

Omenn syndrome is a severe immunodeficiency disease commonly arising from hypomorphic RAG recombinase gene mutations. RAG recombinase mediates V(D)J joining during early B-lymphocyte development in the bone marrow (BM). V(D)J recombination and class switch recombination are thought to partition between the BM and secondary lymphoid organs. Kenter and colleagues show that V(D)J joining and switching are interchangeably inducible in the BM. This study has important implications for the development of Omenn syndrome, autoimmune diseases, and leukemia.
Keywords: B-cell development, Ag gene rearrangement, gene expression
Abstract
V(D)J joining is mediated by RAG recombinase during early B-lymphocyte development in the bone marrow (BM). Activation-induced deaminase initiates isotype switching in mature B cells of secondary lymphoid structures. Previous studies questioned the strict ontological partitioning of these processes. We show that pro-B cells undergo robust switching to a subset of immunoglobulin H (IgH) isotypes. Chromatin studies reveal that in pro-B cells, the spatial organization of the Igh locus may restrict switching to this subset of isotypes. We demonstrate that in the BM, V(D)J joining and switching are interchangeably inducible, providing an explanation for the hyper-IgE phenotype of Omenn syndrome.
Humoral immunity is dependent on antigen receptors that are assembled from immunoglobulin (Ig) heavy chain VH, D, and JH segments during early B-cell development. RAG recombinase mediates V(D)J gene segment assembly in the bone marrow (BM) (Zhang et al. 2010; Schatz and Ji 2011). In mature B cells located in secondary lymphoid structures, activation-induced deaminase (AID) initiates a switch from IgM expression to secondary Ig classes (IgG3, IgG1, IgG2b, IgG2a, IgE, and IgA) that have specialized effector functions (Stavnezer 1996, 2000). Class switch recombination (CSR) occurs via an intrachromosomal deletional process while maintaining the original V(D)J assembly (Kenter 2012). The failure to assemble V(D)J exons or signal through the B-cell receptor (BCR) creates a block in the developmental progression of pro-B cells (Jankovic et al. 2004; von Boehmer and Melchers 2010). However, the strict ontological separation of V(D)J joining and CSR may break down under some circumstances (Milili et al. 1991; Rolink et al. 1996; Weller et al. 2001; Dudley et al. 2002; Mao et al. 2004; Han et al. 2007; Ueda et al. 2007; Scheeren et al. 2008; Kuraoka et al. 2009; Wesemann et al. 2011). For example, in μMT mice, membrane Igμ is not expressed, and B-cell development is halted at the pro-B-cell stage; nevertheless, IgA is selectively expressed (Melamed et al. 2000; Macpherson et al. 2001), implying an alternative pathway for CSR that can circumvent the pro-B-cell block. However, it is unclear where and under what circumstances the switch to IgA occurs.
Omenn syndrome is a severe combined immunodeficiency disease associated with autoimmunity and atopy, most commonly arising from hypomorphic mutations in the Rag recombinase genes. The syndrome is characterized by the severe paucity of B and T lymphocytes, hypo-γ-globulinemia, and, paradoxically, elevated IgE levels (Wong and Roth 2007; Ozcan et al. 2008). To account for hyper-IgE in Omenn syndrome, we hypothesized that (1) CSR occurs in BM pro-B cells prior to V(D)J joining, (2) CSR is predisposed to IgE, and (3) V(D)J joining events can follow CSR. Here, we show that Rag-deficient or Mb1-deficient pro-B cells can be induced to undergo robust CSR, albeit to a restricted subset of Ig constant (CH) region genes, including IgG2b and IgE, prior to or following V(D)J joining. Immunization of Rag1−/− mice with LPS promotes IgG2b switching in BM pro-B cells. We also demonstrate that V(D)J joining can follow CSR in Abelson transformed pro-B-cell and pre-B-cell lines. Studies of Igh locus chromatin structure revealed a unique stage-specific organization in pro-B cells that is correlated with preferential IgG2b and IgE switching. The flexible expression of the V(D)J recombination and CSR programs has implications for the genesis of Omenn syndrome hyper-IgE, autoimmune repertoire development, and leukemagenesis via the coexpression of the RAG and AID recombinases (Tsai et al. 2008).
