more quickly from forward suppression than the firing in ACx neurons does if pairs of sounds are delivered at Δt = 3–4 s (Wehr and Zador, 2005). Because those data were obtained using loose cell-attached recordings that were filtered and thresholded during analysis, we hypothesized that the whole-cell recordings used in our study can reveal more APs during the burst. This hypothesis was based on the observation that, during the burst, the first AP is always larger than successive APs (Figs. 1h, 3a, 7c, 8a). To test this hypothesis, we performed simultaneous whole-cell and cell-attached recordings (seal resistance of ∼1 GΩ) from thalamic neurons (Fig. 8d). Pairs of brief depolarizing pulses consistently evoked the burst–1AP pattern of firing in whole-cell recordings (Fig. 8e). The first AP was substantially larger than successive APs during the burst and comparable with the AP evoked by a second depolarizing pulse. However, in cell-attached recordings, the burst pattern of firing was less obvious. Thus, it is very likely that, by applying a threshold to cell-attached recordings, only a 1AP–1AP pattern instead of the burst–1AP pattern of firing is detected (Fig. 8e). Together, these results indicate that thalamic neurons have the intrinsic ability to fire a burst of APs, which depends on Cav3.1 T-type calcium channels, and this property contributes to the PPD of synaptic transmission at TC synapses and forward suppression of sound-evoked responses in the ACx.
Discussion
Here we report new findings that expand our understanding of decreased responsiveness of the ACx to a series of sounds. First, this long-lasting forward suppression is correlated with synaptic depression at TC projections. Second, TC synaptic depression is caused in part by a switch in the firing mode of thalamic neurons. During the first stimulus, thalamic neurons respond with a burst of APs, and during the second, they respond with only a single AP. Thus, thalamic neurons respond to a pair of brief identical stimuli with the burst–1AP pattern of firing. Third, this switch in firing depends on Cav3.1 T-type calcium channels that are predominantly expressed in thalamic relay neurons. Inhibition or knockdown of Cav3.1 T-type channels specifically in the thalamus substantially diminishes TC synaptic depression and forward suppression of sound-evoked responses in the ACx.
On the basis of our data and results from previous studies, we conclude that forward suppression in the ACx involves multiple mechanisms. It was initially thought that GABAA feedforward cortical inhibition was responsible for forward suppression (Calford and Semple, 1995; Brosch and Schreiner, 1997; Tan et al., 2004). However, recordings in the rat ACx revealed that sound-evoked IPSCs can last no longer than 100 ms (Wehr and Zador, 2005). Thus, GABAergic mechanisms can explain forward suppression in the ACx but only at short ISIs. Here we show that thalamic neurons can switch their firing patterns between the bursting mode and a single AP during two stimuli, and this switch can be observed at ISIs as long as 1 s. This burst–1AP pattern of firing of thalamic neurons depends on thalamus-enriched Cav3.1 T-type calcium channels. Although our data show convincingly that these channels contribute substantially to forward suppression, pharmacologic inhibition or knockdown of these channels does not completely eliminate the forward suppression. These results suggest that other mechanisms are also involved in forward suppression.
Several reports suggest that short-term depression at TC synapses underlies forward suppression in sensory cortices (Castro-Alamancos, 1997; Carandini et al., 2002; Chung et al., 2002; Freeman et al., 2002). Indeed, a pair of identical stimulations causes TC synapses to undergo robust and reliable short-term depression. In contrast, most excitatory synapses, including CC synapses, respond to the same experimental paradigm with short-term facilitation (Llinás and Jahnsen, 1982; Coulter et al., 1989; Stratford et al., 1996; Huang and Kandel, 1998; Zakharenko et al., 2001; Beierlein and Connors, 2002; Beierlein et al., 2002). In this study, we confirmed in the ACx that TC (but not CC) synapses respond to pairs of identical stimulations with PPD. From only this phenomenological point of view, we argue that short-term plasticity at TC synapses is better suited than CC mechanisms to explain forward suppression in the ACx. This argument is strengthened by the fact that facilitation at CC synapses can last as long as 300–400 ms, whereas depression at TC synapses lasts as long as 1 s, similar to what is observed for forward suppression in the ACx in vivo.
