

Abstract
Objectives:
To review clinical characteristics and response to immunomodulation therapy in autoimmune encephalitis presenting with status epilepticus (SE), epilepsy, and cognitive decline.
Design:
Observational, prospective case series.
Setting:
All India Institute of Medical Sciences, New Delhi, India.
Materials and Methods:
Prospective analysis of 15 patients, who presented with SE, epilepsy, cognitive decline, and other neurological symptoms with positive autoantibodies. Demographic and clinical characteristics were recorded. Brain magnetic resonance imaging (MRI), cerebrospinal-fluid analysis (CSF), and tumor screening were done periodically. Treatment received and responses (categorized as per patients and treating doctor's information) were noted.
Results:
There were 15 (males = 10) patients of autoimmune encephalitis. The mean age of presentation was 24 years (range: 2-64 years). The most common onset was subacute (64%) and four (29%) patients presented as SE. Predominant clinical presentations were seizures (100%) almost of every semiology. CSF was done in 10 patients; it was normal in 60%. Brain MRI was done in all patients, in six (40%) it was normal, six (40%) showed T2W and FLAIR hyperintensities in bilateral limbic areas. Antibodies found were the N-methyl-D-aspartate receptor antibody in seven (50%), voltage-gated potassium channel antibody in five (36%), two of antiglutamic acid decarboxylase, and one patient with double stranded DNA (dsDNA) antibodies. None showed evidence of malignancy. Patients received immunotherapy, either steroids, intravenous immunoglobulin, or both. Follow-up showed significant improvement in majority of cases, neither further seizures nor relapse in nine (67%) cases. One death occurred, due to delayed presentation.
Conclusions:
Uncommon but potentially reversible causes of SE, epilepsy, and cognitive decline may be immune-related and high index of suspicion will prevent missing the diagnosis.
Key Words: Autoimmune encephalitis, cognitive decline, drug refractory epilepsy, seizures, status epilepticus
Introduction
Seizures clusters and status epilepticus (SE) are common medical emergencies. They have to be identified early and treated at the earliest. Common causes of SE are acute events (brain injury, stroke, infection, tumor, or childhood febrile SE) or occur in patients with a prior history of epilepsy. A smaller proportion of patients with or without seizures due to epilepsy and SE are likely to be due to intercurrent illness such as systemic infections or metabolic disturbances or changes in antiepileptic medication or some remote symptomatic etiology. There is an emergence in the recognition of immune-mediated epilepsy of paraneoplastic or nonparaneoplastic etiology with or without encephalitis.[1,2] Some of these immune-mediated encephalitis have identified antibodies and others are associated with syndromes having yet unidentified/unknown antibodies. Autoantibodies of paraneoplastic limbic encephalitis include antineuronal nuclear antibody type 1, collapsin response-mediator protein 5 (CRMP-5), and Ma2. Voltage-gated potassium channel (VGKC) complex and glutamic acid decarboxylase 65 antibodies, often nonparaneoplastic in etiology, have been reported in patients with limbic encephalitis[3,4,5,6] and idiopathic epilepsy with anti epileptic drug (AED)-resistant seizures.[7,8,9,10,11,12] Newly identified autoantibody specificities that strongly correlate with clinical seizures include N-methyl-D-aspartate (NMDA),[13] gamma aminobutyric acid B,[14] and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors.[15] Identification of immune-mediated mechanisms in SE, epilepsy, cognitive decline, and other neurological symptoms is important to provide patients a benefit of immunomudulatory therapy, which may albeit be slow, but potentially completely reverse the epilepsy and other associated neurological manifestations. The antibodies are mainly of two types targeting neuronal cell surface antigen and the other targeting intracellular antigens. Antineuronal antibodies affect the neuronal signaling or synaptic transmission, the evidence to this is supported by benefit of immunomodulation and in vitro studies.[13,15,16,17,18] The intracellular antibodies mediated by cytotoxic T cell, is supported by autopsies showing inflammatory infiltrates of mononuclear cells, including CD4 and CD8 cells, which predominate in symptomatic areas of the nervous system.[19,20,21] These onconeural antibodies can be absent or be present in variable titers in patients with or without cancers.[22,23,24,25] Presence of antibodies, therefore, should not be the only defining feature of neurologic syndromes of autoimmune encephalitis. If an antibody present is usually associated with the particular neurologic syndrome of the patient, then the patient should be evaluated for said symptoms and treated.[26,27]
We describe herewith 15 cases of immune-mediated encephalitis all having antibodies positivity and presenting with seizures. The intention of this article is to raise awareness of immune-mediated epilepsy as a potentially reversible cause of drug refractory epilepsy and SE.
