

While Cdc7 kinase plays an established role in DNA replication, its role in DNA repair and recombination is poorly understood. In this study, Yamada et al. elucidate how the Cdc7–ASK (Dbf4) kinase complex is regulated in response to stalled DNA replication. The authors show that stabilization of this complex depends on ATR–Chk1-mediated replication checkpoint signaling and its interplay with the ubiquitin–proteasome machinery. This study defines a novel mechanism to guard against replication collapse under conditions of replication stress.
Keywords: Cdc7, Chk1, APC/CCdh1, RAD18, replication stress, DNA lesion bypass
Abstract
Cdc7 kinase regulates DNA replication. However, its role in DNA repair and recombination is poorly understood. Here we describe a pathway that stabilizes the human Cdc7–ASK (activator of S-phase kinase; also called Dbf4), its regulation, and its function in cellular responses to compromised DNA replication. Stalled DNA replication evoked stabilization of the Cdc7–ASK (Dbf4) complex in a manner dependent on ATR–Chk1-mediated checkpoint signaling and its interplay with the anaphase-promoting complex/cyclosomeCdh1 (APC/CCdh1) ubiquitin ligase. Mechanistically, Chk1 kinase inactivates APC/CCdh1 through degradation of Cdh1 upon replication block, thereby stabilizing APC/CCdh1 substrates, including Cdc7–ASK (Dbf4). Furthermore, motif C of ASK (Dbf4) interacts with the N-terminal region of RAD18 ubiquitin ligase, and this interaction is required for chromatin binding of RAD18. Impaired interaction of ASK (Dbf4) with RAD18 disables foci formation by RAD18 and hinders chromatin loading of translesion DNA polymerase η. These findings define a novel mechanism that orchestrates replication checkpoint signaling and ubiquitin–proteasome machinery with the DNA damage bypass pathway to guard against replication collapse under conditions of replication stress.
Cdc7 was originally identified in budding yeast as a kinase essential for initiation of chromosomal DNA replication (Johnston et al. 1999; Masai and Arai 2002; Labib 2010). The Cdc7 kinase is conserved in evolution (Jiang and Hunter 1997; Sato et al. 1997; Kim et al. 1998), and, by analogy to yeast, mammalian Cdc7 interacts with a regulatory subunit called ASK (activator of S-phase kinase) or Dbf4 (referred to as ASK in this study) and phosphorylates proteins of the MCM2–7 complex, a component of the replicative helicase (Jiang et al. 1999; Kumagai et al. 1999; Yamashita et al. 2005). Phosphorylation of MCM proteins by Cdc7 has been implicated in triggering DNA replication origin firing through unwinding chromosomal DNA (Masai et al. 2010).
The level of yeast Dbf4 protein oscillates during the cell cycle, being absent during G1 and accumulating during the S and G2 phases. This oscillation is accomplished by both transcriptional and post-transcriptional regulation. In budding yeast, protein stability of Dbf4 is regulated by the cell cycle regulatory ubiquitin ligase anaphase-promoting complex/cyclosome (APC/C) (Weinreich and Stillman 1999; Ferreira et al. 2000). The level of human ASK protein fluctuates during the cell cycle, partly due to transcriptional regulation (Wu and Lee 2002; Yamada et al. 2002). In contrast, post-transcriptional mechanisms of Cdc7 and ASK protein abundance control are unknown in mammals.
As human Cdc7 phosphorylates Claspin, a factor essential for activation of checkpoint kinase Chk1, and Cdc7 activity contributes to Chk1 activation (Kim et al. 2008), mammalian Cdc7 may be involved in not only unperturbed DNA replication but also other aspects of DNA metabolism, such as DNA repair and recombination. Indeed, at least in yeast, the Cdc7 kinase has already been implicated in DNA repair and mutagenesis. Thus, mutants of cdc7 exhibit varying degrees of mutability in response to genotoxic insults, including UV light, ethylmethanesulfonate, nitrosoguanidine, and nitrogen mustard (Njagi and Kilbey 1982). Interestingly, CDC7 belongs to the RAD6 epistasis group (Njagi and Kilbey 1982; Pessoa-Brandao and Sclafani 2004) of genes that control a relatively poorly understood DNA repair pathway of translesion DNA synthesis and damage tolerance. While kinase activity and protein–protein interactions of the Cdc7 kinase complex have been proposed as candidate regulatory modes for DNA repair, the molecular mechanisms behind such regulation remain to be elucidated.
Overexpression of Cdc7–ASK occurs in various types of cancer (Hess et al. 1998; Nambiar et al. 2007; Bonte et al. 2008; Clarke et al. 2009; Kulkarni et al. 2009) and often correlates with poor prognosis, suggesting that deregulated Cdc7 kinase activity may promote survival of cancer cells and tumor progression. Recently, some small molecule inhibitors of Cdc7 kinase activity have been developed and are being tested in clinical trials as candidate anti-cancer drugs (Ito et al. 2008; Montagnoli et al. 2008; Vanotti et al. 2008; Ermoli et al. 2009).
Given the biological significance of the Cdc7–ASK complex, its emerging involvement in human pathology, and insufficient insights into the molecular mechanisms that regulate and in turn are impacted by the Cdc7 kinase activity, better understanding of Cdc7–ASK regulation and function is highly desirable. Here, we studied human cells to elucidate regulation of the Cdc7–ASK kinase complex and its biological function, with emphasis on cellular responses to DNA replication stress. Our present work revealed a multifaceted regulatory interplay of Cdc7–ASK with key checkpoint signaling and effector pathways, including cell cycle regulatory ubiquitylation and translesion DNA synthesis. The mechanism that we describe helps orchestrate responses of human cells to insults that impair DNA replication and threaten genomic integrity.
Results
Stabilization and chromatin binding of active Cdc7–ASK upon DNA replication block
Previous reports reached mutually contradictory conclusions about changes of chromatin binding, active complex formation, and activity of Cdc7 kinase upon genotoxic stress (Costanzo et al. 2000; Dierov et al. 2004; Tenca et al. 2007; Lee et al. 2012). To address this important issue, we first assessed the kinetics of chromatin binding of Cdc7–ASK in U2OS cells exposed to various genotoxic stresses. The amounts of both Cdc7 and ASK in a chromatin-enriched fraction (P3) increased during hydroxyurea (HU)-induced DNA replication block (Fig. 1A). Flow cytometry analysis confirmed that the HU-treated cells accumulated at the G1/S boundary and early S phase (Supplemental Fig. 1A). Similar results were obtained for two other cell lines treated by HU and upon treatment with another genotoxic agent, MMC, which also causes replication fork stalling but through another mechanism: DNA interstrand cross-links (Supplemental Fig. 1B). In contrast, upon X-ray irradiation, the chromatin-bound fraction of Cdc7–ASK remained unchanged despite a transient change of the cell cycle profile (Supplemental Fig. 1C–E).
Figure 1.

