

Abstract
Background:
Creutzfeldt-Jakob disease is a rapidly progressive, fatal, transmissible neurodegenerative disorder caused by prion protein. It is still considered rare in countries like India. This is probably due to nonavailability of autopsy studies in majority of the center. The recent European diagnostic criterion for sporadic CJD (sCJD) is useful for making an early diagnosis.
Objective:
To report a series of patients of probable sCJD from a neurology institute of eastern India.
Materials and Methods:
Patients of rapidly developing dementia fulfilling the diagnostic criteria for sCJD were included. All were investigated in detail to find out any possible treatable cause including electroencephalography (EEG), magnetic resonance imaging (MRI) of brain, and cerebrospinal fluid analysis.
Results:
A total 10 patients of probable sCJD diagnosed using the European diagnostic criterion between December 2011 and January 2013. The clinical features are consistent with other reported series. While 60% of patients had the classical EEG findings, 100% had typical MRI features. Eight patients died within a mean duration of 4.56 months from the disease onset.
Conclusions:
The clinical features are similar to other reported series. Our observation raises question about the prevalence of this disease in India which needs more elaborate studies.
Key Words: Creutzfeldt-Jakob disease, electroencephalography, magnetic resonance imaging
Introduction
Creutzfeldt-Jakob disease (CJD) is a rapidly progressive, fatal transmissible neurodegenerative disorder caused by accumulation of an abnormally shaped membrane-bound protein, the prion protein, in neurons.[1] In about 85% of cases classified as sporadic CJD (sCJD), no etiology can be identified. The incidence of disease is 0.5-1.5 per million per year with little annual, seasonal or geographic variation.[2] Mean survival of CJD patients reported in the literature is 5 months with over 80% of patients succumbing to the disease by 12 months of onset.[3,4] In India, the disease is still underreported. According to the national CJD registry at National Institute of Mental Health and Neurosciences (NIMHANS), Bangalore, India, there were only 85 recorded cases of CJD till September 2005.[5] Thirty cases including 20 definite and 10 probable cases were reported between 1971 and 1990. Demographic analysis has shown similarities to the previously published reports from other parts of the world. Another series of 10 cases was published by Mehndiratta et al., in 2001 from New Delhi.[6]
Here we report a series of 10 cases from eastern India diagnosed in year 2011-2012 of sCJD on the basis of current European diagnostic criteria.[7]
Materials and Methods
Patients of rapidly developing dementia admitted in the institute between December 2011 and January 2013 were included in this study. The patients were diagnosed on the basis of criteria proposed by magnetic resonance imaging (MRI)-CJD consortium criteria for sCJD. Details of place of residence, occupation, dietary habits, age at onset, duration of symptoms, history of any surgical procedure or head trauma, hypertension, exposure to drugs like lithium, toxins like bismuth, hormone replacement therapy, and any relevant family history were recorded. History of dog bite and vaccination with Semple's vaccine for the same was not known in any of the cases. All patients underwent investigations including complete hemogram, erythrocyte sedimentation rate (ESR), blood sugar, kidney function tests, liver function tests, urinalysis, serum Venereal Disease Research Laboratory (VDRL), human immunodeficiency virus (HIV), serum anti-thyroid peroxidase (anti-TPO) antibodies test, antinuclear antibody (ANA). Cerebrospinal fluid (CSF) studies for cell count, cell type, protein and sugar, bacteriological and virological studies, electrocardiogram (ECG), and chest X-rays, and ultrasonography (USG) abdomen were carried out in all the patients. All patients were subjected to a 1.5 Tesla MRI scan including diffusion weighted and fluid-attenuated inversion recovery (FLAIR) images. Electroencephalography (EEG) was recorded using the international 10-20 System. The periodic polyspike-wave complexes (PSWCs) were defined as diffuse biphasic or triphasic sharp wave complexes with duration between 100 and 600 ms and an intercomplex interval between 500 and 2,000 ms. None of the patients underwent brain biopsy.