Materials and Methods
A prospective detection and follow-up of 15 patients was done (December 2011-June 2013). These patients presented with seizures and other neurological symptoms with positive autoantibodies [Figure 1]. Demographic and clinical characteristics (seizure types, clinical course, and associated symptoms) were recorded. Neuroimaging [brain magnetic resonance imaging (MRI)], cerebrospinal fluid analysis (CSF), and tumor screening was done periodically. All patients underwent thorough clinical examination along with skeletal survey (X-rays), chest and abdomen and pelvis computed tomography scan, positron emission tomography (PET) whole body, prostate-specific antigen in male patients more than 50 years, carcinoembryonic antigen 125 in all women, complete blood count, liver, renal function test, and serum electrophoresis. Serum electrophoresis was done in elderly patients (>50 years). None of the patients showed any evidence of malignancy either at baseline or in annual follow-up with whole body PET scan.
Figure 1.
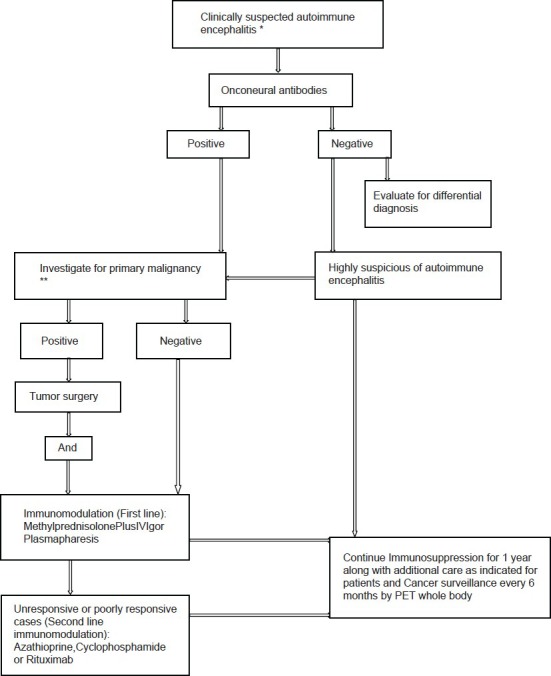
*Acute, Subacute (more commonly), chronic rapidly progressive neurological deficit, exclude alternate differential diagnosis. **Thorough clinical examination along with skeletal survey (X rays), chest and abdomen and pelvis CT (Computed tomography) scan, positron emission tomography (PET) whole body, prostate-specific antigen (PSA) in male patients more than 50 years, carcinoembryonic antigen 125 (CEA-125) in all women, serum electrophoresis was done in elderly patients more than 50 years
Anti-NMDA antibody was measured by radioimmunoassay. Enzyme immunoassay for anti-VGKC antibody and anti-GAD antibodies was done. These were tested in 1:10 dilution. Serum levels of anti-NMDA antibodies were ranging between 1:800 and 3200 either positive or negative in that dilution. Anti-VGKC antibody ranged from 197 to 2800 (>100pM/mL is positive) and anti-GAD antibody 105 to 200 (normal range: 0-10 U/Ml) antinuclear antibodies (ANA), anti-dsDNA, and anti-Ro antibody also had to be significant in titer of >1:40 (immunofluorescence), 65 (normal range: 0-50 IU/mLby ELISA; enzyme-linked immunosorbent assay technique) and 106 U/mL (normal <8 U/mL by ELISA), respectively. Treatment received by patients and response to the treatment was noted. Response to immunotherapy was categorized as per patients and treating doctors information (regarding seizure free/control and other neurological symptom improvement).
Data were expressed as median (range and interquartile range) for continuous variables and counts (percentages) for categorical variables. Detailed statistical analyses was done following entry of data in Microsoft excel 2011.