Stalled DNA replication stabilizes the active Cdc7–ASK kinase complex on the chromatin. (A) Stabilization of Cdc7 and ASK (Dbf4) proteins during HU treatment in U2OS cells. U2OS cells were treated with 2 mM HU for the indicated times, harvested, and subjected to biochemical cell fractionation. (Left) Samples were analyzed by immunoblotting. (Right) Quantification of Cdc7 and ASK protein in P3 fraction upon HU treatment. (B) Stabilization of Cdc7–ASK is not a consequence of S-phase arrest. U2OS cells were treated with 3 mM thymidine for 24 h and then released for 1 h followed by treatment with 0.5 mM HU or mock for the indicated times. Cells were subjected to whole-cell lysis (left) or biochemical fractionation (right) and then analyzed by immunoblotting. (C) The Cdc7–ASK complex on the chromatin after HU treatment. U2OS cells with or without HU treatment were biochemically fractionated, and chromatin-bound proteins were extracted by sonication in immunoprecipitation buffer. Cell lysates of S2 and P3 fractions were immunoprecipitated using control rabbit IgG or anti-ASK antibody, followed by immunoblotting. (*) A nonspecific band. (D) Cdc7 kinase is active under replication stress. Asynchronous U2OS cells were treated or not with 15 μM PHA-767491 for 3 h. U2OS cells were also treated with 2 mM HU for the indicated times with or without 15 μM PHA-767491 for the last 3 h. Whole-cell lysates were analyzed by immunoblotting. (E) Cdc7-dependent phosphorylation of MCM2 upon replication block. U2OS-shCdc7 cells grown with or without doxycycline (Dox) for 3 d were treated or not with 2 mM HU for 24 h. Whole-cell lysates (left) and P3 fractions (right) were analyzed by immunoblotting. (F) Cdc7 kinase activity is required for stabilization of ASK. U2OS cells were preincubated with or without 1.5 μM PHA-767491 for 1h and treated with 2 mM HU or 3 M cisplatin for 24 h, and whole-cell lysates were analyzed by immunoblotting.
To ensure that the stabilization of Cdc7–ASK upon replication block did not simply reflect a higher fraction of S-phase cells, we performed cell cycle synchronization experiments with or without replication block (Fig. 1B). U2OS cells were synchronized at the G1/S boundary by a thymidine block and then allowed to progress into S phase for 1 h followed by treatment with 0.5 mM HU to block DNA replication before the cells were harvested at various time points. As expected, in both whole-cell extracts (Fig. 1B, left) and the chromatin-enriched fraction (Fig. 1B, right), we observed stabilization of Cdc7–ASK under replication stress. Parallel immunoprecipitation experiments confirmed that the abundance of the Cdc7–ASK complex bound to chromatin increased upon HU treatment (Fig. 1C). Notably, ASK remained phosphorylated in cells under replication stress evoked by either HU or MMC (Supplemental Fig. 1F). Given that ASK is mainly autophosphorylated by Cdc7 (Supplemental Fig.1G; Kumagai et al. 1999), this result suggested that the Cdc7 kinase is active on chromatin during DNA replication blockade. We also found that acute inhibition of Cdc7 kinase activity by a small molecule inhibitor (PHA-767491) induced rapid dephosphorylation of ASK and MCM2 at Ser53—both modifications that depend on Cdc7–ASK kinase activity (Supplemental Fig. 1G,H)—in an unperturbed cell cycle as well as under replication stress (Fig. 1D). Furthermore, phosphorylation of MCM2 at Ser53 was increased upon HU treatment but abolished in Cdc7- or ASK-depleted cells (Fig. 1E; Supplemental Fig. 1I). Chemical inhibition or RNAi-mediated knockdown of Cdc7 prevented accumulation of ASK and impaired ASK phosphorylation (Fig. 1D–F). The latter became particularly apparent from accumulation of hypophosphorylated ASK in Cdc7-depleted cells exposed to proteasome inhibition (Supplemental Fig. 1J). From these results, we concluded that, under replication stress conditions, an active form of the Cdc7–ASK kinase complex becomes stabilized and accumulates on chromatin.
ATR–Chk1 signaling is required to stabilize Cdc7–ASK
As the ATR–Chk1 checkpoint signaling is activated and contributes to responses to replication stress (Bartek and Lukas 2003), we asked whether the ATR–Chk1 cascade regulates stabilization of Cdc7–ASK upon replication block. Knockdown of ATR impaired stabilization of Cdc7–ASK under replication stress conditions without major impact on cell cycle profiles compared with control ATR-proficient cells (Fig. 2A; Supplemental Fig. 2A,B). Next, using gene silencing by shRNA or cep-3891, a small molecule inhibitor of Chk1 kinase (Syljuasen et al. 2004, 2005), we observed Chk1 kinase-dependent stabilization of the Cdc7–ASK complex upon HU treatment (Fig. 2B,C), independently of cell cycle profiles (Supplemental Fig. 2C,D). In contrast to ATR–Chk1 signaling, knockdown of Chk2 kinase did not affect accumulation of Cdc7–ASK upon HU or MMC treatment (Supplemental Fig. 2E). These results showed that the ATR–Chk1 pathway is required for stabilization of the Cdc7–ASK complex, including its chromatin fraction, upon replication fork stalling.
Figure 2.

ATR–Chk1 signaling is required for stabilization of Cdc7–ASK upon replication block. (A) Impaired stabilization of Cdc7–ASK in ATR-depleted cells. U2OS-shATR cells were grown with or without Dox for 2 d followed by MMC treatment for the indicated times. Harvested cells were subjected to whole-cell lysis or biochemical fractionation and then analyzed by immunoblotting. (B) Impaired stabilization of Cdc7–ASK in Chk1-depleted cells. U2OS-shChk1 cells were grown with or without Dox for 2 d and then HU-treated, harvested, biochemcally fractionated, and analyzed by immunoblotting. (C) Impaired stabilization of Cdc7–ASK upon inhibition of Chk1 activity. U2OS cells were preincubated or not with cep-3891 for 2 h followed by HU treatment, and harvested cells were biochemically fractionated and analyzed by immunoblotting.
Cdc7–ASK complex interacts with Cdh1 and is targeted for degradation by APC/CCdh1
In budding yeast, protein stability of Dbf4 is regulated by the APC/C ubiquitin–proteasome pathway (Weinreich and Stillman 1999; Ferreira et al. 2000). Therefore, we next examined whether APC/C is involved in the stabilization of Cdc7–ASK upon replication block in human cells. First, we confirmed a moderate increase of ASK upon treatment with a proteasome inhibitor, MG132 (Supplemental Fig. 3A). Since ASK protein is absent in cells during the G0–G1 phase, accumulates at the G1/S transition, and declines between late M and early G1 (Supplemental Fig. 3B,C), we hypothesized that ASK may be a target of the APC/CCdh1 ubiquitin ligase that is active in G0/G1 (Skaar and Pagano 2008) and becomes inactivated through phosphorylation of Cdh1 by cyclin-dependent kinases (Cdks) from the G1/S boundary to early mitosis (Zachariae et al. 1998; Lukas et al. 1999; Listovsky et al. 2000). To test this hypothesis, we assessed a potential interaction between Cdh1 and Cdc7–ASK. As shown in Figure 3A, endogenous Cdc7 and ASK bound to Flag-Cdh1. Next, we showed that depletion of Cdh1 led to increased abundance of both Cdc7 and ASK (Fig. 3B; Supplemental Fig. 3D). Although we observed an increase of S-phase population by ∼20% among Cdh1-depleted cells compared with control cells (Supplemental Fig. 3E,F), the dramatic increase of chromatin-bound ASK in Cdh1-depleted cells (∼2.8 times more, an increase of 180%) could not be attributed to indirect cell cycle effects and suggested that human ASK is a direct target for the APC/CCdh1 pathway, analogous to yeast. In addition, we observed that Cdh1 protein abundance decreased upon HU treatment, likely leading to stabilization of Cdc7–ASK under replication stress (Fig. 3B). Furthermore, using a cell line with inducible expression of myc-Cdh1, we confirmed that ectopic myc-Cdh1 prevented the stabilization of the Cdc7–ASK complex upon HU treatment (Fig. 3C) despite the fact that the ectopic myc-Cdh1 did not dramatically alter the cell cycle profile compared with control cells, likely due to the Cdh1 induction for only a short time and the accumulation of cells at the G1/S boundary upon HU treatment (Supplemental Fig. 3G). Importantly, depletion of Cdh1 stabilized ASK and Cdc7 in U2OS cells, as documented by slower protein turnover upon cycloheximide treatment (Fig. 3D).
Figure 3.