Results
A total 10 cases of probable sCJD were admitted at our center during December 2011 to January 2013 (M:F = 4:6). Table 1 gives details of demographic profile of patients, their clinical, EEG, and MRI findings. All of them were the residents of West Bengal, but came from various districts suggesting no geographical clustering. The mean age of patients at the time of diagnosis was 56.1 (SD = 8.26, range 39-70) years. While six patients presented with behavioral abnormalities, four had ataxia, five had extrapyramidal features, four had visual hallucination, and one with cortical blindness as their presenting symptoms. All of our patients had spontaneous myoclonus during some stage of the disease and of them 5 had stimulus-sensitive myoclonus. All of them became bedbound within 3-4 months from their illness and developed akinetic mutism. One of our patients had known history of surgical procedure (cataract surgery) done 2 years before onset of illness. None of our patients had history of head trauma, intake of lithium, or any hormone replacement therapy. No history suggestive of exposure to toxins like bismuth was noted in any of the cases. Eight patients died with mean duration of illness of 4.56 months from disease onset. We do not have information about two patients. Six patients (60%) had periodic spike wave complexes, while all patients had diffuse slowing of background on EEG [Figure 1]. We could not perform repeat EEG in our patients as they were either discharged or taken away from hospital once a diagnosis was disclosed to their relatives. CSF showed normal to mildly elevated protein in all our patients and only one patient had raised cell count in CSF (20 cells/mm3). The CSF of all patients was negative for viral/bacterial infection. The serum anti-TPO antibodies were positive in three patients with very high titer in one. MRI was performed in all 10 patients including diffusion weighted imaging (DWI) sequence. All patients (100%) had MRI features suggestive for CJD that include bilateral symmetrical hyperintensities in caudate and putamen in T2 and FLAIR sequences with gyral pattern diffusion restriction in bilateral parieto-occipital and temporal regions [Figure 2]. In addition, the “hockey stick sign” was detected in one patient [Figure 3] and “pulvinar sign” in another one. Three patients received high dose methylprednisolone for 5 days with the suspicion of autoimmune encephalitis, but none of them showed improvement.
Table 1.
Demographic profile including disease duration and total duration illness before death, clinical features, EEG, and MRI findings
Figure 1.
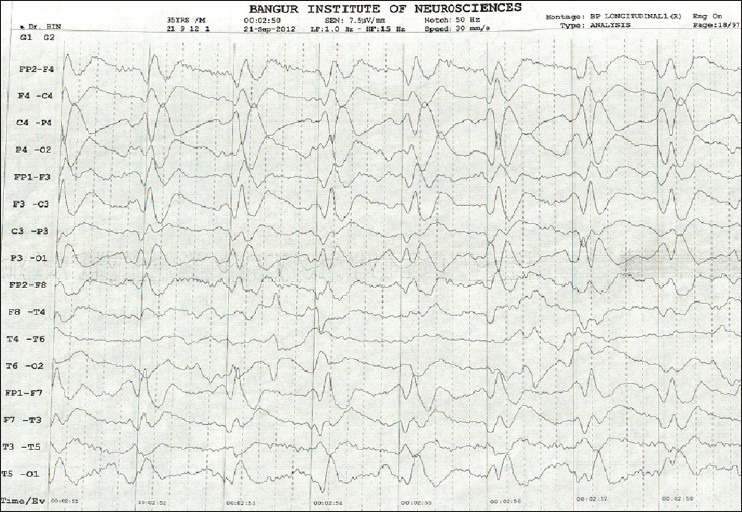
Electroencephalography showing periodic spike and wave discharges
Figure 2.
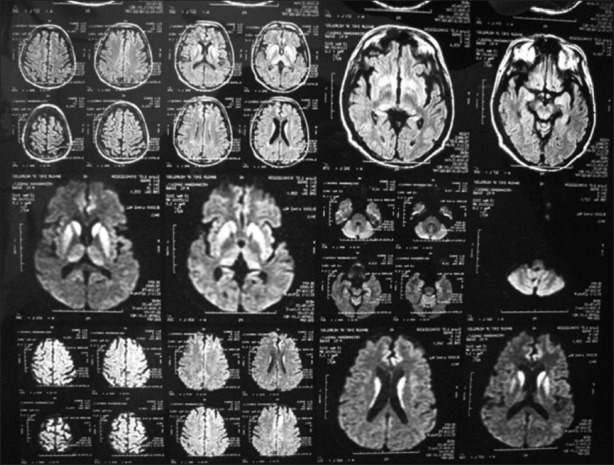
Magnetic resonance imaging diffusion weighted sequence showing restriction in bilateral basal ganglia and gyral pattern of diffusion restriction in bilateral parieto-occipital cortex and bilateral frontoparietal cortex
Figure 3.