Results
There were 15 (males = 10 and females = 5) patients recorded of autoimmune encephalitis. The mean age of presentation was 24 years (age range: 2-64 years). Onset of disease most commonly was subacute (62%) followed by chronic, none of the patients presented with an acute onset. All patients had seizures (100%) and these were of various semiology [Tables 1 and 2]. Other associated clinical features were recorded, as mentioned in Tables 1 and 2. CSF was done in 10 patients and was normal in six (6 of 10; 60%), in the rest of CSF showed variable findings from mild raised protein (75-150 mg/dL) to raised cell counts (10-20/cu mm). CSF was more likely to be done in those who presented with SE and encephalitis like presentation rather than drug refractory epilepsy. Neuroimaging (brain MRI) was done in all patients, six (40%) were normal, six (40%) showed T2W and FLAIR hyperintensities in bilateral limbic areas, one had bilateral basal ganglia atrophy with periventricular hyperintensity on T2W and FLAIR images, one had diffuse cerebral and cerebellar atrophy, and one had right insular cortex hyperintensities with atrophy of the right hippocampus. Some of other abnormalities observed were bilateral perisylvian and bilateral posterior hyperintensities in one [Figure 2a] and another patient showing bilateral hyperintense swollen hippocampus [Figure 2b]. Antibodies were tested in all patients as these patients had no known cause for their seizures and encephalopathy (infective, herpes, malignant, metabolic, etc.) and were basically categorized as unknown etiology of status, epilepsy, and encephalopathy depending on their clinical manifestations. NMDA receptor antibody positivity was found in seven (50%) patients, VGKC antibody in five (36%) patients, and two patients had anti-GAD and in another anti-dsDNA antibodies were found. None of the patients showed any evidence of malignancy in the periodic tumor screening done. Each of patients received treatment in form of immunotherapy, steroid, intravenous immunoglobulin (IVIg), or both. There was a significant improvement in majority of cases, that is, no further seizures or relapse in 10 (67%) of 15 cases. There was one death (case 1), she presented very late, after almost 5 years of duration of illness without receiving any specific treatment and being misdiagnosed with a psychiatric diagnosis. On diagnosis, she received aggressive immunomodulation (steroids, rituximab, immunoglobulin) but developed drug-induced pancytopenia and died of severe sepsis. In case 4, initial workup (concordant clinical, video electroencephalography, MRI brain, and substraction ictal single photon emission computed tomography (SPECT) coregistred with interictal SPECT (SISCOS) study) was concluded as mesial temporal sclerosis of the right side. He was operated upon, but did not show any change in seizures, reevaluation showed his as having NMDA receptor antibody positivity and he showed a dramatic response to oral steroids, earlier his seizure frequency was 25 to 30 per month to the frequency post steroids of 1 to 2 per month. Case 6 had a presentation of seizures clusters, cognitive decline and was almost in a mute state she turned out to be NMDA positive, she was initiated on IVIg followed by pulse steroids and plasmapharesis. But had poor recovery after 2 weeks of immunomodulation, she was initiated upon pulse cyclophosphamide and rituximab. She is gradually recovering in the most recent follow-up. She is able to communicate and has no seizures. Case 14, continued having seizures, although frequency and intensity had reduced, as compared with previous by 70%.
Table 1.
Clinical, treatment, and follow-up profile of patients
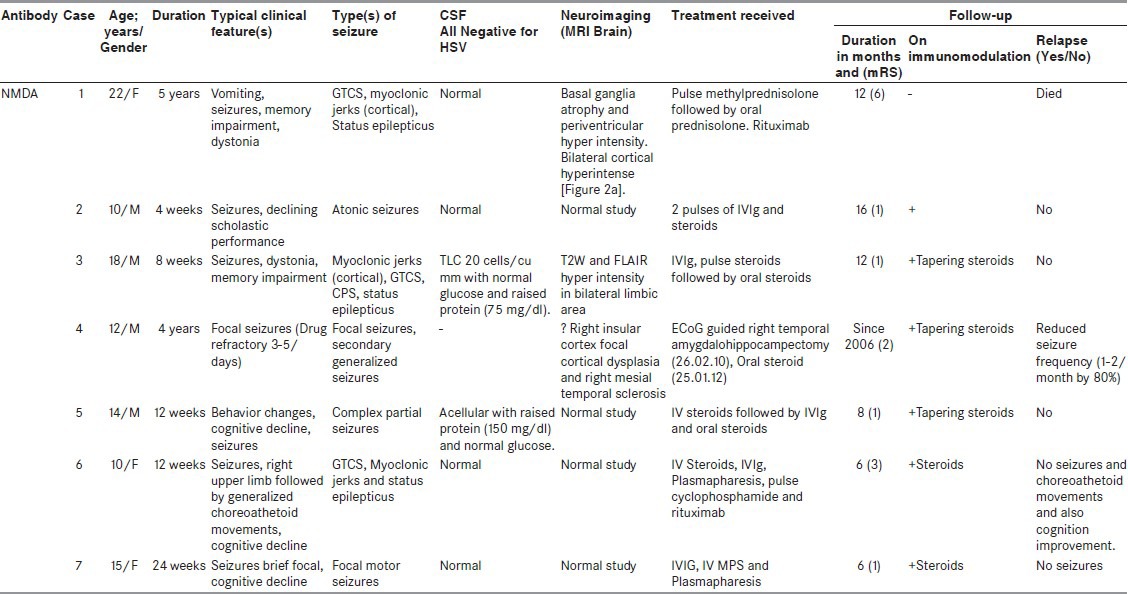
Table 2.