By using calcium imaging at presynaptic terminals, we observed PPD of calcium transients in the TC presynapse. Because calcium concentration is tightly linked to neurotransmitter release, this association may explain PPD of synaptic transmission at TC synapses. In contrast, the presynaptic terminal of cortical neurons responded with facilitation of calcium transients to a pair of APs, which is consistent with PPF of synaptic transmission at these synapses.
The TC PPD of synaptic transmission most likely originates from presynaptic thalamic relay neurons. In our study, this notion was supported by TGU experiments at dendritic spines of thalamorecipient neurons in the ACx, which are the sites for TC inputs. Pairs of identical pulses of exogenous glutamate delivered to these dendritic spines evoked postsynaptic responses of similar amplitudes, indicating that postsynaptic mechanisms are not involved in the mechanisms of TC PPD and suggesting a role for presynaptic mechanisms.
Short-term plasticities, including PPF and PPD, are predominantly explained by presynaptic mechanisms. The most well accepted explanation of PPF of synaptic transmission at excitatory synapses is that residual calcium remains in the presynaptic terminal for some time after the first stimulus (Katz and Miledi, 1968). The calcium transient that is evoked by the second stimulus adds to this residual calcium, thereby producing a larger net calcium level and causing more neurotransmitter release. Although this theory explains PPF, it does not explain PPD. Moreover, it cannot explain why PPD lasts for hundreds of milliseconds, whereas residual calcium stays in the presynaptic terminal for substantially shorter periods.
We showed that TC PPD occurs because of a switch in the firing properties of thalamic relay neurons from a burst of several APs to a single AP. This switch depends on Cav3.1 T-type calcium channels and can last for hundreds of milliseconds. We showed that pharmacologic or genetic inhibition of these channels in thalamic neurons is sufficient to eliminate TC PPD and, most importantly, forward suppression in the ACx. Thus, we argue that Cav3.1 T-type calcium channels in thalamic neurons are responsible for depression of TC synaptic transmission and that this mechanism contributes to the long-lasting reduced responsiveness of the ACx induced by a brief sound.
Why thalamic neurons switch their firing patterns during two stimuli and why this switch lasts so long can be explained by the properties of T-type calcium channels. Several groups have shown that thalamic relay neurons in vivo can fire APs in the burst mode (high-frequency burst of APs) and the tonic mode (individual APs; Llinás and Jahnsen, 1982; Jahnsen and Llinás, 1984; McCormick and Feeser, 1990; Sherman, 2001; Swadlow and Gusev, 2001; Llinás and Steriade, 2006). The burst-firing mode is attributed to a low-threshold calcium conductance that generates AP bursts in thalamic neurons, which is inactivated during membrane depolarization and de-inactivated at membrane potentials hyperpolarized more than −60 mV (Chemin et al., 2002). This conductance, later identified as T-type calcium channels (Coulter et al., 1989; Crunelli et al., 1989; Huguenard and Prince, 1992), produces depolarizing afterpotentials large enough to generate low-threshold spikes (Jahnsen and Llinás, 1984). Thalamic bursts must be preceded by a hyperpolarized membrane potential (more negative than −60 mV) sustained long enough to inactivate T-type calcium channels (Llinás and Steriade, 2006). This is consistent with the observation of AP bursts during the first response in the paired-pulse paradigm, when the first stimulus arrives after sustained silence of thalamic neurons. The second stimulus that follows shortly thereafter arrives when T-type calcium channels are inactivated and cannot support bursting activity. T-type calcium channels slowly recover from inactivation, and reactivation may take several hundreds of milliseconds, depending on the membrane potential (Satin and Cribbs, 2000; Kuo and Yang, 2001). The physiologic implication of this process is that the slow kinetics of T-type calcium channel reactivation may put important constraints on the switch between the burst- and tonic-firing modes of thalamic neurons. This notion is consistent with our data showing a long-lasting switch between firing modes during the paired-pulse paradigm, PPD at TC synapses, and forward suppression in the ACx.