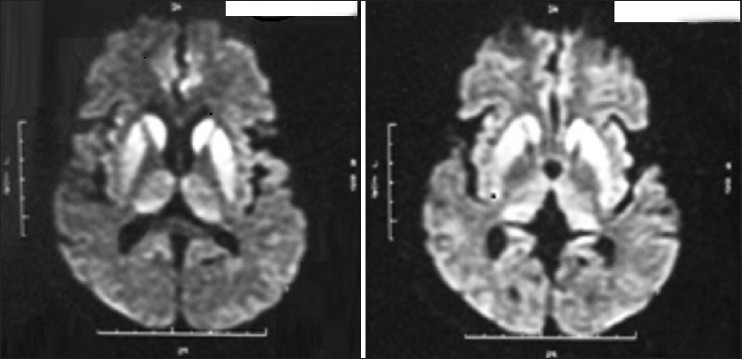
MRI showing “hockey stick sign”
Discussion
This case series consists of 10 cases (M:F = 4:6) of probable sCJD diagnosed on the basis of current European diagnostic criteria. All of our patients had clinical features of rapidly developing dementia with myoclonus. The other clinical features include behavioral abnormalities, ataxia, extrapyramidal features, cortical blindness, etc., All of them were evaluated with detailed investigations to exclude other treatable causes; like metabolic, infective, and autoimmune diseases. Three patients were given high dose methylprednisolone for 5 days with the suspicion of autoimmune encephalitis, without any improvement. All patients showed a positive MRI finding fulfilling the criteria. The EEG was positive for PSWCs in six patients, while all patients showed diffuse slowing of background. Thus, all of our patients fulfilled the diagnosis of probable sCJD according to the criteria.
The mean age of patients at the time of diagnosis was 56.1 years. In the 10 cases of Mehndiratta et al.,[6] the mean age was 53.80 (SD = 7.32) years with M:F of 1:1. All of our patients had myoclonus and similar finding was also reported by Mehndiratta et al.[6] Myoclonus is considered as the most characteristic and constant sign in CJD. Our observation about various presenting symptoms, for example, abnormal behavior, ataxia, and extrapyramidal features are similar to the case series of Mehndiratta et al.[6] All of our patients became bedbound and developed akinetic mute state and eight patients died within a mean period of 4.56 months from the disease onset. This is consistent with other reported series.
While six of our patients had classical EEG changes like PSWCs, all patients had diffuse slowing of background. PSWCs in EEG give a sensitivity of 67% and specificity of 86% for detection of CJD.[8] More than 90% of patients may show periodic complexes if repeated EEG records are taken.[9] We could not perform repeat EEG in our patients, and thus it was not possible to document changes.
The PSWCs on EEG have been used as one of the central diagnostic tests for CJD.[10] PSWCs are recorded usually in middle and late stages of the disease.[11] PSWCs, either lateralized (in earlier stages) or generalized, occur in about two-thirds of patients with sCJD, with a positive predictive value of 95%.[11] However, PSWCs are not always specific for CJD.[11] PSWCs are therefore of limited use for the early diagnosis of CJD.
MRI with DWI and FLAIR sequences is an invaluable modality in supporting the diagnosis of CJD. The detection of the specified high signal abnormalities in FLAIR or DWI MRI is considered to have similar diagnostic importance as PSWCs on the EEG or 14-3-3 protein detection in the CSF.[8] Shiga et al., in their study concluded that diffusion-weighted MRI had higher sensitivity (92%) in the detection of CJD than FLAIR (41-59%), T2 sequences (36-50%), EEG (50-78%), CSF protein 14-3-3 (84%), or neuron-specific enolase (73%).[13] Matoba et al., noted that the hyperintensity in the basal ganglia and cortex during the early stages was more extensive and conspicuous while in the later stages there was disappearance of the abnormal signals in the cortex.[14]
Zerr et al.,[12] modified the clinical diagnostic criteria for sCJD and included MRI findings with detection of either hyperintensity in the basal ganglia (both caudate nucleus and putamen) or in at least two cortical regions (from either the temporal, parietal, or occipital cerebral cortices). This implies that the detection of the specified high signal abnormalities in FLAIR or diffusion weighted MRI is considered at the same level of diagnostic importance as PSWCs on the EEG or 14-3-3 protein detection in the CSF.