Details of case series
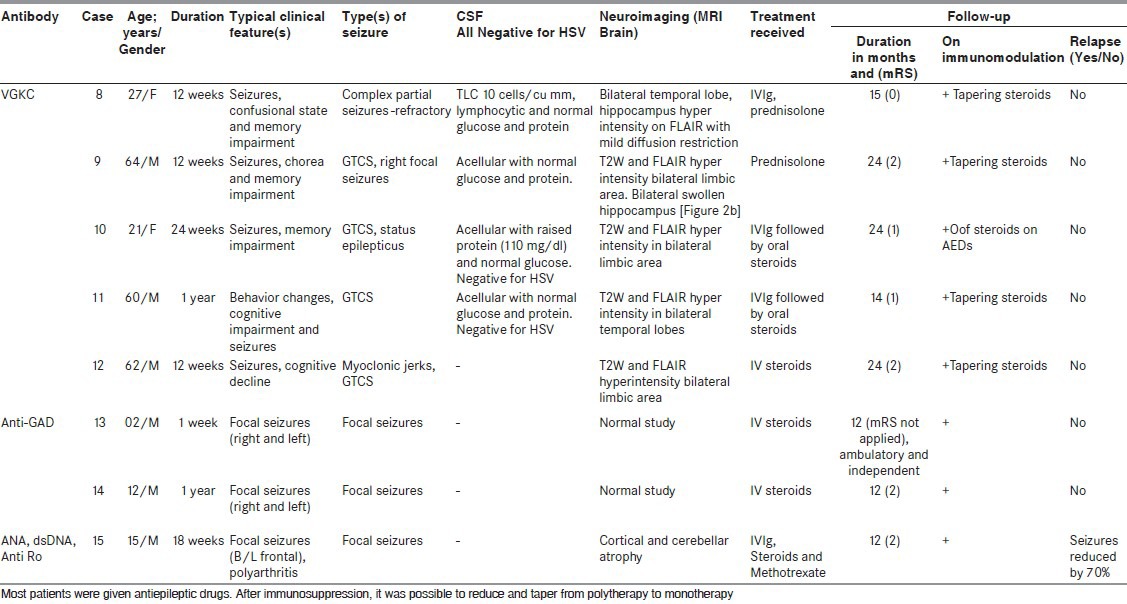
Figure 2.
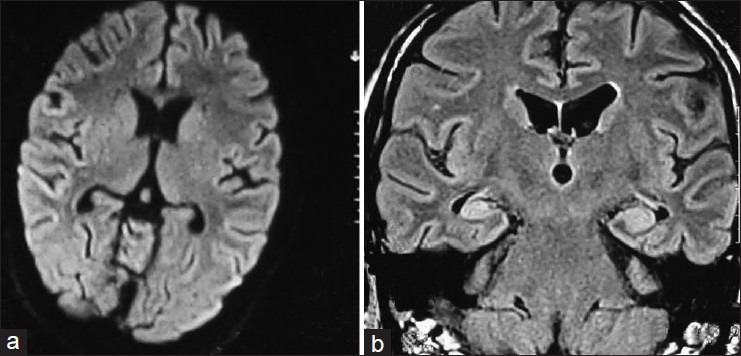
(a) Bilateral posterior parietal cortical hyperintensity on FLAIR magnetic resonance imaging (anti-sN-methyl-D-aspartate antibody positive) (b) Bilateral hyper intense swollen hippocampus (antivoltage-gated potassium channel antibody)
Discussion
Our study is prospective observation of 15 persons of suspected and subsequently confirmed autoimmune encephalitis. Seizures were the predominant and main presenting clinical feature present in all the cases (100%). We could find a very significant, no relapse and complete seizure freedom in 67% cases and overall there was significant improvement in the neurological status in 94% of our patients. Our series reflects that the diagnosis of autoimmune etiology should be considered in all persons with an unknown etiology of status, encephalopathy, cognitive, and behavior changes when other common causes have been ruled out. Our series is similar to previous observations on autoimmune encephalitis.[8,9,10,11,28,29,30]
An autoimmune etiology is identified most readily in patients who present with the full syndrome of limbic encephalitis, characterized by subacute memory impairment with affective changes and temporal lobe seizures. The diagnosis of autoimmune limbic encephalitis is aided by detection of neural autoantibodies with radiological or pathological evidence of temporomedial inflammation and in some cases a history of neoplasia in the preceding 5 years.[31]
One of our patients had undergone temporal lobectomy with a misdiagnosis of hippocampal sclerosis; this has also been observed by previous authors reporting limbic encephalitis to be an antecedent in adult onset temporal lobe epilepsy.[32,33] Another of our patient reached late to us, a psychiatric diagnosis of catatonia, drug-induced movement disorder, seizures and schizophrenia had been made. Other series too have a variation in diagnosis, for example, adult onset temporal lobe epilepsy in anti-NMDA receptor antibodies.[32]
The diagnosis of autoimmune encephalitis presenting as SE, epilepsy, cognitive impairment, and other neurological symptoms, requires a high index of suspicion at initial evaluation.