Given the timescales of Cav3.1 T-type calcium channel-dependent forward suppression in TC synapses, this mechanism may be involved in mechanisms of temporal separation of sounds (Bregman, 1990; Moore, 1995) and phenomena such as mismatch negativity and stimulus-specific adaptation (SSA), which are thought to underlie mechanisms of novelty detection, recognition of acoustic objects, scene analysis, and auditory memory (Ulanovsky et al., 2003, 2004; Nelken, 2004). In SSA, there is a specific decrease in the response to a frequent (standard) stimulus, which does not generalize to another rare stimulus (deviant). In the context of SSA, the burst–1AP switch may be involved in mechanisms of adaptation of cortical responses to standard tones as well as in the presentation of deviants alone (Lazar and Metherate, 2003; Taaseh et al., 2011). Given the properties of PPD of APs in thalamic neurons, we expect that tones repeatedly delivered at ISIs shorter than 1 s will produce Cav3.1 channel inactivation and reduction of bursting in thalamic neurons. SSA measured in the ACx can diminish at ISIs longer than 1 s (Ulanovsky et al., 2003), indicating that PPD of APs in thalamic neurons and SSA in the ACx operate at the same timescale. However, given that SSA is detected in different parts of the auditory system, including the inferior colliculus (Pérez-González et al., 2005; Malmierca et al., 2009), the auditory thalamus (Anderson et al., 2009; Antunes et al., 2010; Antunes and Malmierca, 2011), the ACx (Ulanovsky et al., 2003), and auditory localization pathways (Netser et al., 2011), it is most likely that the burst–1AP switch observed here in thalamic neurons is only a part of the complex SSA machinery.
In summary, we show that thalamic relay neurons in the auditory thalamus switch their firing modes between bursting and tonic. This switch depends on Cav3.1 T-type calcium channels and contributes to PPD at TC projections and the long-lasting period of decreased responsiveness of the ACx.
Footnotes
This work was supported in part by the National Institute of Mental Health Grants R01 DC012833 and R01 MH097742 and by the American Lebanese Syrian Associated Charities. We thank Melissa Johnson, Dr. Christopher Calabrese, and the St. Jude Small Animal Imaging Center for assistance with animal surgeries; Drs. Sungkun Chun and Victoria Frohlich and the St. Jude Cellular Imaging Shared Resource for help with imaging experiments; Drs. John Gray and Junming Yue and St. Jude and University of Tennessee Vector Development and Production Resources for production of the viruses; and Drs. Angela McArthur and Vani Shanker for editing this manuscript. We also thank Dr. Jay Blundon and other members of the Zakharenko laboratory for their constructive comments on early versions of this manuscript.
The authors declare no competing financial interests.
References
- Anderson LA, Christianson GB, Linden JF. Stimulus-specific adaptation occurs in the auditory thalamus. J Neurosci. 2009;29:7359–7363. doi: 10.1523/JNEUROSCI.0793-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MP, Mochizuki T, Xie J, Fischler W, Manger JP, Talley EM, Scammell TE, Tonegawa S. Thalamic Cav3.1 T-type Ca2+ channel plays a crucial role in stabilizing sleep. Proc Natl Acad Sci U S A. 2005;102:1743–1748. doi: 10.1073/pnas.0409644102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes FM, Malmierca MS. Effect of auditory cortex deactivation on stimulus-specific adaptation in the medial geniculate body. J Neurosci. 2011;31:17306–17316. doi: 10.1523/JNEUROSCI.1915-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes FM, Nelken I, Covey E, Malmierca MS. Stimulus-specific adaptation in the auditory thalamus of the anesthetized rat. PLoS One. 2010;5:e14071. doi: 10.1371/journal.pone.0014071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett EL, Smith PH. Effects of paired-pulse and repetitive stimulation on neurons in the rat medial geniculate body. Neuroscience. 