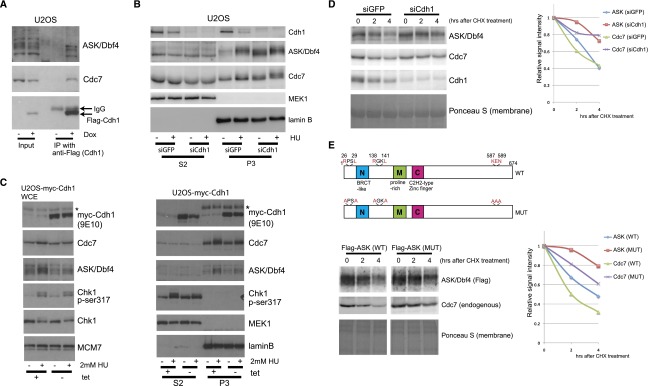
Stabilization of Cdc7–ASK is regulated by APC/CCdh1. (A) Interaction of Cdh1 with Cdc7–ASK. Lysates from a stable U2OS cell line inducibly expressing Flag-Cdh1, grown with or without Dox for 2 d, were immunoprecipitated using anti-Flag antibody and analyzed by immunoblotting. (B) Chromatin-bound ASK increases in Cdh1-depleted cells. U2OS cells were transfected with control siRNA (siGFP) or siRNA for Cdh1 (siCdh1); 24 h later, the cells were treated or not with 2 mM HU for 24 h and then biochemically fractionated and analyzed by immunoblotting. (C) Impaired stabilization of Cdc7–ASK by ectopic Cdh1. U2OS cells inducibly expressing myc-Cdh1, grown with or without tetracycline for 2 d, were treated or not with 2 mM HU for 24 h, subjected to whole-cell lysis (left) or biochemical fractionation (right), and analyzed by immunoblotting. (*) Endogenous c-myc. (D) Cdc7 and ASK proteins are stabilized in Cdh1-depleted cells. U2OS cells transfected with siGFP or siCdh1 for 24 h were treated with 1 μg/mL cycloheximide (CHX) for the indicated times followed by cell lysis and immunoblot analysis. (Left) Ponceau S staining served as a loading control. (Right) Quantification of the signal intensity. (E) ASK protein mutated in two D boxes and the KEN box is more stable. (Top panel) Schematic presentation of D boxes and the KEN box in human ASK. (Left) U2OS cells transfected to express Flag-tagged ASK (wild-type or mutant) were treated with 1 μg/mL CHX for the indicated times, lysed, and analyzed by immunoblotting. (Right) Quantification of the signal intensity.
Substrates of APC/CCdh1 harbor so-called D-box and/or KEN-box recognition sequences (van Leuken et al. 2008). Indeed, we identified two putative D boxes and one KEN box in ASK (Fig. 2E, top panel) and validated these by demonstrating that ASK protein mutated in all three recognition sequences was more stable than wild-type ASK (Fig. 3E, bottom panel). Interestingly, endogenous Cdc7 protein was also more stable in cells expressing such mutant ASK, probably due to complex formation with the more stable ASK. These results showed that the human Cdc7–ASK complex is a new target for the APC/CCdh1 pathway as well as for the ATR–Chk1 signaling.
Chk1 kinase activity is required for inactivation of APC/CCdh1 upon replication stress
Given the paucity of knowledge about any relationship between replication checkpoint signaling and APC/CCdh1-mediated proteolysis and our observation of decreased Cdh1 upon HU treatment (Fig. 3B), we next analyzed the kinetics of Cdh1 protein abundance in response to replication stress. Abundance of Cdh1 in U2OS cells decreased during treatments with HU (Fig. 4A, left), MMC, cisplatin (CIS), and camptothecin (CPT) (Fig. 4A, right), and the decrease of Cdh1 upon HU treatment was also confirmed in MRC5 and HeLa cells (Fig. 4B). In these experiments, checkpoint impact was documented by inactivation of Cdks due to degradation of Cdc25A phosphatase and phosphorylation of Cdks at Tyr15 (Fig. 4B).
Figure 4.

Chk1 kinase activity stabilizes the Cdc7–ASK complex through inactivation of APC/CCdh1 upon replication block. (A) Decreased Cdh1 protein level upon replication block. (Left) The cell lysates used also in Figure 1A were used for immunoblotting. (Right) U2OS cells were treated with 1 μM MMC, 2 μM CIS, or 1 μM CPT for 24 h; lysates were analyzed by immunoblotting. (B) Activation of the replication checkpoint pathway in MRC5 and HeLa cells. MRC5 (left) and HeLa (right) cells were treated with 2 mM HU for the indicated times. Whole-cell lysates were analyzed by immunoblotting. (C) The APC/C pathway in Cdh1 degradation upon replication block. U2OS cells were transfected with GFP siRNA or Cdc27 siRNA. After 24 h, cells were exposed to 2 mM HU for 24 h. Whole-cell lysates were analyzed by immunoblotting. (D) Chk1 kinase activity is required for degradation of Cdh1 upon replication block. U2OS-shChk1 cells were grown with or without Dox for 2 d and then treated with 2 mM HU for 24 h. (Left) Whole-cell lysates were analyzed by immunoblotting. HeLa cells preincubated with or without cep-3891 for 2 h were treated or not with 2mM HU for 24 h. (Right) Whole-cell lysates were analyzed by immunoblotting. (E) Stabilization of Cdc7–ASK requires Chk1 activation. U2OS-shChk1 cells reconstituted with HA-tagged wild-type (left) or mutant Chk1 (S317A [middle] or S345A [right]) were grown with or without Dox for 2 d and then treated or not with HU for 24 h, and cell lysates were analyzed by immunoblotting. (F) Depletion of Cdh1 restabilizes Cdc7–ASK in Chk1-depleted cells. U2OS-shChk1 cells were transfected with siGFP or siCdh1. After 24 h, cells were grown with or without Dox for 2 d and then treated or not with HU for 24 h, and cell lysates were analyzed by immunoblotting.
Since Cdh1 can promote its own degradation during unperturbed cell cycles (Listovsky et al. 2004), we examined such a scenario upon replication block. Indeed, knockdown of Cdc27, an essential component of APC/C, impaired the HU-evoked decrease of Cdh1 protein abundance (Fig. 4C), while similar cell cycle profiles were seen in control siRNA (siGFP)-treated and siCdc27-treated cells, likely due to a relatively short-term Cdc27 depletion combined with S-phase accumulation of HU-treated cells (Supplemental Fig. 4A). These results strongly suggested that the activated checkpoint inhibits the APC/CCdh1 pathway by promoting Cdh1 autodegradation upon replication stress because otherwise the APC/CCdh1 could potentially become active even in S phase due to checkpoint-induced inhibition of Cdk kinases. Indeed, we found that shRNA-mediated Chk1 knockdown or chemical inhibition of Chk1 activity impaired the decrease of Cdh1 in response to HU or CIS (Fig. 4D; Supplemental Fig. 4B). Consistently, known substrates of APC/CCdh1, including ASK, Cdc6, and Cyclin A, did not accumulate in these cells upon HU treatment. In contrast, Cyclin E, which is not a target of APC/CCdh1, remained unaffected by Chk1 inhibition (Fig. 4D). In complementation assays, inducible ectopic HA-tagged wild-type Chk1, but not Chk1 mutants defective in activatory phosphorylations (S317A or S345A), could fully rescue the ability to inactivate APC/CCdh1 in cells depleted of endogenous Chk1 (Fig. 4E). Furthermore, Cdh1 depletion restored the stabilization of Cdc7–ASK upon replication block in Chk1-depleted cells (Fig. 4F). These findings indicate that Chk1 signaling in response to replication stress promotes APC/C-mediated degradation of Cdh1, thereby inactivating the APC/CCdh1 pathway.