The combination of FLAIR and DWI has a sensitivity, specificity, and accuracy of over 90% in differentiating CJD from other dementias.[15] It is argued that the multifocal cortical and subcortical hyperintensities in the grey matter showing restricted diffusion on MRI may be more useful than the CSF protein 14-3-3 analysis.[16] Zerr et al.,[12] proposed that high signal abnormalities in caudate nucleus and putamen or at least two cortical regions (temporal, parietal, or occipital lobes) either in DWI or FLAIR together with typical clinical signs can be diagnostic for probable sCJD. Based partly upon their report, ‘high signal in caudate/putamen on MRI brain scan’ has been used as one of the laboratory findings in the diagnostic criteria for probable sCJD in the European CJD Surveillance System (EUROCJD) since January 2010.[7] However, their criteria did not distinguish DWI and FLAIR, thereby maintaining ambiguity about the diagnostic values of MRI in situations where DWI is not available. More recently, Vitali et al., reported that hyperintensity greater on DWI than FLAIR is diagnostic for sCJD, whereas hyperintensity greater on FLAIR than DWI is characteristic for nonprion rapidly progressive dementia.[17] Furthermore, reduction of apparent diffusion coefficient in subcortical (striatum) hyperintensity regions on DWI is supportive for sCJD.[17] In another recent study, Fujita et al., argued that FLAIR without DWI is unreliable for the diagnosis of sCJD.[18] On the other hand, high signals in the cerebral cortex have not been regarded as diagnostic in the EUROCJD criteria, probably because cortical abnormalities are less reliable on conventional MRI. They suggest that, using standardized or variable DWI but not FLAIR, cortical signals can also be used as a diagnostic marker.[18] All of our patients had bilateral symmetrical hyperintensities in caudate and putamen in T2 and FLAIR sequences with gyral pattern diffusion restriction in bilateral parieto-occipital and temporal regions. One of them also had the “hockey stick sign” and another one had the “pulvinar sign”.
This case series is single center experience over a period of 1 year. The disease is still considered a rare entity in India. Barring NIMHANS registry and case series from New Delhi, there are few case reports from various parts of India. Mehndiratta et al., reported 10 cases from a tertiary care center in north India, gathered over 9 years (1990-1998). While their patients came from four different states of north India, all our patients are resident of West Bengal. However, we did not find any geographical clustering. The observation of so many patients within 1 year period is an important observation. This demands rethinking about the disease prevalence in our country. However, it may also because of availability of new diagnostic criteria and easier availability of MRI in recent years, that helped us to make a diagnosis even without autopsy and availability of sophisticated test like estimation of CSF protein 14-3-3.
Conclusion
CJD is a fatal spongiform encephalopathy. Diagnosis is usually suspected in a patient with rapidly progressive dementia with myoclonus. Additional signs include visual hallucination, cerebellar or extrapyramidal features, akinetic rigid state, and mutism. These clinical features with a typical EEG finding of periodic PSWCs and MRI features of T2 and FLAIR hyperintensities in cortical or subcortical grey matters and diffusion restriction are almost diagnostic of this disease. Early diagnosis may be possible on basis of MRI brain scan and this has been used as one of the laboratory findings in the diagnostic criteria for probable sCJD in the EUROCJD since January 2010. Our study highlights the increasing prevalence of the disease in India, which needs to be validated by studies from other parts of the country.