2002;113:957–974. doi: 10.1016/S0306-4522(02)00240-3. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Connors BW. Short-term dynamics of thalamocortical and intracortical synapses onto layer 6 neurons in neocortex. J Neurophysiol. 2002;88:1924–1932. doi: 10.1152/jn.2002.88.4.1924. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Fall CP, Rinzel J, Yuste R. Thalamocortical bursts trigger recurrent activity in neocortical networks: layer 4 as a frequency-dependent gate. J Neurosci. 2002;22:9885–9894. doi: 10.1523/JNEUROSCI.22-22-09885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundon JA, Zakharenko SS. Presynaptic gating of postsynaptic synaptic plasticity: a plasticity filter in the adult auditory cortex. Neuroscientist. 2013;19:465–478. doi: 10.1177/1073858413482983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundon JA, Bayazitov IT, Zakharenko SS. Presynaptic gating of postsynaptically expressed plasticity at mature thalamocortical synapses. J Neurosci. 2011;31:16012–16025. doi: 10.1523/JNEUROSCI.3281-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Calcium influx and transmitter release in a fast CNS synapse. Nature. 1996;383:431–434. doi: 10.1038/383431a0. [DOI] [PubMed] [Google Scholar]
- Bregman AS. Auditory scene analysis: the perceptual organization of sound. Cambridge, MA: Massachusetts Institute of Technology; 1990. [Google Scholar]
- Brosch M, Schreiner CE. Time course of forward masking tuning curves in cat primary auditory cortex. J Neurophysiol. 1997;77:923–943. doi: 10.1152/jn.1997.77.2.923. [DOI] [PubMed] [Google Scholar]
- Calford MB, Semple MN. Monaural inhibition in cat auditory cortex. J Neurophysiol. 1995;73:1876–1891. doi: 10.1152/jn.1995.73.5.1876. [DOI] [PubMed] [Google Scholar]
- Carandini M, Heeger DJ, Senn W. A synaptic explanation of suppression in visual cortex. J Neurosci. 2002;22:10053–10065. doi: 10.1523/JNEUROSCI.22-22-10053.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA. Short-term plasticity in thalamocortical pathways: cellular mechanisms and functional roles. Rev Neurosci. 1997;8:95–116. doi: 10.1515/revneuro.1997.8.2.95. [DOI] [PubMed] [Google Scholar]
- Chemin J, Monteil A, Perez-Reyes E, Bourinet E, Nargeot J, Lory P. Specific contribution of human T-type calcium channel isotypes (alpha(1G), alpha(1H) and alpha(1I)) to neuronal excitability. J Physiol. 2002;540:3–14. doi: 10.1113/jphysiol.2001.013269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S, Bayazitov IT, Blundon JA, Zakharenko SS. Thalamocortical long-term potentiation becomes gated after the early critical period in the auditory cortex. J Neurosci. 2013;33:7345–7357. doi: 10.1523/JNEUROSCI.4500-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S, Li X, Nelson SB. Short-term depression at thalamocortical synapses contributes to rapid adaptation of cortical sensory responses in vivo. Neuron. 2002;34:437–446. doi: 10.1016/S0896-6273(02)00659-1. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Huguenard JR, Prince DA. Calcium currents in rat thalamocortical relay neurones: kinetic properties of the transient, low-threshold current. J Physiol. 1989;414:587–604. doi: 10.1113/jphysiol.1989.sp017705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creutzfeldt O, Hellweg FC, Schreiner C. Thalamocortical transformation of responses to complex auditory stimuli. Exp Brain Res. 1980;39:87–104. doi: 10.1007/BF00237072. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Rose HJ, Metherate R. Auditory thalamocortical synaptic transmission in vitro. J Neurophysiol. 2002;87:361–384. doi: 10.1152/jn.00549.2001. [DOI] [PubMed] [Google Scholar]
- Crunelli V, Lightowler S, Pollard CE. A T-type Ca2+ current underlies low-threshold Ca2+ potentials in cells of the cat and rat lateral geniculate nucleus. J Physiol. 1989;413:543–561. doi: 10.1113/jphysiol.1989.sp017668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman ML, Peters A. The forms of non-pyramidal neurons in the visual cortex of the rat. J Comp Neurol. 1978;179:761–793. doi: 10.1002/cne.901790406. [DOI] [PubMed] [Google Scholar]