Cdc7–ASK kinase is required for activation of DNA damage bypass
Reports on the involvement of yeast Cdc7 in translesion synthesis (Njagi and Kilbey 1982; Pessoa-Brandao and Sclafani 2004; Dolan et al. 2010) and Cdc7-dependent phosphorylation of RAD18 being required for recruitment of DNA polymerase η (Pol η) to stalled replication forks (Day et al. 2010) led us to examine the potential link between Cdc7–ASK and translesion synthesis in human cells. First, we confirmed that endogenous Cdc7–ASK interacted with RAD18 in the chromatin (Fig. 5A), consistent with a previous study (Day et al. 2010). In 293T cells, Cdc7 showed only weak interaction with RAD18 compared with a robust ASK–RAD18 complex (Supplemental Fig. 5A). Coexpression of ASK enhanced the complex formation between Cdc7 and RAD18 (Supplemental Fig. 5B), suggesting that Cdc7–ASK and RAD18 form a complex through the interaction between ASK and RAD18.
Figure 5.
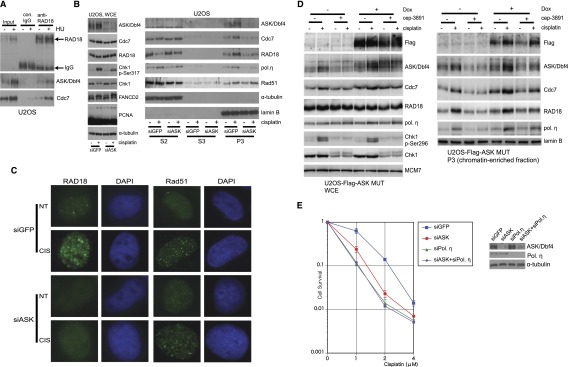
ASK interacts with RAD18 and is required for chromatin accumulation of Rad18 and DNA Pol η upon replication block. (A) Endogenous Cdc7–ASK and RAD18 interact in vivo. U2OS cells treated or not with 2 mM HU for 24 h were lysed with buffer A, and nuclei were collected, suspended in immunoprecipitation buffer, sonicated, and centrifuged. The lysates were immunoprecipitated using control rabbit IgG or anti-RAD18 antibody. Samples were analyzed by immunoblotting. (B) Depletion of ASK impairs chromatin binding of RAD18 and Pol η. U2OS cells were transfected with siGFP or siASK (siASK-a). After 48 h, cells were treated or not with 3 μM CIS for 24 h, and whole-cell lysates (left) or biochemical fractions (right) were analyzed by immunoblotting. (C) Depletion of ASK impairs RAD18 foci formation. U2OS cells were transfected with siGFP or siASK, replated for 48 h, treated or not with 3 μM CIS for 24 h, pre-extracted with buffer A, fixed with 4% formaldehyde, and stained for RAD18 or Rad51. Nuclei were stained with DAPI. (D) Overexpression of stable ASK mutant partly rescues chromatin binding of RAD18 and Pol η in Chk1-inhibited cells. U2OS-Flag-ASKMUT cells were grown with or without Dox for 24 h and then treated with 6 μM CIS with or without cep-3891 for 24 h and subjected to whole-cell lysis (left) or biochemical fractionation (right). The samples were analyzed by immunoblotting. Inhibition of Chk1 activity was confirmed by impaired autophosphorylation of Chk1 at Ser296. (E) ASK-depleted cells are hypersensitive to CIS treatment. U2OS cells were transfected with siGFP, siASK, siPol η, or siASK and siPol η (siASK+siPol η). After 48 h, cells were replated at a density of 1 × 103 per 6-cm dish and treated with CIS for 7 d. Viable cells were counted; values were normalized to that of concentration 0. (Left) Experiments were repeated three times (n = 3, average ± SD). (Right) Confirmation of depletion of ASK and Pol η.
Next, we established that monoubiquitination of PCNA upon CIS treatment and the abundance of RAD18 remained unchanged in ASK-depleted cells (Fig. 5B, left). Notably, chromatin binding of RAD18 was diminished upon ASK knockdown (Fig. 5B, right; Supplemental Fig. 5C). Thus, whereas RAD18 was redistributed from the soluble fraction (S2) to the chromatin-enriched fraction (P3) upon replication block evoked by CIS or MMC treatment in control cells, such redistribution was impaired in ASK-depleted cells, indicating that the ASK–RAD18 interaction is required for chromatin accumulation of RAD18 upon replication stress. Conversely, overexpression of ASK in U2OS cells increased chromatin binding of RAD18 without significantly affecting the cell cycle profile (Supplemental Fig. 5D,E). Furthermore, ASK depletion also impaired chromatin loading of Pol η upon CIS or MMC treatment (Fig. 5B, right; Supplemental Fig. 5C) and foci formation of RAD18 in such cells (Fig. 5C; Supplemental Fig. 5F). In contrast, chromatin binding and foci formation of Rad51, an essential factor for homologous recombination (HR), were not diminished in ASK-depleted cells (Fig. 5B,C; Supplemental Fig. 5G). In fact, ASK depletion resulted in a modest but significant enhancement of spontaneous Rad51 foci formation (Supplemental Fig. 5G).
Addressing the role of Chk1 signaling, we noticed that chromatin loading of both RAD18 and Pol η was impaired upon depletion of Chk1 (Supplemental Fig. 5H, right) despite the fact that monoubiquitination of PCNA, which is necessary for Pol η recruitment, was even enhanced both with and without CIS treatment (Supplemental Fig. 5H, left). These results were consistent with previous reports (Watanabe et al. 2004; Day et al. 2010) in that binding of Pol η to monoubiquitinated PCNA is necessary but not sufficient for chromatin binding of Pol η, for which interaction with RAD18 is also required. Looking for a mechanistic explanation of these results, we found that ectopic expression of a stable mutant ASK protein in which the two D boxes and the KEN box were mutated (Fig. 3E) partially rescued chromatin loading of RAD18 and Pol η upon CIS treatment in Chk1-inhibited cells (Fig. 5D). This finding suggested that Chk1-dependent stabilization of ASK plays a pivotal role in DNA lesion bypass. Importantly, while ASK-depleted cells were more sensitive to CIS treatment than control cells, hypersensitivity of Pol η-depleted cells was not enhanced by codepletion of ASK (Fig. 5E), indicating an epistatic relationship. We propose that the hypersensitivity of ASK-depleted cells reflects the inefficient chromatin loading of Pol η, a key component ensuring cellular tolerance to CIS treatment (Albertella et al. 2005; Chen et al. 2006; Hicks et al. 2010). It should be noted that ASK depletion itself did not induce any pronounced cell death phenotype (data not shown). Given that Cdc7 kinase activity is required for stabilization of ASK (Fig. 1E,F; Supplemental Fig. 1H,J), our data suggest that Chk1-dependent stabilization of an active Cdc7–ASK kinase selectively regulates RAD18-dependent DNA damage bypass without affecting the Rad51-dependent HR pathway.