[34] The clinical presentations in our patients were heterogeneous, but some general observations can be made. Very brief frequent seizures, faciobrachial seizures, tonic seizures associated with movement disorder and behavioral changes. Data from the current cohort suggest that autoimmune investigation should be considered in the presence of one or more of the following: Subacute onset of neurological symptoms (epilepsy, movement disorder, behavioral changes, and cognitive decline), an unusually high seizure frequency, very brief seizures, faciobrachial seizures, intra individual seizure variability with seizure clusters or multifocality, antiepileptic drug resistance with a nonsurgical substrate, personal or family history of autoimmunity [either organ-specific (e.g., thyroid disease, diabetes mellitus, pernicious anemia, or celiac disease) or nonorgan-specific (rheumatoid arthritis or systemic lupus erythematosus)], or recent or past neoplasia. Serological testing is increasingly valuable as an aid to establishing the diagnosis of an autoimmune etiology.
Half (7 of 15; 47%) of our patients were anti-NMDA receptor antibody positive with mean age of 14 years (range: 10-22 years), having seizures both focal (2) and myoclonic jerks (1) in three of seven patients with memory impairment, dystonia, choreoathetoid movements, reduced scholastic performance, behavioral changes, and vomiting. In comparison to a series of 100 patients[13] our observation varied having mean age lesser (14 years), as compared to 23 years in Dalmau's series, a contrast in gender preponderance was also observed. Clinical features observed in anti-NMDA receptor encephalitis were similar to other series which have reported; seizures of any type in 76%, dystonia and choreoathetoid movement in 47% each, additional neuropsychiatric manifestations, autonomic instability and central hypoventilation. Dalmau (2008) also observed prodromal symptoms, that is, viral-like symptoms in 72 of 84 patients. A total of six of our patients had undergone CSF study which was normal in half, rest half showed raised protein and one together with mild lymphocytic pleocytosis. In Dalmau's series 95 CSF studies were abnormal, but main abnormalities were similar, in form of raised protein and lymphocytic pleocytosis. We had only three (43%) abnormal MRIs and four (57%) normal MRIs in anti-NMDA receptor antibody positive cases. Whereas Dalmau's series about 55% had abnormal MRIs. The abnormalities we observed were basal ganglia atrophy, periventricular hyperintensities, and bilateral limbic region hyperintensity on T2 and FLAIR images. One case showed features of mesial temporal sclerosis. Normal MRI brain is not unusual for autoimmune epilepsies. We had no patients with tumor on screening at baseline or till now- all having 1 year of follow up with sequential whole body PET and ultrasound abdomen and pelvis.