Results and Discussion
To test the proposition that robust CSR can occur in BM pro-B cells prior to V(D)J recombination, we evaluated Rag2, AID, and isotype-specific germline transcript (GLT) expression in an E2A−/− pre-pro-B cell line, in pro-B cells isolated from Rag1- or Mb1-deficient mice, and in a panel of Abelson transformed pro-B-cell lines (Rag-deficient R2K2 and 445.3) and pre-B-cell lines (PA112.1, PA112.2, PA48.1, A70.2, and ATM2A). E2A and Rag deficiencies preclude V(D)J joining, whereas Mb1-deficient pro-B cells assemble V(D)J exons but cannot signal through the pre-BCR and are blocked at the pro-B-cell stage (Pelanda et al. 2002). Elevated Rag2 but not AID transcripts are detected in all strains of unstimulated pre-pro-B and pro-B cells but not in the Abelson transformed cell lines, as expected (Fig. 1A, top; Supplemental Fig. S1; Muljo and Schlissel 2003). Upon activation with the CSR inducers LPS+CD40L or LPS+CD40L+IL4, Rag2 expression is reduced in E2A−/− pre-pro-B cells and Rag1-deficient and Mb1-deficient pro-B cells (Fig. 1A). In contrast, AID expression is markedly induced in Rag1-deficient and Mb1-deficient pro-B cells but not E2A−/− pre-pro-B cells (Fig. 1A, bottom). Similarly, in R2K2 and PA112.2 as well as other Abelson transformed cell lines, AID expression substantially increases in response to CSR stimuli (Fig. 1A, bottom; Supplemental Fig. S1A,B). E2A−/− pre-pro-B cells were not further considered because AID is not expressed. Previous studies of BM pro-B cells concluded that AID transcription was undetectable (Crouch et al. 2007) but did not evaluate expression in response to CSR activators.
Figure 1.

Pro-B cells undergo inducible CSR. E2A-deficient pre-pro-B cells, Rag1-deficient or Mb1-deficient pro-B cells, Abelson transformed pro-B-cell lines (R2K2), and pre-B-cell lines (PA112.2) were unstimulated (US) or activated with LPS+CD40L (LC) or LPS+CD40L+IL4 (LCI), and wild-type (WT) resting splenic B cells were unstimulated or activated with LPS (L) or LPS+IL4 (LI). (*) P ≤ 0.05; (**) P ≤ 0.001–0.0001. (A,B) Quantitative RT–PCR assays for Rag2 and AID (A) and GLTs γ3, γ1, γ2b, and ɛ (B) from five samples from three to five independent experiments were normalized to the rRNA 18S transcript. (C, top) DC-PCR assay schematic, EcoRI (RI) sites, and nAChR gene loading control. (Bottom) DC-PCR analyses of μ → γ3, μ → γ1, μ → γ2b, and μ → ɛ CSR are representative of three independent experiments. (D) DC-PCR analysis of pro-B cells from Rag1−/− mice injected with LPS or PBS. Wild-type or AID−/− splenic B cells activated with LPS or no template control (C) are representative of three independent experiments.
We next examined the pattern of GLT expression in pro-B cells. CH genes are organized in transcription units that include a noncoding intronic (I) exon, the switch (S) region, and the CH exons. Downstream S regions are targeted for recombination with Sμ by selective activation of GLT promoters in response to specific stimuli. GLTs initiate from a promoter located upstream of each I exon and terminate 3′ of the CH region (Stavnezer 2000). Typically, GLTs γ3 and γ2b are induced by LPS or LPS+CD40L, whereas GLTs γ1 and ɛ also require IL4 in mature B cells (Fig. 1B). However, in response to the appropriate CSR stimuli, Rag1-deficient and Mb1-deficient pro-B cells and the Abelson transformed cell lines expressed GLT γ2b and ɛ but not γ1 and γ3 (Fig. 1B; Supplemental Fig. S1A). Accordingly, expression of GLT ɛ and not γ1 was noted for immature B cells (Wesemann et al. 2011).