The above results prompted us to clarify the mechanism for ASK-dependent chromatin accumulation of RAD18. To identify the region of ASK that interacts with RAD18, we coexpressed RAD18-myc together with various deletion mutants of Flag-tagged ASK in human cells (Fig. 6A) and found that motif C of ASK was required for the ASK–RAD18 binding (Fig. 6B,C). Motif C is highly conserved throughout evolution and interacts with Cdc7 (Kumagai et al. 1999; Masai and Arai 2000; Ogino et al. 2001; Sato et al. 2003). In addition, yeast mutants in motif C are hypersensitive to the alkylating agent methylmethansulphonate (Fung et al. 2002; Gabrielse et al. 2006; Dolan et al. 2010). Next, we examined whether Flag-ASKmotif C, Cdc7, and RAD18 can form a trimeric complex. As shown in Supplemental Figure 6A, interaction between Cdc7 and RAD18 was enhanced by coexpression of Flag-ASKmotif C. Notably, Cdc7 did not compete with RAD18 for binding to ASKmotif C (Supplemental Fig. 6B), suggesting that Cdc7 and RAD18 can bind to motif C of ASK simultaneously. In a reciprocal approach, we also identified the region of RAD18 required for ASK binding using a series of RAD18 deletion mutants (Supplemental Fig. 6C, top panel) and found that the N-terminal region containing the RING finger domain is responsible for ASK binding (Supplemental Fig. 6C, bottom panel). To clarify the significance of the interaction between RAD18 and motif C of ASK, we established a cell line inducibly expressing the Flag-ASKmotif C fragment to see whether chromatin binding of RAD18 might be impaired by a dominant-negative effect of such an ectopically expressed motif C. Whole-cell lysate analyses showed that the ASK level and phosphorylation of MCM2 at Ser53 remained largely unaffected by expression of the Flag-ASKmotif C fragment (Fig. 6D, left), suggesting that endogenous Cdc7 kinase activity is not impaired by the ectopic motif C fragment. Interestingly, the Cdc7 protein level was increased by expression of the Flag-ASKmotif C fragment. Biochemical fractionation revealed that the Flag-ASKmotif C fragment was exclusively localized in the S2 fraction and that both redistribution of RAD18 from S2 to P3 and chromatin binding of Pol η upon CIS treatment were impaired (Fig. 6D, right). As we confirmed that the Flag-ASKmotif C fragment localized in nuclei by immunostaining (Supplemental Fig. 6D) and that this did not affect the cell cycle profile (Supplemental Fig. 6E), our results suggest that the Flag-ASKmotif C fragment prevents chromatin binding of RAD18. We confirmed an interaction between Flag-ASKmotif C and endogenous RAD18 (Fig. 6E), supporting our idea that interaction between RAD18 and ASK via its motif C is required for chromatin binding of RAD18 upon replication block. As a control, we also established a cell line inducibly expressing the Flag-ASKmotif N (Supplemental Fig. 6F), a fragment that did not interact with RAD18 (Supplemental Fig. 6G). As shown in Supplemental Figure 6H, ectopic expression of Flag-ASKmotif N did not affect chromatin binding of RAD18 and Pol η. Finally, we found that Flag-ASKmotif C-expressing cells were hypersensitive to CIS treatment (Fig. 6F), likely due to impaired RAD18–Pol η-dependent damage bypass (Yamashita et al. 2002; Albertella et al. 2005; Nojima et al. 2005; Chen et al. 2006). Such interpretation was consistent with the observed epistatic impact of Pol η depletion and expression of Flag-ASKmotif C on hypersensitivity to CIS treatment in human cells (Supplemental Fig. 6I).
Figure 6.

Motif C of ASK binds to RAD18, and ectopic Flag-motif C inhibits chromatin binding of RAD18. (A) Schematic representation of human ASK and its deletion mutants. (B) Mapping the region in ASK for interaction with RAD18. 293T cells were transfected with pCAGGS-RAD18-myc alone or with pcDNA4/TO-Flag-ASK or a deletion mutant; cells harvested 48 h later were subjected to immunoprecipitation using anti-Flag antibody. Samples were analyzed by immunoblotting. (C) Motif C of ASK interacts with RAD18. 293T cells were transfected with pCAGGS-RAD18-myc alone or with pcDNA4/TO-Flag-ASKmotif C (Flag-motif C) and harvested 48 h later, and lysates were immunoprecipitated using anti-Flag antibody. Samples were analyzed by immunoblotting. (D) Ectopically expressed Flag-motif C inhibits chromatin binding of RAD18. U2OS-Flag-ASKmotif C cells were grown with or without Dox for 3 d, treated or not with 3 μM CIS for 24 h, and lysed with 1× Laemmli sample buffer (LSB) (left) or biochemically fractionated (right). Samples were analyzed by immunoblotting. (E) Ectopically expressed Flag-motif C interacts with endogenous RAD18. U2OS-Flag-ASKmotif C cells were grown with or without Dox for 3 d, lysed with immunoprecipitation buffer without sonication, and immunoprecipitated using anti-Flag antibody. Samples were analyzed by immunoblotting. (F) Flag-motif C-expressing cells are hypersensitive to CIS. U2OS-Flag-ASKmotif C cells were grown with or without Dox for 3 d, replated at a density of 1 × 103 per 6-cm dish, and treated with CIS for 7 d, and viable cells were counted. Values were normalized to that of concentration 0. Experiments were repeated three times (n = 3, average ± SD). (G) The proposed model of orchestrated response to replication stress (see the text for details).
Discussion
The results of this study advance our understanding of cellular responses to replication stress by providing insights into the following three issues: (1) the currently conflicting topic of the activity of the Cdc7–ASK (Dbf4) kinase upon replication block, (2) identification of ATR–Chk1-mediated silencing of the APC/CCdh1 protein turnover pathway and thereby stabilization of ASK by the replication checkpoint, and (3) discovery of an unexpected positive role of Cdc7–ASK in the DNA lesion bypass pathway through chromatin recruitment of RAD18. The model that integrates these findings and their significance within the broader context of the field are discussed below.
Cdc7 kinase activity in human cells under replication stress
Given its essential role in initiation and progression of DNA replication, it is reasonable to assume that the kinase activity of Cdc7 is down-regulated by the DNA replication checkpoint to prevent late origin firing. Indeed, several laboratories reported that the Cdc7 kinase activity is decreased upon replication fork stalling in yeast and Xenopus egg extracts (Weinreich and Stillman 1999; Costanzo et al. 2003; Zegerman and Diffley 2010). Here we show that, somewhat unexpectedly, in human somatic cells, an active Cdc7–ASK kinase complex resides on chromatin upon DNA replication block. Our data are consistent with some previous observations (Tenca et al. 2007; Lee et al. 2012). For example, Lee et al. (2012) reported that human ASK is phosphorylated by ATM/ATR upon various genotoxic insults, and such phosphorylation is involved in the checkpoint response; however, the investigators failed to detect suppression of Cdc7 kinase activity. Collectively, these studies raise the intriguing question of how such seemingly conflicting results can be reconciled. While interspecies differences and distinct properties of oocytes versus somatic cells cannot be entirely excluded, we propose an alternative explanation, also considering the high degree of conservation of the Cdc7 kinase complex throughout evolution. Thus, we speculate that at least two spatiotemporally and functionally distinct fractions of Cdc7 kinase may exist in cells exposed to replication stress: (1) a mobile fraction that is inhibited and thereby contributes to the silencing of late replication origin firing (Zegerman and Diffley 2010) and (2) a chromatin-bound active Cdc7–ASK (Dbf4) complex that locally promotes translesion synthesis at stalled replication forks (this study). It is also possible that the global Cdc7–ASK kinase activity becomes transiently inhibited during the initial response to replication stress to prevent late origin firing, yet a fraction of the Cdc7–ASK complex may be reactivated during prolonged replication stress to facilitate, for example, dormant origin firing and damage bypass. In either case, it is intriguing that the activity of the ATR/Chk1 (and their yeast orthologs) would directly phosphorylate Cdc7/ASK (Dbf4) to inhibit its activity in one fraction while also triggering degradation of Cdh1 and hence stabilization of ASK and the activity of the chromatin-bound Cdc7/ASK described in our present study. Experimental validation of the concept of a dual role of spatiotemporally distinct subsets of Cdc7 could explain the apparent discrepancies in the field.