We had five patients of autoimmune epilepsy with anti-VGKC antibody positivity with male preponderance (60%) and mean age of 47 years (range: 21-64 years). All these patients had seizures and memory impairment. Few patients had additional confusional state, behavioral changes, and chorea. In a series of 51 cases by Tan et al.,[35] which had a predominant female distribution (73%) the mean age at diagnosis was 66 years. Our observation differed in gender distribution, although the age distribution was nearly similar. Predominant clinical features[35] observed were cognitive impairment in 71%, seizures (faciobrachial) in 58%, autonomic instability in 33%, myoclonus in 29%, sleep disturbances in 26%, and extrapyramidal features in 21% (chorea in 4% cases) cases. Also noted were hyponatremia (36%), cranial nerve or brainstem involvement (19%), peripheral nerve hyperexcitability (17%), neuropathy (14%), headache (6%), cerebellar features (8%), and Morvan syndrome (3%). Our patients were similar in clinical findings. Our patients had CSF (two of four cases) abnormality in form of raised protein in one case and mild lymphocytic pleocytosis in one. However, magnetic resonance of the brain was abnormal in all the cases. In the series of Tan et al., 57% CSF (27 of 47 available CSF studies) and 54% (26 of 48 available MRIs) of MRIs were abnormal. None of our patients had a tumor at baseline or followup.
We had two children with anti-GAD antibody positivity these children were in their first decade with refractory focal epilepsy. They had a normal brain MRI and had a very excellent response to steroids, and had complete seizure freedom. Only one case of autoimmune epilepsy had ANA, anti-dsDNA, and anti-Ro positive, he was a 15 years boy with recurrent focal seizures and polyarthritis.
Our study shows excellent prognosis of being seizure-free and no further relapse of symptoms along with resolution of associated neurological deficits in 70%. As noted in 22 of 27 (81%) patients the immunomodulation therapy led to favorable outcome (P < 0.05).[36] In a recent observation of anti-NMDA encephalitis;[37] various immunotherapies used were steroids as first line, IVIg, and plasmapharesis, regarding second-line therapy cyclophosphamide and rituximab was used. There is a significant benefit in the use of both first-line and second-line immunotherapy, although this study was not a randomized trial. Hence, it is advisable for such patients to get immunomodulation in form of IVIg or MPS; however, future relapses may occur and if there is no improvement more aggressive immunomodulation with cyclophosphamide and rituximab is warranted. There have been no placebo-controlled clinical trials; the treatment is based on few prospective series that have been reported in literature.
On follow-up of antibody levels, three had reduced titers and these were absent in two cases at 6 months of follow-up and absent levels in five cases at 12 months of follow-up (one case had reduced levels at 6 months had absent levels at 12 months). It was not possible to repeat titers more frequently, as it would increase costs of management.
To conclude, rare causes of SE, epilepsy, and cognitive decline may be immune-related and a high index of suspicion should be kept, as these are reversible in many cases. The clinical accompaniments in these patients of seizures associated with movement disorders; cognitive, behavioral features will help us making an early diagnosis and prevent neurological morbidity.
Footnotes
Source of Support: The antibodies were tested free by a research grant by AIIMS research section and the DBT
Conflict of Interest: None declared
References
- 1.Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol. 2008;7:327–40. doi: 10.1016/S1474-4422(08)70060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tüzün E, Dalmau J. Limbic encephalitis and variants: Classification, diagnosis and treatment. Neurologist. 2007;13:261–71. doi: 10.1097/NRL.0b013e31813e34a5. [DOI] [PubMed] [Google Scholar]