Activation of AID expression and germline transcription prompted us to investigate whether CSR occurs in pro-B or pre-B cells. We used semiquantitative digestion circularization PCR (DC-PCR) to detect CSR events, since pro-B cells do not express surface Ig (Fig. 1C, top). The nonrearranging Acetylcholine receptor (nAcR) gene was used as a loading control (Fig. 1C). DC-PCR products representing μ → γ2b and μ → ɛ CSR events are detected in the Rag1-deficient and Mb1-deficient pro-B cells and Abelson transformed cell lines, whereas μ → γ3 or μ → γ1 events are not detected in either case (Fig. 1C, bottom; Supplemental Fig. S2). In contrast, in mature splenic B cells activated with LPS or LPS+IL4, a full spectrum of μ → γ3/γ2b and μ → γ1/ɛ CSR events is detected, respectively (Fig. 1C). This restricted pattern of CSR in the pro-B versus mature B cells parallels their GLT expression profiles. Strikingly, the levels of DC-PCR products in primary pro-B cells and activated splenic B cells were similar, indicating robust CSR in early B cells (Fig. 1C; Supplemental Fig. S2).
High-frequency μ → γ2b/ɛ CSR in pro-B cells was confirmed by several independent methods, including analysis of post-switch transcripts (PSTs), circle transcript PCR (CT-PCR) assays, and S/S junction analyses. Following CSR, the Iμ exon is in close proximity with a new downstream CH region, thereby permitting PST expression that was detected using a forward primer in Iμ together with a reverse primer in the relevant CH exon. PST γ2b and ɛ are induced in Rag1-deficient and Mb1-deficient pro-B cells and Abelson transformed cell lines upon activation with CSR stimuli (Supplemental Fig. S3). CT-PCR assays indicate that μ → γ2b or μ → ɛ CSR occurs dynamically in Rag1−/− pro-B cells and Abelson transformed cell lines (Supplemental Fig. S4). We sequenced S/S recombination junctions from appropriately activated Rag1−/− pro-B cells or R2K2 cells and found that Sμ joined to a variety of sites in Sγ2b or Sɛ (Supplemental Figs. S5–S8). Switch junctions were blunt or contained short junctional microhomologies or occasional insertions in normal proportions (Stavnezer et al. 2010) and confirmed that the mechanism of CSR in pro-B cells is operationally indistinguishable from that observed in mature B cells.
To determine whether CSR can be induced in vivo, Rag1−/− mice were injected with LPS or PBS, and pro-B cells were isolated directly from the BM and tested for CSR by DC-PCR. Wild-type or AID-deficient splenic B cells that were activated with LPS in ex vivo culture served as positive and negative controls for CSR, respectively. nAChR amplification is shown as a loading control, and the absence of PCR products in reactions devoid of template demonstrate PCR specificity. Under these activation conditions, μ → ɛ CSR is negligible. Switching μ → γ2b but not μ → γ3 was observed in 67% (six out of nine) of LPS immunized mice (P = 0.007; χ2) but not (zero out of six) for PBS-injected controls from three independent experiments (Fig. 1D). Targeting of CSR to the γ2b but not the γ3 locus reflects the established GLT expression pattern that we observed for pro-B cells activated in ex vivo culture (Fig. 1B,C). We conclude that CSR can occur in BM Rag1−/− pro-B cells prior to V(D)J recombination.
To address the question of whether a B cell that experienced CSR can subsequently undergo V(D)J joining, switched clones from the R2K2 pro-B-cell line were isolated by limiting dilution. CSR was confirmed in clones B5 (μ → γ2b) and A2 (μ → ɛ) by DC-PCR, and clonality was demonstrated by virtue of unique intra-Sμ region rearrangements (Fig. 2A; Supplemental Fig. S9). Treatment of these cell lines with STI571, a pharmacologic inhibitor of Abl kinase activity, resulted in activation of V(D)J joining and differentiation to the late pre-B-cell state (Muljo and Schlissel 2003). STI571 stimulation of B5 and A2 clones led to induction of the V(D)J joining gene program (Fig. 2B); Muljo and Schlissel 2003). The Rag2+ but not the empty expression construct induced diverse DDFL16.1–JH1 coding joins (CJs), while PST expression persisted (Fig. 2C; Supplemental Figs. S10, S11). Thus, pro-B-cell clones that experienced CSR retain the ability to undergo V(D)J recombination. Importantly, evidence indicates that BM pro-B cells expressing IgG1 (Waisman et al. 2007; Dougan et al. 2012) or transgenic Ig-γ2b chains (Storb et al. 1994) are capable of mature B-cell development that is indistinguishable from wild type.