Links among ATR–Chk1 signaling, Cdc7–ASK, and APC/CCdh1-mediated protein turnover
One of the novel findings in our present study is the identification of a signaling pathway that connects the ATR–Chk1 checkpoint to the APC/CCdh1 protein degradation machinery. In unperturbed cell cycles, APC/CCdh1 is inactivated by Cdk-mediated phosphorylation of Cdh1 from the G1/S boundary to early mitosis (Zachariae et al. 1998; Lukas et al. 1999; Listovsky et al. 2000; Sorensen et al. 2001). When the replication fork is stalled, the replication checkpoint becomes activated, and, subsequently, Cdk activity is down-regulated through targeting of the Cdc25A phosphatase (Bartek et al. 2004). In the latter scenario of Cdk silencing, the APC/CCdh1 would be expected to become activated during S phase, when it is normally inactive. We show that such “inappropriate” activation of APC/CCdh1 is prevented by ATR/Chk1 activity-promoted degradation of Cdh1 (see the model in Fig. 6G), as documented by the impact of the Chk1 inhibitor and phenotypes of the Chk1 phospho-mutants S317A or S345A. Our data implicate Chk1 activity in autodegradation of Cdh1, likely through phosphorylation of components of the APC/C complex, including Cdh1 itself. This scenario is supported by the recent identification of APC1 as a Chk1 substrate (Blasius et al. 2011). In contrast to the replication checkpoint, the DNA damage checkpoint, induced by ionizing radiation (IR), activates APC/CCdh1 during S/G2 in order to prevent mitotic entry of cells with damaged DNA (Sudo et al. 2001; Bassermann et al. 2008). Consistently, we did not observe stabilization of Cdc7–ASK upon IR despite ATR–Chk1 activation. It is unclear why the APC/CCdh1 pathway is differently regulated upon replication stress and IR-inflicted DNA double-strand breaks, but we speculate that the APC/CCdh1 pathway may contribute to the choice of DNA damage repair pathways. RAD18 regulates not only lesion bypass but also the HR pathway (Szuts et al. 2006; Saberi et al. 2007; Huang et al. 2009). Since our results show that stabilization of active Cdc7–ASK kinase through inactivation of APC/CCdh1 is required for RAD18-dependent lesion bypass, activation of APC/CCdh1 upon IR may be required for the RAD18-dependent HR. Interestingly, factors involved in DNA repair choice, such as 53BP1 and Rap80, are among candidate targets for APC/CCdh1 (Adams and Carpenter 2006; Cho et al. 2012). Relevant to human disease, down-regulation of Cdh1 correlates with poor survival of glioma patients, possibly due to the impaired G2 checkpoint (Bassermann et al. 2008). We show here that depletion of Cdh1 leads to increased abundance of chromatin-bound ASK (Fig. 3B), and overexpression of ASK enhances chromatin binding of RAD18 (Supplemental Fig. 5D). Based on these results, we propose that cancer-associated overabundance/activity of ASK may contribute to poor survival of cancer patients through accentuation of the RAD18-dependent error-prone damage bypass pathway, thereby increasing the mutation rate and hence the adaptability and treatment resistance of tumor cells.
Cdc7–ASK kinase regulates the DNA damage bypass pathway
Another conceptually important finding in our present study is the mechanistic insight into orchestration of the replication checkpoint and the DNA damage bypass pathway. Our data strongly indicate that the ATR–Chk1 cascade and RAD18-dependent damage bypass are linked through the Cdc7–ASK kinase in human cells (Fig. 6G). Genetic studies in yeast suggest that Cdc7 activity is required for DNA repair; however, the basis of such a link in yeast and any such connection in mammalian cells has remained speculative. Recently, Cdc7 was reported to phosphorylate a serine cluster in the Pol η-binding motif of RAD18, which is necessary for recruitment of Pol η to the sites of replication fork stalling (Day et al. 2010). The latter study concluded that the Cdc7-dependent RAD18 phosphorylation is not required for chromatin loading of RAD18. Our present results established that an interaction between the ASKmotif C and the N-terminal region of RAD18 is essential for chromatin association of RAD18. These results strongly indicate that Cdc7, ASK, and RAD18 form a trimeric complex in which the ASKmotif C provides an interaction platform for both Cdc7 and RAD18. This interaction likely facilitates not only a Cdc7–ASK-mediated phosphorylation of RAD18 to prime the latter for interaction with Pol η but also the recruitment of the RAD18–Pol η complex to stalled replication forks. In addition, our finding that the N-terminal region of RAD18 containing the RING domain interacts with ASK is quite intriguing given that FANCD2 and Rad51C also interact with the RING domain of RAD18 (Huang et al. 2009; Williams et al. 2011). It is possible that a binding partner of RAD18 may determine the optimal DNA damage response pathway upon distinct genotoxic insults. Overall, our results and those of Day et al. (2010) help elucidate the regulatory mechanism of the Cdc7–RAD18-dependent lesion bypass. In contrast, foci formation of Rad51 in response to CIS treatment remained intact in ASK-depleted cells, suggesting that Cdc7–ASK may not be involved in homologous recombinational repair upon replication block evoked by CIS. Rather, we found that spontaneous formation of Rad51 foci was slightly increased in ASK-depleted cells, consistent with the previous report that conditional knockout of the cdc7 gene in mouse embryonic stem cells induced spontaneous Rad51 foci formation (Kim et al. 2002). Since Rad18-dependent damage bypass is important during chronic low-dose UV exposure to prevent G2 arrest by DNA damage checkpoint activation and Rad52-dependent HR repair in yeast (Hishida et al. 2009), Cdc7 kinase may promote DNA damage bypass to prevent fork collapse caused by prolonged replication fork stalling.
Potential exploitation of Cdc7 kinase inhibitors in cancer therapy
DNA cross-link-inducing drugs such as MMC and CIS are commonly used as anti-cancer drugs; however, such treatment, albeit initially often effective, is almost invariably followed by recurrent growth of more aggressive and drug-resistant tumors. It has been postulated that mutation rate is one of the critical determinants of tumor chemoresistance and that chemotherapy itself promotes mutations and subsequent selection of therapy-resistant clones. Recently, translesion synthesis polymerases have been implicated in acquired chemoresistance (Doles et al. 2010; Xie et al. 2010). In the light of our results in the damage bypass-promoting role of Cdc7–ASK, we propose that a combination of, e.g., platinum drugs to evoke replication blockade and a small molecule inhibitor of Cdc7 kinase might offer an innovative strategy to help prevent the emergence of drug-resistant recurrent cancer cells.
Materials and methods
Cell culture
Human U2OS, HeLa, MRC5, and HEK293T cells were cultured in DMEM with 10% fetal bovine serum. The U2OS/Myc-Cdh1 cell line and U2OS-shATR cell line were described previously (Sorensen et al. 2000; Rendtlew Danielsen et al. 2009). The U2OS/Flag-Cdh1 cell line was established by cotransfection of pcDNA6/TR and pcDNA4/TO-Flag-Cdh1 and clonal selection with 5 μg/mL blasticidin S (InvivoGen) and 400 μg/mL Zeocin (Invitrogen). Inducible knockdown cell lines for Cdc7 and Chk1 were established by cotransfection of pcDNA6/TR and the respective shRNA vectors described in the Supplemental Material into U2OS cells and clonal selection with 1 μg/mL puromycin (Sigma-Aldrich) and blasticidin S. Other drugs used in this study included doxycycline (BD Biosciences), tetracycline (Calbiochem), HU (Sigma-Aldrich), MMC (Sigma-Aldrich), CIS (Hospira), cycloheximide (Sigma-Aldrich), PHA-767491 (Calbiochem), and cep-3891 (Cephalon).
Biochemical analysis
Whole-cell extracts were prepared by direct lysis with 1× Laemmli sample buffer (LSB). Biochemical cell fractionation was performed as described (Mendez and Stillman 2000). Cell lysis buffer for immunoprecipitation was as follows: 5 mM Hepes-KOH (pH 7.9), 120 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol, 0.25% Triton X-100, 1 mM NaF, 1 mM β-glycerophosphate, and protease inhibitor cocktail (Roche). Information about antibodies used in this study can be found in the Supplemental Material. For immunoprecipitation, we used an anti-Flag M2 affinity gel (Sigma-Aldrich).