- Flagmeyer I, Haas HL, Stevens DR. Adenosine A1 receptor-mediated depression of corticostriatal and thalamostriatal glutamatergic synaptic potentials in vitro. Brain Res. 1997;778:178–185. doi: 10.1016/S0006-8993(97)01060-3. [DOI] [PubMed] [Google Scholar]
- Freeman TC, Durand S, Kiper DC, Carandini M. Suppression without inhibition in visual cortex. Neuron. 2002;35:759–771. doi: 10.1016/S0896-6273(02)00819-X. [DOI] [PubMed] [Google Scholar]
- Gil Z, Connors BW, Amitai Y. Efficacy of thalamocortical and intracortical synaptic connections: quanta, innervation, and reliability. Neuron. 1999;23:385–397. doi: 10.1016/S0896-6273(00)80788-6. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron. 1998;21:169–178. doi: 10.1016/S0896-6273(00)80524-3. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA. A novel T-type current underlies prolonged Ca2+-dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. J Neurosci. 1992;12:3804–3817. doi: 10.1523/JNEUROSCI.12-10-03804.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnsen H, Llinás R. Electrophysiological properties of guinea-pig thalamic neurones: an in vitro study. J Physiol. 1984;349:205–226. doi: 10.1113/jphysiol.1984.sp015153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol. 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CC, Yang S. Recovery from inactivation of t-type Ca2+ channels in rat thalamic neurons. J Neurosci. 2001;21:1884–1892. doi: 10.1523/JNEUROSCI.21-06-01884.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar R, Metherate R. Spectral interactions, but no mismatch negativity, in auditory cortex of anesthetized rat. Hear Res. 2003;181:51–56. doi: 10.1016/S0378-5955(03)00166-7. [DOI] [PubMed] [Google Scholar]
- Llinás R, Jahnsen H. Electrophysiology of mammalian thalamic neurones in vitro. Nature. 1982;297:406–408. doi: 10.1038/297406a0. [DOI] [PubMed] [Google Scholar]
- Llinás RR, Steriade M. Bursting of thalamic neurons and states of vigilance. J Neurophysiol. 2006;95:3297–3308. doi: 10.1152/jn.00166.2006. [DOI] [PubMed] [Google Scholar]
- Lund JS. Organization of neurons in the visual cortex, area 17, of the monkey (Macaca mulatta) J Comp Neurol. 1973;147:455–496. doi: 10.1002/cne.901470404. [DOI] [PubMed] [Google Scholar]
- Malmierca MS, Cristaudo S, Pérez-González D, Covey E. Stimulus-specific adaptation in the inferior colliculus of the anesthetized rat. J Neurosci. 2009;29:5483–5493. doi: 10.1523/JNEUROSCI.4153-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Feeser HR. Functional implications of burst firing and single spike activity in lateral geniculate relay neurons. Neuroscience. 1990;39:103–113. doi: 10.1016/0306-4522(90)90225-S. [DOI] [PubMed] [Google Scholar]
- Miller LM, Escabí MA, Read HL, Schreiner CE. Spectrotemporal receptive fields in the lemniscal auditory thalamus and cortex. J Neurophysiol. 2002;87:516–527. doi: 10.1152/jn.00395.2001. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Moore BCJ. Hearing. San Diego: Academic; 1995. [Google Scholar]
- Nelken I. Processing of complex stimuli and natural scenes in the auditory cortex. Curr Opin Neurobiol. 2004;14:474–480. doi: 10.1016/j.conb.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Netser S, Zahar Y, Gutfreund Y. Stimulus-specific adaptation: can it be a neural correlate of behavioral habituation? J Neurosci. 2011;31:17811–17820. doi: 10.1523/JNEUROSCI.4790-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-González D, Malmierca MS, Covey E. Novelty detector neurons in the mammalian auditory midbrain. Eur J Neurosci. 2005;22:2879–2885. doi: 10.1111/j.1460-9568.2005.04472.x. [DOI] [PubMed] [Google Scholar]