Figure 2.

V(D)J joining can follow CSR in early B-cell lines. Cells were untreated (U) or activated with LPS+CD40L (LC) or LPS+CD40L+IL4 (LCI). Results are representative of three to nine samples from three independent experiments. (A) DC-PCR analysis of CSR in R2K2 and in clones A2 and B5. (B) STI571 treatment induces Rag1, Igκ GLT, and Irf4 expression as assessed by quantitative RT–PCR in B5 and A2 clones. (C) DDFL16 → JH1 CJs assessed in A2 and B5 clones transfected with empty (E) or Rag2 (R) expression constructs and the Mb1 loading control. (D, top) Treatment timeline in PA112.2 cells. (Bottom) Quantitative RT–PCR for AID and Rag2 expression. (E) Semiquantitative RT–PCR for PST γ2b and ɛ or Hprt. (F) Vκ6–23 → Jκ1 CJs and Mb1 assessed by semiquantitative PCR.
To determine whether class-switched pre-B cells could complete the developmental program of recombination, we assayed V–J joining at the light chain Igκ locus. PA112.2 cells (strain 129; Lκ, germline) were exposed to CSR inducers for 48 h, and switching was activated as indicated by increased expression of AID and PSTs γ2b or ɛ, which persisted for 8 h following withdrawal of the CSR inducers (Fig. 2D,E). In cells exposed to STI571, AID diminished, and Rag2 concomitantly increased, accompanied by active Vκ6–23–Jκ1 CJ formation (Fig. 2D,F) with appropriate junctional diversity (Supplemental Fig. S12). CSR induction had no effect on subsequent generation of Vκ6–23–Jκ1 CJs (Supplemental Fig. S13). Active PST expression following STI571 treatment indicates that populations of switched cells are fully capable of V(D)J joining (Fig. 2D,E). We conclude that B cells expressing BCR with secondary isotypes may emerge directly from the BM. Intriguingly, the CSR or V(D)J gene programs can be alternately expressed in response to specific inducers during early B-cell ontogeny.
The pattern of CSR in pro-B cells is skewed toward IgG2b and IgE and is atypical of mature B cells. Germline transcription in activated mature B cells is regulated by the spatial organization of the Igh locus through long-range chromatin contacts between the Eμ and 3′Eα enhancers and GLT promoters (Supplemental Fig. S14A; Wuerffel et al. 2007; Sellars et al. 2009). Could γ3:γ1 contacts in a developmental stage-specific chromatin structure restrict CSR in pro-B cells (Supplemental Fig. S14B)? Using the chromosome conformation capture (3C) carbon copy (5C) method for unbiased detection of chromatin contacts (Dostie et al. 2006), we simultaneously examined a matrix of 12,656 possible looping interactions in parallel across the entire Igh locus and focused here on the 220-kb CH domain spanning Eμ and 3′Eα (Supplemental Material; Supplemental Fig. S15; Supplemental Tables S2, S3). We detected abundant γ3:γ1 chromatin interactions (Fig. 3, orange circle), indicating a compact conformation in Rag2−/− pro-B cells that is absent in resting splenic B cells (Fig. 3). We examined the log2 ratio pro-B/resting B 5C interactions (represented by red [pro-B] and blue [resting B] in Fig. 3) and confirmed that γ3:γ1 interactions are elevated in pro-B cells (Fig. 3). We found that Eμ:γ2b−ɛ and Eμ:γ3–γ1 contacts are associated with pro-B and resting B cells, respectively, in accord with the pattern of potential GLT expression and CSR. In contrast, few long-range interactions were detected for a gene desert region on chromosome 5 (Supplemental Fig. S16).
Figure 3.
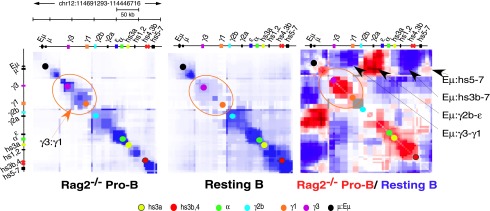
A developmentally stage-specific chromatin configuration of the Igh locus. 5C analyses of the Igh locus in Rag 2−/− pro-B cells and resting splenic B cells are represented in heat maps of 5C data (bin size 30 kb; step size 3.0 kb). Interaction frequencies are plotted against genomic distance. Blue intensity represents the contact frequency between genomic coordinates. The diagonal represents frequent proximal intrachromosomal interactions along the linear Igh locus. 5C signals off the diagonal indicate looping interactions. (Right panel) Log2 ratio of pro-B/resting B 5C interactions where pro-B (red) and resting B (blue) contact.