Immunofluorescence
Cells grown on the coverslips were pre-extracted with buffer A (Mendez and Stillman 2000) for 10 min on ice and fixed with 4% formaldehyde for 10 min at room temperature. The fixed cells were stained with anti-RAD18 antibody or anti-Rad51 antibody. Samples were analyzed by Scan R (Olympus) for quantification of the foci.
Cell survival assay
Cells were plated at a density of 1 × 103 per 6-cm dish and treated with CIS at the indicated concentrations for 7 d. The number of viable cells was counted. The values were normalized to those of parallel mock-treated cells. Experiments were repeated three times (n = 3, average ± SD)
Acknowledgments
We thank members of our laboratories in Copenhagen and Olomouc for support. This work was funded by the Strategic Japan-Denmark Cooperative Programme from the Danish Agency for Science, Technology, and Innovation (DASTI); Japan Science and Technology Agency (JST); the Danish Cancer Society; the Lundbeck Foundation; the Novo Nordisk Foundation; the Grant Agency of the Czech Republic (no. 301/11/P554); and the European Commission (projects InflaCare, Biomedreg, and DDResponse).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.224568.113.
References
- Adams MM, Carpenter PB 2006. Tying the loose ends together in DNA double strand break repair with 53BP1. Cell Div 31: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertella MR, Green CM, Lehmann AR, O’Connor MJ 2005. A role for polymerase η in the cellular tolerance to cisplatin-induced damage. Cancer Res 65: 9799–9806 [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas J 2003. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3: 421–429 [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas C, Lukas J 2004. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol 5: 792–804 [DOI] [PubMed] [Google Scholar]
- Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M 2008. The Cdc14B–Cdh1–Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 134: 256–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius M, Forment JV, Thakkar N, Wagner SA, Choudhary C, Jackson SP 2011. A phospho-proteomic screen identifies substrates of the checkpoint kinase Chk1. Genome Biol 12: R78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonte D, Lindvall C, Liu H, Dykema K, Furge K, Weinreich M 2008. Cdc7-Dbf4 kinase overexpression in multiple cancers and tumor cell lines is correlated with p53 inactivation. Neoplasia 10: 920–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YW, Cleaver JE, Hanaoka F, Chang CF, Chou KM 2006. A novel role of DNA polymerase η in modulating cellular sensitivity to chemotherapeutic agents. Mol Cancer Res 4: 257–265 [DOI] [PubMed] [Google Scholar]
- Cho HJ, Lee EH, Han SH, Chung HJ, Jeong JH, Kwon J, Kim H 2012. Degradation of human RAP80 is cell cycle regulated by Cdc20 and Cdh1 ubiquitin ligases. Mol Cancer Res 10: 615–625 [DOI] [PubMed] [Google Scholar]
- Clarke LE, Fountaine TJ, Hennessy J, Bruggeman RD, Clarke JT, Mauger DT, Helm KF 2009. Cdc7 expression in melanomas, Spitz tumors and melanocytic nevi. J Cutan Pathol 36: 433–438 [DOI] [PubMed] [Google Scholar]
- Costanzo V, Robertson K, Ying CY, Kim E, Avvedimento E, Gottesman M, Grieco D, Gautier J 2000. Reconstitution of an ATM-dependent checkpoint that inhibits chromosomal DNA replication following DNA damage. Mol Cell 6: 649–659 [DOI] [PubMed] [Google Scholar]
- Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J 2003. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell 11: 203–213 [DOI] [PubMed] [Google Scholar]
- Day TA, Palle K, Barkley LR, Kakusho N, Zou Y, Tateishi S, Verreault A, Masai H, Vaziri C 2010. Phosphorylated Rad18 directs DNA polymerase η to sites of stalled replication. J Cell Biol 191: 953–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierov J, Dierova R, Carroll M 2004. BCR/ABL translocates to the nucleus and disrupts an ATR-dependent intra-S phase checkpoint. Cancer Cell 5: 275–285 [DOI] [PubMed] [Google Scholar]
- Dolan WP, Le AH, Schmidt H, Yuan JP, Green M, Forsburg SL 2010. Fission yeast Hsk1 (Cdc7) kinase is required after replication initiation for induced mutagenesis and proper response to DNA alkylation damage. Genetics 185: 39–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doles J, Oliver TG, Cameron ER, Hsu G, Jacks T, Walker GC, Hemann MT 2010. Suppression of Rev3, the catalytic subunit of Polζ, sensitizes drug-resistant lung tumors to chemotherapy. Proc Natl Acad Sci 107: 20786–20791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermoli A, Bargiotti A, Brasca MG, Ciavolella A, Colombo N, Fachin G, Isacchi A, Menichincheri M, Molinari A, Montagnoli A, et al. 2009. Cell division cycle 7 kinase inhibitors: 1H-pyrrolo[2,3-b]pyridines, synthesis and structure-activity relationships. J Med Chem 52: 4380–4390 [DOI] [PubMed] [Google Scholar]
- Ferreira MF, Santocanale C, Drury LS, Diffley JF 2000. Dbf4p, an essential S phase-promoting factor, is targeted for degradation by the anaphase-promoting complex. Mol Cell Biol 20: 242–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung AD, Ou J, Bueler S, Brown GW 2002. A conserved domain of Schizosaccharomyces pombe dfp1+ is uniquely required for chromosome stability following alkylation damage during S phase. Mol Cell Biol 22: 4477–4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielse C, Miller CT, McConnell KH, DeWard A, Fox CA, Weinreich M 2006. A Dbf4p BRCA1 C-terminal-like domain required for the response to replication fork arrest in budding yeast. Genetics 173: 541–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess GF, Drong RF, Weiland KL, Slightom JL, Sclafani RA, Hollingsworth RE 1998. A human homolog of the yeast CDC7 gene is overexpressed in some tumors and transformed cell lines. Gene 211: 133–140 [DOI] [PubMed] [Google Scholar]
- Hicks JK, Chute CL, Paulsen MT, Ragland RL, Howlett NG, Gueranger Q, Glover TW, Canman CE 2010. Differential roles for DNA polymerases η, ζ, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links. Mol Cell Biol 30: 1217–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishida T, Kubota Y, Carr AM, Iwasaki H 2009. RAD6–RAD18–RAD5-pathway-dependent tolerance to chronic low-dose ultraviolet light. Nature 457: 612–615 [DOI] [PubMed] [Google Scholar]
- Huang J, Huen MS, Kim H, Leung CC, Glover JN, Yu X, Chen J 2009. RAD18 transmits DNA damage signalling to elicit homologous recombination repair. Nat Cell Biol 11: 592–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Taniyami C, Arai N, Masai H 2008. Cdc7 as a potential new target for cancer therapy. Drug News Perspect 21: 481–488 [DOI] [PubMed] [Google Scholar]
- Jiang W, Hunter T 1997. Identification and characterization of a human protein kinase related to budding yeast Cdc7p. Proc Natl Acad Sci 94: 14320–14325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, McDonald D, Hope TJ, Hunter T 1999. Mammalian Cdc7–Dbf4 protein kinase complex is essential for initiation of DNA replication. EMBO J 18: 5703–5713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LH, Masai H, Sugino A 1999. First the CDKs, now the DDKs. Trends Cell Biol 9: 249–252 [DOI] [PubMed] [Google Scholar]
- Kim JM, Sato N, Yamada M, Arai K, Masai H 1998. Growth regulation of the expression of mouse cDNA and gene encoding a serine/threonine kinase related to Saccharomyces cerevisiae CDC7 essential for G1/S transition. Structure, chromosomal localization, and expression of mouse gene for S. cerevisiae Cdc7-related kinase. J Biol Chem 273: 23248–23257 [DOI] [PubMed] [Google Scholar]