- 3.Vincent A, Buckley C, Schott JM, Baker I, Dewar BK, Detert N, et al. Potassium channel antibody-associated encephalopathy: A potentially immunotherapy-responsive form of limbic encephalitis. Brain. 2004;127(Pt 3):701–12. doi: 10.1093/brain/awh077. [DOI] [PubMed] [Google Scholar]
- 4.Thieben MJ, Lennon VA, Boeve BF, Aksamit AJ, Keegan M, Vernino S. Potentially reversible autoimmune limbic encephalitis with neuronal potassium channel antibody. Neurology. 2004;62:1177–82. doi: 10.1212/01.wnl.0000122648.19196.02. [DOI] [PubMed] [Google Scholar]
- 5.Pittock SJ, Yoshikawa H, Ahlskog JE, Tisch SH, Benarroch EE, Kryzer TJ, et al. Glutamic acid decarboxylase autoimmunity with brainstem, extrapyramidal, and spinal cord dysfunction. Mayo Clin Proc. 2006;81:1207–14. doi: 10.4065/81.9.1207. [DOI] [PubMed] [Google Scholar]
- 6.Saiz A, Blanco Y, Sabater L, González F, Bataller L, Casamitjana R, et al. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: Diagnostic clues for this association. Brain. 2008;131(Pt 10):2553–63. doi: 10.1093/brain/awn183. [DOI] [PubMed] [Google Scholar]
- 7.Barajas RF, Collins DE, Cha S, Geschwind MD. Adult-onset drug-refractory seizure disorder associated with anti-voltage-gated potassium-channel antibody. Epilepsia. 2010;51:473–7. doi: 10.1111/j.1528-1167.2009.02287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Majoie HJ, de Baets M, Renier W, Lang B, Vincent A. Antibodies to voltage-gated potassium and calcium channels in epilepsy. Epilepsy Res. 2006;71:135–41. doi: 10.1016/j.eplepsyres.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 9.Irani SR, Buckley C, Vincent A, Cockerell OC, Rudge P, Johnson MR, et al. Immunotherapy-responsive seizure-like episodes with potassium channel antibodies. Neurology. 2008;71:1647–8. doi: 10.1212/01.wnl.0000326572.93762.51. [DOI] [PubMed] [Google Scholar]
- 10.Liimatainen S, Peltola M, Sabater L, Fallah M, Kharazmi E, Haapala AM, et al. Clinical significance of glutamic acid decarboxylase antibodies in patients with epilepsy. Epilepsia. 2010;51:760–7. doi: 10.1111/j.1528-1167.2009.02325.x. [DOI] [PubMed] [Google Scholar]
- 11.Peltola J, Kulmala P, Isojärvi J, Saiz A, Latvala K, Palmio J, et al. Autoantibodies to glutamic acid decarboxylase in patients with therapy-resistant epilepsy. Neurology. 2000;55:46–50. doi: 10.1212/wnl.55.1.46. [DOI] [PubMed] [Google Scholar]
- 12.Irani SR, Michell AW, Lang B, Pettingill P, Waters P, Johnson MR, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol. 2011;69:892–900. doi: 10.1002/ana.22307. [DOI] [PubMed] [Google Scholar]
- 13.Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, et al. Anti-NMDA-receptor encephalitis: Case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7:1091–8. doi: 10.1016/S1474-4422(08)70224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, et al. Antibodies to the GABA (B) receptor in limbic encephalitis with seizures: Case series and characterisation of the antigen. Lancet Neurol. 2010;9:67–76. doi: 10.1016/S1474-4422(09)70324-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol. 2009;65:424–34. doi: 10.1002/ana.21589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Giorgio R, Bovara M, Barbara G, Canossa M, Sarnelli G, De Ponti F, et al. Anti-HuD-induced neuronal apoptosis underlying paraneoplastic gut dysmotility. Gastroenterology. 2003;125:70–9. doi: 10.1016/s0016-5085(03)00664-4. [DOI] [PubMed] [Google Scholar]
- 17.Sommer C, Weishaupt A, Brinkhoff J, Biko L, Wessig C, Gold R, et al. Paraneoplastic stiff-person syndrome: Passive transfer to rats by means of IgG antibodies to amphiphysin. Lancet. 2005;365:1406–11. doi: 10.1016/S0140-6736(05)66376-3. [DOI] [PubMed] [Google Scholar]
- 18.Manto MU, Laute MA, Aguera M, Rogemond V, Pandolfo M, Honnorat J. Effects of anti-glutamic acid decarboxylase antibodies associated with neurological diseases. Ann Neurol. 2007;61:544–51. doi: 10.1002/ana.21123. [DOI] [PubMed] [Google Scholar]
- 19.Voltz R, Gultekin SH, Rosenfeld MR, Gerstner E, Eichen J, Posner JB, et al. A serologic marker of paraneoplastic limbic and brain-stem encephalitis in patients with testicular cancer. N Engl J Med. 1999;340:1788–95. doi: 10.1056/NEJM199906103402303. [DOI] [PubMed] [Google Scholar]