3C studies confirmed abundant γ3:γ1a (1.7-fold, P = 0.053) and γ3:γ1b (twofold, P = 0.01) chromatin contacts in Rag2−/− pro-B cells as compared with resting B cells or ConA-activated splenic T cells, where γ1a and γ1b are opposite ends of Hind III fragment C (Fig. 4A,B). Rag2−/− pro-B cells and the 445.3 pro-B-cell line display a similar profile of γ3:γ1 interactions, indicating that in early B cells, a compacted higher-order chromatin structure involving γ3:γ1 may limit that accessibility to the CSR machinery (Supplemental Fig. S17). In contrast, γ3:γ2b contacts observed in both Rag2−/− pro-B cells and wild-type resting B cells are similar to each other and may be related to a spatial organization that is absent in T cells (Fig. 4B).
Figure 4.

Absence of IgG3 and IgG1 CSR in pro-B cells is related to looping interactions between the γ3 and γ1 loci. (A) A segment of the Igh locus spanning 220 kb (shown at the top) in which 3C HindIII restriction fragments (A, B, C, D, and H) are shown. (B,C) 3C assays using chromatin from Rag2−/− pro-B cells, unstimulated (US) or LPS+CD40L (LC)-stimulated 445.3 cells, wild-type (WT) resting splenic B cells, or ConA-stimulated T cells. (*) P ≤ 0.05; (ns) Not significant. (B) 3C assays anchored at γ3 were analyzed for interactions with γ1 or γ2b. (C) Hs3b,4 (3′Eα) anchor was analyzed for interactions with Eμ, γ3, or γ2b.
Because interaction of distal Igh enhancers with the targeted CH locus is essential to bring participating S regions into close proximity (Wuerffel et al. 2007), we examined Rag2−/− pro-B and 445.3 cells for these chromatin contacts in 3C assays. 3′Eα:Eμ (H-A) interactions in Rag2−/− pro-B cells and resting splenic B cells are similarly abundant and significantly higher than in ConA-stimulated splenic T cells (Fig. 4C, left panel). When CSR is induced in LPS+IL4-activated B cells, 3′Eα:Eμ (H-A) interactions become elevated relative to resting B cells (Fig. 4C, left panel). Notably, when the 445.3 pro-B-cell line is activated with LPS+CD40L to initiate CSR, 3′Eα:Eμ (H-A) (P < 0.006) and hs3b,4:γ2b (P < 0.002) interactions significantly increase, whereas γ3:3′Eα interactions remain unchanged (Fig. 4C, right panel). Thus, our results demonstrate a unique three-dimensional chromatin structure in pro-B cells that supports isotype-specific CSR and appears to preclude γ3 and γ1 GLT expression and CSR in a developmental stage-specific fashion (Supplemental Fig. S14B).
Our findings establish that pro-B cells in the BM can undergo robust CSR, biased toward IgG2b and IgE, in response to specific stimuli followed by V(D)J recombination. Thus, pro-B cells that are delayed in the BM by genetic predisposition (as in Omenn syndrome) or environmental cues have the capacity to undergo CSR in response to bacterial infections and to eventually populate the periphery following V(D)J joining. Two additional conclusions flow from our findings. The origin of DNA damage reminiscent of both RAG and AID activity in some human leukemias (Tsai et al. 2008) has been difficult to explain. This footprint of DNA damage in leukemia may be related to coexpression of AID and RAG1/2 that we observed in pro-B cells induced to switch. Most notably, alternating expression of CSR versus V(D)J joining in early B cells reveals an unanticipated flexibility of these gene programs, with implications for our understanding of developmental and lineage commitment.