- Perkins KL. Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J Neurosci Methods. 2006;154:1–18. doi: 10.1016/j.jneumeth.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RJ, Blundon JA, Bayazitov IT, Zakharenko SS. Connectivity patterns revealed by mapping of active inputs on dendrites of thalamorecipient neurons in the auditory cortex. J Neurosci. 2009;29:6406–6417. doi: 10.1523/JNEUROSCI.0258-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose HJ, Metherate R. Auditory thalamocortical transmission is reliable and temporally precise. J Neurophysiol. 2005;94:2019–2030. doi: 10.1152/jn.00860.2004. [DOI] [PubMed] [Google Scholar]
- Satin J, Cribbs LL. Identification of a T-type Ca(2+) channel isoform in murine atrial myocytes (AT-1 cells) Circ Res. 2000;86:636–642. doi: 10.1161/01.RES.86.6.636. [DOI] [PubMed] [Google Scholar]
- Sherman SM. Tonic and burst firing: dual modes of thalamocortical relay. Trends Neurosci. 2001;24:122–126. doi: 10.1016/S0166-2236(00)01714-8. [DOI] [PubMed] [Google Scholar]
- Smith PH, Populin LC. Fundamental differences between the thalamocortical recipient layers of the cat auditory and visual cortices. J Comp Neurol. 2001;436:508–519. doi: 10.1002/cne.1084. [DOI] [PubMed] [Google Scholar]
- Stratford KJ, Tarczy-Hornoch K, Martin KA, Bannister NJ, Jack JJ. Excitatory synaptic inputs to spiny stellate cells in cat visual cortex. Nature. 1996;382:258–261. doi: 10.1038/382258a0. [DOI] [PubMed] [Google Scholar]
- Swadlow HA, Gusev AG. The impact of “bursting” thalamic impulses at a neocortical synapse. Nat Neurosci. 2001;4:402–408. doi: 10.1038/86054. [DOI] [PubMed] [Google Scholar]
- Taaseh N, Yaron A, Nelken I. Stimulus-specific adaptation and deviance detection in the rat auditory cortex. PLoS One. 2011;6:e23369. doi: 10.1371/journal.pone.0023369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19:1895–1911. doi: 10.1523/JNEUROSCI.19-06-01895.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AY, Zhang LI, Merzenich MM, Schreiner CE. Tone-evoked excitatory and inhibitory synaptic conductances of primary auditory cortex neurons. J Neurophysiol. 2004;92:630–643. doi: 10.1152/jn.01020.2003. [DOI] [PubMed] [Google Scholar]
- Trussell LO, Zhang S, Raman IM. Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron. 1993;10:1185–1196. doi: 10.1016/0896-6273(93)90066-Z. [DOI] [PubMed] [Google Scholar]
- Ulanovsky N, Las L, Nelken I. Processing of low-probability sounds by cortical neurons. Nat Neurosci. 2003;6:391–398. doi: 10.1038/nn1032. [DOI] [PubMed] [Google Scholar]
- Ulanovsky N, Las L, Farkas D, Nelken I. Multiple time scales of adaptation in auditory cortex neurons. J Neurosci. 2004;24:10440–10453. doi: 10.1523/JNEUROSCI.1905-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viaene AN, Petrof I, Sherman SM. Synaptic properties of thalamic input to the subgranular layers of primary somatosensory and auditory cortices in the mouse. J Neurosci. 2011;31:12738–12747. doi: 10.1523/JNEUROSCI.1565-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr M, Zador AM. Synaptic mechanisms of forward suppression in rat auditory cortex. Neuron. 2005;47:437–445. doi: 10.1016/j.neuron.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Weston MC, Nehring RB, Wojcik SM, Rosenmund C. Interplay between VGLUT isoforms and endophilin A1 regulates neurotransmitter release and short-term plasticity. Neuron. 2011;69:1147–1159. doi: 10.1016/j.neuron.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Nimchinsky EA, Scheuss V, Pologruto TA, Oertner TG, Sabatini BL, Svoboda K. Imaging calcium concentration dynamics in small neuronal compartments. Sci STKE. 2004;2004:l5. doi: 10.1126/stke.2192004pl5. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA. Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat Neurosci. 2001;4:711–717. doi: 10.1038/89498. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, Morozov A. Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA3-CA1 synapses. Neuron. 2003;39:975–990. doi: 10.1016/S0896-6273(03)00543-9. [DOI] [PubMed] [Google Scholar]