

Abstract
Pulmonary alveolar proteinosis represents a rare syndrome characterized by the accumulation of proteinaceous phospholipid-laden material in the alveoli. This leads to impaired gas exchange and arterial hypoxemia of varying degrees. The diagnosis is confirmed by lung biopsy. Sequential whole-lung lavage (WLL) first described in 1963 is the standard of care. We report a case of a male diagnosed of having pulmonary alveolar proteinosis (PAP) on transbroncial lung biopsy (TBLB). He was treated with sequential WLL (Left followed by right, Left being more involved on chest X-ray) followed by recombinant GM-CSF, with good result.
Keywords: Granulocyte macrophage colony stimulating factor, pulmonary alveolar proteinosis, Trans Bronchial Lung Biopsy, whole-lung lavage
Introduction
Pulmonary alveolar proteinosis (PAP) is characterized by accumulation of surfactant proteins in the alveoli due to defective surfactant clearance by alveolar macrophages. The clinical course of the disease is variable, ranging from respiratory failure to spontaneous resolution. A crazy-paving pattern is suggestive of, but not specific, to PAP. Lung biopsy (by open-lung surgery, video-assisted thoracoscopic surgery or TBLB) is the gold standard for the diagnosis of PAP.[1]
Case Report
A 54-year-old male, non-smoker, non-alcoholic, known diabetic and hypertensive, presented with fever, dry cough, and progressive breathlessness for one month. Initial evaluation in a different hospital confirmed hypoxia and PFT (pulmonary function tests) was suggestive of a restrictive pattern. A CT chest demonstrated characteristic crazy-paving pattern. [Figure 1a]. Sputum for PAS was negative. He was labeled as having interstitial lung disease and was discharged on oxygen and steroids.
Figure 1a.
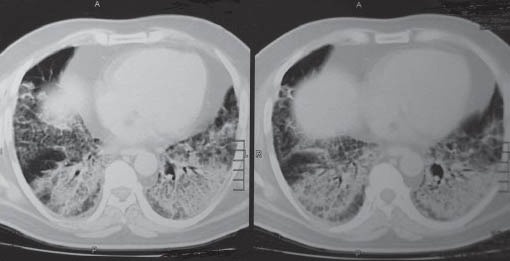
CT chest showing and #x0022; Crazy Pavement and #x0022; appearance
His symptoms worsened gradually and he presented to our Institution on high flow oxygen and was found to be PaO2-42 on FIO2 of 1. There was no history suggestive of a connective tissue disorder. On examination, he had bilateral basal crepitations on the chest. Patient was admitted in ICU, blood gases showed type I respiratory failure with a paO2 of 53 mm of Hg on FIO2 of 0.6. Rest of the routine investigations were normal. The chest X-ray [Figure 1b] showed bilateral reticular shadows affecting the lower zones (left > right). Bronchoscopy with lavage was done; the patient did not consent to lung biopsy. BAL microbiology was negative. After counseling, a repeat bronchoscopy with TBLB was done, which showed pulmonary alveolar proteinosis on histopathology (PAS stain was strongly positive) [Figure 2]. As the patient was deteriorating, it was decided to treat him with whole lung lavage (WLL).
Figure 1b.
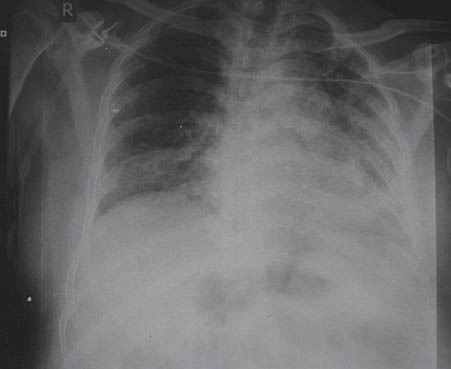
Chest X-ray at presentation
Figure 2.