Limitations of the study
We could not perform the CSF 14-3-3 protein analysis because of the lack of the availability of the test at our institute. Diagnosis can only be confirmed by histological examination of brain tissue obtained either by the brain biopsy or after autopsy which was not performed at our center.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil
References
- 1.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–83. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson RT, Gibbs CJ., Jr Creutzfeldt-Jakob disease and related transmissible spongiform encephalopathies. N Engl J Med. 1998;339:1994–2004. doi: 10.1056/NEJM199812313392707. [DOI] [PubMed] [Google Scholar]
- 3.Brown P, Gibbs CJ, Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, et al. Human spongiform encephalopathy: The National Institute of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513–29. doi: 10.1002/ana.410350504. [DOI] [PubMed] [Google Scholar]
- 4.Will RG, Matthews WB. A retrospective study of Creutzfeldt-Jakob disease in England and Wales 1970 − 79.I: Clinical features. J Neurol Neurosurg Psychiatry. 1984;47:134–40. doi: 10.1136/jnnp.47.2.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shankar SK, Satishchandra P. Did BSE in the UK originate from Indian continent? Lancet. 2005;366:790–1. doi: 10.1016/S0140-6736(05)67193-0. [DOI] [PubMed] [Google Scholar]
- 6.Mehndiratta MM, Bajaj BK, Gupta M, Anand R, Tatke M, Seryam S, et al. Creutzfeldt-Jakob disease: Report of 10 cases from North India. Neurol India. 2001;49:338–41. [PubMed] [Google Scholar]
- 7.Diagnostic criteria for sporadic CJD from 1 January 2010. National Creutzfeldt–Jakob Disease Surveillance Diagnostic Criteria [online] [Last accessed on 2013 May 8]. Available from http://www.cjd.ed.ac.uk/criteria.htm .
- 8.Steinhoff BJ, Racker S, Herrendorf G, Poser S, Grosche S, Zerr I, et al. Accuracy and reliability of periodic sharp wave complexes in Creutzfeldt-Jakob disease. Arch Neurol. 1996;53:162–6. doi: 10.1001/archneur.1996.00550020074017. [DOI] [PubMed] [Google Scholar]
- 9.Chiofalo N, Fuentes A, Gálvez S. Serial EEG findings in 27 cases of Creutzfeldt-Jakob disease. Arch Neurol. 1980;37:143–5. doi: 10.1001/archneur.1980.00500520041005. [DOI] [PubMed] [Google Scholar]
- 10.Zerr I, Pocchiari M, Collins S, Brandel JP, de Pedro Cuesta J, Knight RS, et al. Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurology. 2000;55:811–5. doi: 10.1212/wnl.55.6.811. [DOI] [PubMed] [Google Scholar]
- 11.Wieser HG, Schindler K, Zumsteg D. EEG in Creutzfeldt-Jakob disease. Clin Neurophysiol. 2006;117:935–51. doi: 10.1016/j.clinph.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 12.Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt–Jakob disease. Brain. 2009;132:2659–68. doi: 10.1093/brain/awp191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shiga Y, Miyazawak K, Sato S, Fukushima R, Shibuya S, Sato Y, et al. Diffusion-weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt-Jakob disease. Neurology. 2004;63:443–9. doi: 10.1212/01.wnl.0000134555.59460.5d. [DOI] [PubMed] [Google Scholar]
- 14.Matoba M, Tonami H, Miyagi H, Yokota H, Yamamoto I. Creutzfeldt-Jakob disease: Serial changes on diffusion weighted MRI. J Comput Assist Tomogr. 2001;25:274–7. doi: 10.1097/00004728-200103000-00022. [DOI] [PubMed] [Google Scholar]
- 15.Young GS, Geschwind DM, Fischbein NJ, Martindale LJ, Henry RG, Liu S, et al. Diffusion-weighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: High sensitivity and specificity for diagnosis. AJNR Am J Neuroradiol. 2005;26:1551–62. [PMC free article] [PubMed] [Google Scholar]
- 16.Mendez OE, Shang J, Jungreis CA, Kaufer DI. Diffusion-weighted MRI in Creutzfeldt-Jackob disease: A better diagnostic marker than CSF protein 14-3-3? J Neuroimaging. 2003;13:147–51. [PubMed] [Google Scholar]
- 17.Vitali P, Maccagnano E, Caverzasi E, Henry RG, Haman A, Torres-Chae C, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology. 2011;76:1711–9. doi: 10.1212/WNL.0b013e31821a4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujita K, Harada M, Sasaki M, Yuasa T, Sakai K, Hamaguchi T, et al. Multicentre multi-observer study of diffusion-weighted and fluid-attenuated inversion recovery MRI for the diagnosis of sporadic Creutzfeldt–Jakob disease: A reliability and agreement study. BMJ Open. 2012;2 doi: 10.1136/bmjopen-2011-000649. e000649. [DOI] [PMC free article] [PubMed] [Google Scholar]