Materials and methods
In brief, mice were handled according to institutional and National Institutes of Health (NIH) guidelines. Standard protocols were used for cell culture, quantitative PCR, RT–PCR, DC-PCR, CT-PCR, retroviral transductions, statistical analyses, and switch, Vκ–Jκ, and DH–JH junction cloning. Abelson-MuLV transformed pro-B-cell or pre-B-cell lines were kindly provided by Dr. B. Sleckman (Washington University, St. Louis). 3C assays were carried out (Wuerffel et al. 2007) in combination with 5′FAM/ 3′BHQ1-modified probes. 5C primers were designed using online tools (http://my5C.umassmed.edu). 5C library construction was performed as described (Dostie et al. 2006). Additional methods are available in the Supplemental Material.
Acknowledgments
We thank A. Baumgart (The Scripps Research Institute), K.G. Becker, and W.H. Wood (Research Resources Branch, National Institute on Aging/National Institutes of Health) for technical help, and Dr. B. Sleckman (Washington University) for Abelson transformed cell lines. This work was supported by the National Institutes of Health (AI052400 to A.L.K., AI082918 to A.J.F., and HG003143 and HG00459 to J.D), the W.M. Keck Foundation (to J.D), and the Intramural Research Program of the National Institute on Aging (Baltimore, MD) (to R.S.).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.227165.113.
References
- Crouch EE, Li Z, Takizawa M, Fichtner-Feigl S, Gourzi P, Montano C, Feigenbaum L, Wilson P, Janz S, Papavasiliou FN, et al. 2007. Regulation of AID expression in the immune response. J Exp Med 204: 1145–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, et al. 2006. Chromosome conformation capture carbon copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Res 16: 1299–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan SK, Ogata S, Hu CC, Grotenbreg GM, Guillen E, Jaenisch R, Ploegh HL 2012. IgG1+ ovalbumin-specific B-cell transnuclear mice show class switch recombination in rare allelically included B cells. Proc Natl Acad Sci 109: 13739–13744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley DD, Manis JP, Zarrin AA, Kaylor L, Tian M, Alt FW 2002. Internal IgH class switch region deletions are position-independent and enhanced by AID expression. Proc Natl Acad Sci 99: 9984–9989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JH, Akira S, Calame K, Beutler B, Selsing E, Imanishi-Kari T 2007. Class switch recombination and somatic hypermutation in early mouse B cells are mediated by B cell and Toll-like receptors. Immunity 27: 64–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic M, Casellas R, Yannoutsos N, Wardemann H, Nussenzweig MC 2004. RAGs and regulation of autoantibodies. Annu Rev Immunol 22: 485–501 [DOI] [PubMed] [Google Scholar]
- Kenter AL 2012. AID targeting is dependent on RNA polymerase II pausing. Semin Immunol 24: 281–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraoka M, Liao D, Yang K, Allgood SD, Levesque MC, Kelsoe G, Ueda Y 2009. Activation-induced cytidine deaminase expression and activity in the absence of germinal centers: Insights into hyper-IgM syndrome. J Immunol 183: 3237–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson AJ, Lamarre A, McCoy K, Harriman GR, Odermatt B, Dougan G, Hengartner H, Zinkernagel RM 2001. IgA production without μ or δ chain expression in developing B cells. Nat Immunol 2: 625–631 [DOI] [PubMed] [Google Scholar]
- Mao C, Jiang L, Melo-Jorge M, Puthenveetil M, Zhang X, Carroll MC, Imanishi-Kari T 2004. T cell-independent somatic hypermutation in murine B cells with an immature phenotype. Immunity 20: 133–144 [DOI] [PubMed] [Google Scholar]
- Melamed D, Miri E, Leider N, Nemazee D 2000. Unexpected autoantibody production in membrane Ig-μ-deficient/lpr mice. J Immunol 165: 4353–4358 [DOI] [PubMed] [Google Scholar]
- Milili M, Fougereau M, Guglielmi P, Schiff C 1991. Early occurrence of immunoglobulin isotype switching in human fetal liver. Mol Immunol 28: 753–761 [DOI] [PubMed] [Google Scholar]
- Muljo SA, Schlissel MS 2003. A small molecule Abl kinase inhibitor induces differentiation of Abelson virus-transformed pre-B cell lines. Nat Immunol 4: 31–37 [DOI] [PubMed] [Google Scholar]