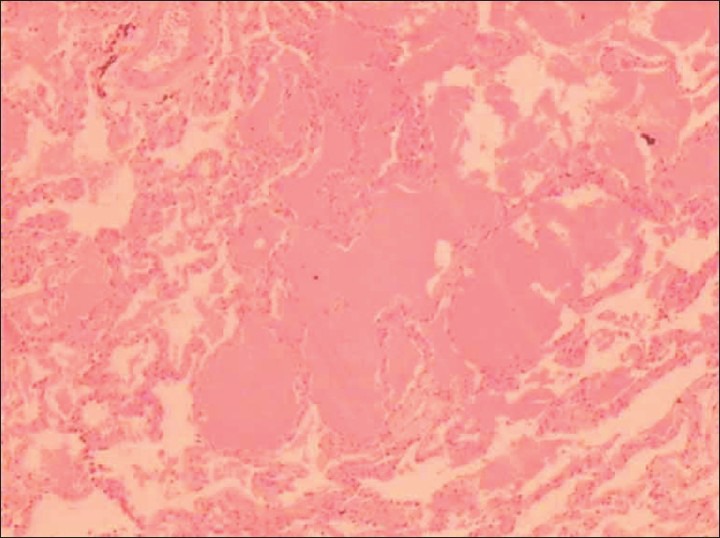
PAS stain positivity on histopathology
Patient was intubated with a double-lumen endotracheal tube for WLL. The left lung was lavaged first with 15.5 L of warmed (at 37°C) normal saline using aliquots of 500 mL. The procedure was terminated once the effluent had cleared significantly. The effluent fluid was collected in sequentially numbered glass bottles [Figure 3]. It had characteristic opaque, milky appearance with dense sediments on standing, which were strongly positive for PAS stain.
Figure 3.

Characteristic milky effluent with sediments
Patient's oxygen requirement decreased to 2-3 liters/min post-extubation. ABG showed pH-7.44, PaO2-62 on FIO2 of 0.4, and PaCO2- 44. CXR showed marked clearing of left side [Figure 4a]. Subsequently, the procedure was repeated on the right lung with 20 liters saline. Patient had marked improvement symptomatically and objectively. His PaO2 on room air was 68 mm of Hg. Repeat HRCT showed marked clearance. Blood samples were sent to University of Cleveland (USA) for anti-GM-CSF antibody titers, which were reported to be highly positive, so recombinant GM-CSF was started. The patient was discharged on N-acytyl cystine nebulization and rGM-CSF therapy for 3 months. His chest X-ray [Figure 4b] after 8 months was almost normal. His oxygen saturation on room air was found to be 92%, and he was carrying out his routine activities including work without getting breathless.
Figure 4a.
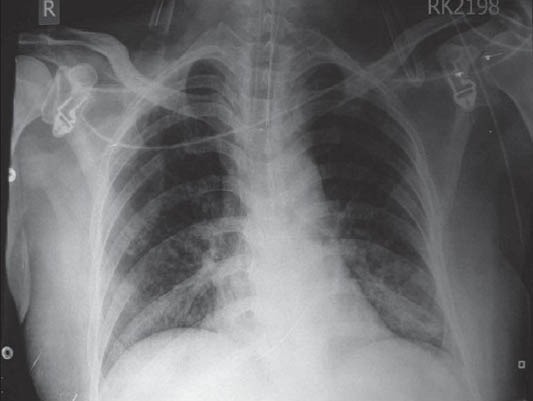
Chest X-ray after left WLL
Figure 4b.
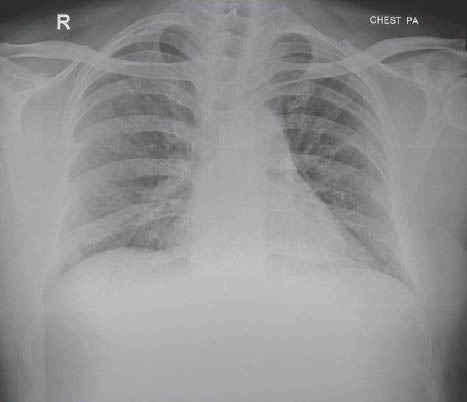
Follow up chest X-ray
Discussion
Pulmonary alveolar proteinosis (PAP) is a rare disorder, in which lipoproteinaceous material accumulates within alveoli.[1] PAP occurs in three clinically distinct forms: Genetic, auto-immune (also called idiopathic or primary), and secondary.[2] The genetic form comprises a heterogeneous group of disorders caused by mutations in the genes encoding surfactant protein B or C or the receptor for GM-CSF. Primary PAP is associated with presence of anti- GM-CSF antibodies preventing uptake of surfactant proteins by alveolar macrophages resulting in their accumulation.
Secondary pulmonary alveolar proteinosis develops in association with conditions involving functional impairment or reduced numbers of alveolar macrophages. Such conditions include some hematologic cancers, inhalation of inorganic dust or toxic fumes and certain infections.
Idiopathic PAP has been a fascinating disorder since its initial description in 1958. Incidence reported in the literature has been estimated to be 0.37 per 100,000 persons.[3]
Primary PAP is seen in more than 90% of cases. The median age at diagnosis is 39 years.[4] Patients usually present with progressive dyspnea and cough, mostly dry in nature.[1] Less common symptoms include fever, chest pain, or hemoptysis, more so in the presence of secondary infections.