- 20.Jean WC, Dalmau J, Ho A, Posner JB. Analysis of the IgG subclass distribution and inflammatory infiltrates in patients with anti-Hu-associated paraneoplastic encephalomyelitis. Neurology. 1994;44:140–7. doi: 10.1212/wnl.44.1.140. [DOI] [PubMed] [Google Scholar]
- 21.Bernal F, Graus F, Pifarré A, Saiz A, Benyahia B, Ribalta T. Immunohistochemical analysis of anti-Hu-associated paraneoplastic encephalomyelitis. ActaNeuropathol. 2002;103:509–15. doi: 10.1007/s00401-001-0498-0. [DOI] [PubMed] [Google Scholar]
- 22.Bataller L, Wade DF, Graus F, Stacey HD, Rosenfeld MR, Dalmau J. Antibodies to Zic4 in paraneoplastic neurologic disorders and small-cell lung cancer. Neurology. 2004;62:778–82. doi: 10.1212/01.wnl.0000113749.77217.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drlicek M, Bianchi G, Bogliun G, Casati B, Grisold W, Kolig C, et al. Antibodies of the anti-Yo and anti-Ri type in the absence of paraneoplastic neurological syndromes: A long-term survey of ovarian cancer patients. J Neurol. 1997;244:85–9. doi: 10.1007/s004150050054. [DOI] [PubMed] [Google Scholar]
- 24.Buckley C, Vincent A. Autoimmune channelopathies. Nat ClinPract Neurol. 2005;1:22–33. doi: 10.1038/ncpneuro0033. [DOI] [PubMed] [Google Scholar]
- 25.Graus F, Dalmou J, Reñé R, Tora M, Malats N, Verschuuren JJ, et al. Anti-Hu antibodies in patients with small-cell lung cancer: Association with complete response to therapy and improved survival. J Clin Oncol. 1997;15:2866–72. doi: 10.1200/JCO.1997.15.8.2866. [DOI] [PubMed] [Google Scholar]
- 26.Miyamoto K, Kato T, Watanabe H, Miyamoto E, Suzuki S. A case of paraneoplastic syndrome accompanied by two types of cancer. J Neurol Neurosurg Psychiatry. 2002;72:408–9. doi: 10.1136/jnnp.72.3.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graus F, Keime-Guibert F, Reñe R, Benyahia B, Ribalta T, Ascaso C, et al. Anti-Hu-associated paraneoplastic encephalomyelitis: Analysis of 200 patients. Brain. 2001;124(Pt 6):1138–48. doi: 10.1093/brain/124.6.1138. [DOI] [PubMed] [Google Scholar]
- 28.Vincent A, Irani SR, Lang B. The growing recognition of immunotherapy-responsive seizure disorders with autoantibodies to specific neuronal proteins. Curr Opin Neurol. 2010;23:144–50. doi: 10.1097/WCO.0b013e32833735fe. [DOI] [PubMed] [Google Scholar]
- 29.McKnight K, Jiang Y, Hart Y, Cavey A, Wroe S, Blank M, et al. Serum antibodies in epilepsy and seizure-associated disorders. Neurology. 2005;65:1730–6. doi: 10.1212/01.wnl.0000187129.66353.13. [DOI] [PubMed] [Google Scholar]
- 30.Irani SR, Bien CG, Lang B. Autoimmune epilepsies. Curr Opin Neurol. 2011;24:146–53. doi: 10.1097/WCO.0b013e3283446f05. [DOI] [PubMed] [Google Scholar]
- 31.Bien CG, Elger CE. Limbic encephalitis: A cause of temporal lobe epilepsy with onset in adult life. Epilepsy Behav. 2007;10:529–38. doi: 10.1016/j.yebeh.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 32.Bien CG, Urbach H, Schramm J, Soeder BM, Becker AJ, Voltz R, et al. Limbic encephalitis as a precipitating event in adult-onset temporal lobe epilepsy. Neurology. 2007;69:1236–44. doi: 10.1212/01.wnl.0000276946.08412.ef. [DOI] [PubMed] [Google Scholar]
- 33.Bien CG, Schulze-Bonhage A, Deckert M, Urbach H, Helmstaedter C, Grunwald T, et al. Limbic encephalitis not associated with neoplasm as a cause of temporal lobe epilepsy. Neurology. 2000;55:1823–8. doi: 10.1212/wnl.55.12.1823. [DOI] [PubMed] [Google Scholar]
- 34.Quek AM, Britton JW, McKeon A, So E, Lennon VA, Shin C, et al. Autoimmune epilepsy: Clinical characteristics and response to immunotherapy. Arch Neurol. 2012;69:582–93. doi: 10.1001/archneurol.2011.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology. 2008;70:1883–90. doi: 10.1212/01.wnl.0000312275.04260.a0. [DOI] [PubMed] [Google Scholar]
- 36.McKeon A, Lennon VA, Pittock SJ. Immunotherapy-responsive dementias and encephalopathies. Continuum (MinneapMinn) 2010;16(2 Dementia):80–101. doi: 10.1212/01.CON.0000368213.63964.34. [DOI] [PubMed] [Google Scholar]
- 37.Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: An observational cohort study. Lancet Neurol. 2013;12:157–65. doi: 10.1016/S1474-4422(12)70310-1. [DOI] [PMC free article] [PubMed] [Google Scholar]