- Ozcan E, Notarangelo LD, Geha RS 2008. Primary immune deficiencies with aberrant IgE production. J Allergy Clin Immunol 122: 1054–1062 [DOI] [PubMed] [Google Scholar]
- Pelanda R, Braun U, Hobeika E, Nussenzweig MC, Reth M 2002. B cell progenitors are arrested in maturation but have intact VDJ recombination in the absence of Ig-α and Ig-β. J Immunol 169: 865–872 [DOI] [PubMed] [Google Scholar]
- Rolink A, Melchers F, Andersson J 1996. The SCID but not the RAG-2 gene product is required for Sμ–Sɛ heavy chain class switching. Immunity 5: 319–330 [DOI] [PubMed] [Google Scholar]
- Schatz DG, Ji Y 2011. Recombination centres and the orchestration of V(D)J recombination. Nat Rev Immunol 11: 251–263 [DOI] [PubMed] [Google Scholar]
- Scheeren FA, Nagasawa M, Weijer K, Cupedo T, Kirberg J, Legrand N, Spits H 2008. T cell-independent development and induction of somatic hypermutation in human IgM+ IgD+ CD27+ B cells. J Exp Med 205: 2033–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellars M, Reina-San-Martin B, Kastner P, Chan S 2009. Ikaros controls isotype selection during immunoglobulin class switch recombination. J Exp Med 206: 1073–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J 1996. Antibody class switching. Adv Immunol 61: 79–146 [DOI] [PubMed] [Google Scholar]
- Stavnezer J 2000. Molecular processes that regulate class switching. Curr Top Microbiol Immunol 245: 127–168 [DOI] [PubMed] [Google Scholar]
- Stavnezer J, Bjorkman A, Du L, Cagigi A, Pan-Hammarstrom Q 2010. Mapping of switch recombination junctions, a tool for studying DNA repair pathways during immunoglobulin class switching. Adv Immunol 108: 45–109 [DOI] [PubMed] [Google Scholar]
- Storb U, Roth P, Kurtz BK 1994. γ2b transgenic mice as a model for the role of immunoglobulins in B cell development. Immunol Res 13: 291–298 [DOI] [PubMed] [Google Scholar]
- Tsai AG, Lu H, Raghavan SC, Muschen M, Hsieh CL, Lieber MR 2008. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell 135: 1130–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y, Liao D, Yang K, Patel A, Kelsoe G 2007. T-independent activation-induced cytidine deaminase expression, class-switch recombination, and antibody production by immature/transitional 1 B cells. J Immunol 178: 3593–3601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Boehmer H, Melchers F 2010. Checkpoints in lymphocyte development and autoimmune disease. Nat Immunol 11: 14–20 [DOI] [PubMed] [Google Scholar]
- Waisman A, Kraus M, Seagal J, Ghosh S, Melamed D, Song J, Sasaki Y, Classen S, Lutz C, Brombacher F, et al. 2007. IgG1 B cell receptor signaling is inhibited by CD22 and promotes the development of B cells whose survival is less dependent on Igα/β. J Exp Med 204: 747–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller S, Faili A, Garcia C, Braun MC, Le Deist FF, de Saint Basile GG, Hermine O, Fischer A, Reynaud CA, Weill JC 2001. CD40–CD40L independent Ig gene hypermutation suggests a second B cell diversification pathway in humans. Proc Natl Acad Sci 98: 1166–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesemann DR, Magee JM, Boboila C, Calado DP, Gallagher MP, Portuguese AJ, Manis JP, Zhou X, Recher M, Rajewsky K, et al. 2011. Immature B cells preferentially switch to IgE with increased direct Sμ to Sɛ recombination. J Exp Med 208: 2733–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SY, Roth DB 2007. Murine models of Omenn syndrome. J Clin Invest 117: 1213–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuerffel R, Wang L, Grigera F, Manis J, Selsing E, Perlot T, Alt FW, Cogne M, Pinaud E, Kenter AL 2007. S–S synapsis during class switch recombination is promoted by distantly located transcriptional elements and activation-induced deaminase. Immunity 27: 711–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gostissa M, Hildebrand DG, Becker MS, Boboila C, Chiarle R, Lewis S, Alt FW 2010. The role of mechanistic factors in promoting chromosomal translocations found in lymphoid and other cancers. Adv Immunol 106: 93–133 [DOI] [PMC free article] [PubMed] [Google Scholar]