The signs on physical examination can be unremarkable, but there are inspiratory crackles, cyanosis, and digital clubbing in a small percentage of patients. In uncomplicated cases, the chest radiograph usually reveals bilateral ill-defined nodular or confluent pattern, suggestive of pulmonary edema but without other radiographic signs of left-sided heart failure.[1] High-resolution CT shows patchy, ground-glass opacification with superimposed interlobular septal and intralobular thickening, a pattern commonly referred to as “crazy paving.”[5] Routine investigations are usually normal.
PFT can be normal but typically shows a restrictive ventilatory defect with a disproportionate and severe reduction of the carbon monoxide diffusing capacity.[4] The impairment of gas exchange is secondary to filling of the alveoli with phospholipoproteinaceous material leading to ventilation and perfusion mismatch.
Clinical and radiographic findings often suggest the diagnosis of PAP in suspected cases,[1] while findings on examination of a BAL specimen can establish the diagnosis.[6] The lavage fluid in patients with this disorder has an opaque, milky appearance. It contains large and foamy alveolar macrophages and increased numbers of lymphocytes.
There are also large, acellular, eosinophilic bodies in a diffuse background of granular material that stains with PAS as well as elevated levels of surfactant proteins.[6]
On light-microscopy, the architecture of the lung parenchyma is found to be preserved unless there is infection. Immunohistochemical staining reveals abundant accumulation of surfactant protein. Idiopathic PAP occurs due to disturbed surfactant homeostasis.[7]
Current therapy for all types of PAP remains WLL, although for the congenital form of the disorder, successful lung transplantation has been reported.[8] Therapy for secondary pulmonary alveolar proteinosis generally involves WLL and the treatment of the underlying condition. Primary PAP has been treated successfully since the early 1960s by WLL, and this procedure remains the standard of care today.[9] Such therapy also improves survival. The median duration of clinical benefit from lavage has been reported to be 15 months. Patients usually respond to one procedure of WLL (both lungs), but a few may require repeated WLL.[4]
Several prospective phase 2 trials of GM-CSF therapy for idiopathic pulmonary alveolar proteinosis have been undertaken. The study conducted by Seymour et al,[10] evaluated the effectiveness of GM-CSF in PAP, the results have been encouraging, but the mechanism of the effect of GM-CSF treatment is not entirely clear.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med. 1958;258:1123–42. doi: 10.1056/NEJM195806052582301. [DOI] [PubMed] [Google Scholar]
- 2.Borie R, Danel C, Debray MP, Taille C, Dombert MC, Aubier M, et al. Pulmonary alveolar proteinosis. Eur Respir Rev. 2011;20:98–107. doi: 10.1183/09059180.00001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ben-Dov I, Kishinevski Y, Roznman J, Soliman A, Bishara H, Zelligson, et al. Pulmonary alveolar proteinosis in Israel: Ethnic clustering. Isr Med Assoc J. 1999;1:75–8. [PubMed] [Google Scholar]
- 4.Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: Progress in the first 44 years. Am J Respir Crit Care Med. 2002;166:215–35. doi: 10.1164/rccm.2109105. [DOI] [PubMed] [Google Scholar]
- 5.Johkoh T, Itoh H, Muller NL, Ichikado K, Nakamura H, Ikezoe J, et al. Crazy-paving appearance at thin-section CT: Spectrum of disease and pathologic findings. Radiology. 1999;211:155–60. doi: 10.1148/radiology.211.1.r99ap10155. [DOI] [PubMed] [Google Scholar]
- 6.Wang BM, Stern EJ, Schmidt RA, Pierson DJ. Diagnosing pulmonary alveolar proteinosis: A review and an update. Chest. 1997;111:460–6. doi: 10.1378/chest.111.2.460. [DOI] [PubMed] [Google Scholar]
- 7.Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med. 2003;349:2527–39. doi: 10.1056/NEJMra023226. [DOI] [PubMed] [Google Scholar]
- 8.Hamvas A, Nogee LM, Mallory GB, Jr, Spray TL, Huddleston CB, August A, et al. Lung transplantation for treatment of infants with surfactant protein B deficiency. J Pediatr. 1997;130:231–9. doi: 10.1016/s0022-3476(97)70348-2. [DOI] [PubMed] [Google Scholar]
- 9.Du Bois RM, McAllister WA, Branthwaite MA. Alveolar proteinosis: Diagnosis and treatment over a 10-year period. Thorax. 1983;38:360–3. doi: 10.1136/thx.38.5.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seymour JF, Presneill JJ, Schoch OD, Downie GH, Moore PE, Doyle IR, et al. Therapeutic efficacy of granulocyte-macrophage colony-stimulating factor in patients with idiopathic acquired alveolar proteinosis. Am J Respir Crit Care Med. 2001;163:524–31. doi: 10.1164/ajrccm.163.2.2003146. [DOI] [PubMed] [Google Scholar]