- Kim JM, Nakao K, Nakamura K, Saito I, Katsuki M, Arai K, Masai H 2002. Inactivation of Cdc7 kinase in mouse ES cells results in S-phase arrest and p53-dependent cell death. EMBO J 21: 2168–2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Kakusho N, Yamada M, Kanoh Y, Takemoto N, Masai H 2008. Cdc7 kinase mediates Claspin phosphorylation in DNA replication checkpoint. Oncogene 27: 3475–3482 [DOI] [PubMed] [Google Scholar]
- Kulkarni AA, Kingsbury SR, Tudzarova S, Hong HK, Loddo M, Rashid M, Rodriguez-Acebes S, Prevost AT, Ledermann JA, Stoeber K, et al. 2009. Cdc7 kinase is a predictor of survival and a novel therapeutic target in epithelial ovarian carcinoma. Clin Cancer Res 15: 2417–2425 [DOI] [PubMed] [Google Scholar]
- Kumagai H, Sato N, Yamada M, Mahony D, Seghezzi W, Lees E, Arai K, Masai H 1999. A novel growth- and cell cycle-regulated protein, ASK, activates human Cdc7-related kinase and is essential for G1/S transition in mammalian cells. Mol Cell Biol 19: 5083–5095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labib K 2010. How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev 24: 1208–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AY, Chiba T, Truong LN, Cheng AN, Do J, Cho MJ, Chen L, Wu X 2012. Dbf4 is direct downstream target of ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) protein to regulate intra-S-phase checkpoint. J Biol Chem 287: 2531–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listovsky T, Zor A, Laronne A, Brandeis M 2000. Cdk1 is essential for mammalian cyclosome/APC regulation. Exp Cell Res 255: 184–191 [DOI] [PubMed] [Google Scholar]
- Listovsky T, Oren YS, Yudkovsky Y, Mahbubani HM, Weiss AM, Lebendiker M, Brandeis M 2004. Mammalian Cdh1/Fzr mediates its own degradation. EMBO J 23: 1619–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas C, Sorensen CS, Kramer E, Santoni-Rugiu E, Lindeneg C, Peters JM, Bartek J, Lukas J 1999. Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature 401: 815–818 [DOI] [PubMed] [Google Scholar]
- Masai H, Arai K 2000. Dbf4 motifs: Conserved motifs in activation subunits for Cdc7 kinases essential for S-phase. Biochem Biophys Res Commun 275: 228–232 [DOI] [PubMed] [Google Scholar]
- Masai H, Arai K 2002. Cdc7 kinase complex: A key regulator in the initiation of DNA replication. J Cell Physiol 190: 287–296 [DOI] [PubMed] [Google Scholar]
- Masai H, Matsumoto S, You Z, Yoshizawa-Sugata N, Oda M 2010. Eukaryotic chromosome DNA replication: Where, when, and how? Annu Rev Biochem 79: 89–130 [DOI] [PubMed] [Google Scholar]
- Mendez J, Stillman B 2000. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: Assembly of prereplication complexes in late mitosis. Mol Cell Biol 20: 8602–8612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagnoli A, Valsasina B, Croci V, Menichincheri M, Rainoldi S, Marchesi V, Tibolla M, Tenca P, Brotherton D, Albanese C, et al. 2008. A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat Chem Biol 4: 357–365 [DOI] [PubMed] [Google Scholar]
- Nambiar S, Mirmohammadsadegh A, Hassan M, Mota R, Marini A, Alaoui A, Tannapfel A, Hegemann JH, Hengge UR 2007. Identification and functional characterization of ASK/Dbf4, a novel cell survival gene in cutaneous melanoma with prognostic relevance. Carcinogenesis 28: 2501–2510 [DOI] [PubMed] [Google Scholar]
- Njagi GD, Kilbey BJ 1982. cdc7-1 a temperature sensitive cell-cycle mutant which interferes with induced mutagenesis in Saccharomyces cerevisiae. Mol Gen Genet 186: 478–481 [DOI] [PubMed] [Google Scholar]
- Nojima K, Hochegger H, Saberi A, Fukushima T, Kikuchi K, Yoshimura M, Orelli BJ, Bishop DK, Hirano S, Ohzeki M, et al. 2005. Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells. Cancer Res 65: 11704–11711 [DOI] [PubMed] [Google Scholar]
- Ogino K, Takeda T, Matsui E, Iiyama H, Taniyama C, Arai K, Masai H 2001. Bipartite binding of a kinase activator activates Cdc7-related kinase essential for S phase. J Biol Chem 276: 31376–31387 [DOI] [PubMed] [Google Scholar]
- Pessoa-Brandao L, Sclafani RA 2004. CDC7/DBF4 functions in the translesion synthesis branch of the RAD6 epistasis group in Saccharomyces cerevisiae. Genetics 167: 1597–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendtlew Danielsen JM, Larsen DH, Schou KB, Freire R, Falck J, Bartek J, Lukas J 2009. HCLK2 is required for activity of the DNA damage response kinase ATR. J Biol Chem 284: 4140–4147 [DOI] [PubMed] [Google Scholar]
- Saberi A, Hochegger H, Szuts D, Lan L, Yasui A, Sale JE, Taniguchi Y, Murakawa Y, Zeng W, Yokomori K, et al. 2007. RAD18 and poly(ADP-ribose) polymerase independently suppress the access of nonhomologous end joining to double-strand breaks and facilitate homologous recombination-mediated repair. Mol Cell Biol 27: 2562–2571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Arai K, Masai H 1997. Human and Xenopus cDNAs encoding budding yeast Cdc7-related kinases: In vitro phosphorylation of MCM subunits by a putative human homologue of Cdc7. EMBO J 16: 4340–4351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Sato M, Nakayama M, Saitoh R, Arai K, Masai H 2003. Cell cycle regulation of chromatin binding and nuclear localization of human Cdc7–ASK kinase complex. Genes Cells 8: 451–463 [DOI] [PubMed] [Google Scholar]
- Skaar JR, Pagano M 2008. Cdh1: A master G0/G1 regulator. Nat Cell Biol 10: 755–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen CS, Lukas C, Kramer ER, Peters JM, Bartek J, Lukas J 2000. Nonperiodic activity of the human anaphase-promoting complex-Cdh1 ubiquitin ligase results in continuous DNA synthesis uncoupled from mitosis. Mol Cell Biol 20: 7613–7623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen CS, Lukas C, Kramer ER, Peters JM, Bartek J, Lukas J 2001. A conserved cyclin-binding domain determines functional interplay between anaphase-promoting complex–Cdh1 and cyclin A–Cdk2 during cell cycle progression. Mol Cell Biol 21: 3692–3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudo T, Ota Y, Kotani S, Nakao M, Takami Y, Takeda S, Saya H 2001. Activation of Cdh1-dependent APC is required for G1 cell cycle arrest and DNA damage-induced G2 checkpoint in vertebrate cells. EMBO J 20: 6499–6508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syljuasen RG, Sorensen CS, Nylandsted J, Lukas C, Lukas J, Bartek J 2004. Inhibition of Chk1 by CEP-3891 accelerates mitotic nuclear fragmentation in response to ionizing radiation. Cancer Res 64: 9035–9040 [DOI] [PubMed] [Google Scholar]
- Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, Helleday T, Sehested M, Lukas J, Bartek J 2005. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol 25: 3553–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szuts D, Simpson LJ, Kabani S, Yamazoe M, Sale JE 2006. Role for RAD18 in homologous recombination in DT40 cells. Mol Cell Biol 26: 8032–8041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenca P, Brotherton D, Montagnoli A, Rainoldi S, Albanese C, Santocanale C 2007. Cdc7 is an active kinase in human cancer cells undergoing replication stress. J Biol Chem 282: 208–215 [DOI] [PubMed] [Google Scholar]
- van Leuken R, Clijsters L, Wolthuis R 2008. To cell cycle, swing the APC/C. Biochim Biophys Acta 1786: 49–59 [DOI] [PubMed] [Google Scholar]
- Vanotti E, Amici R, Bargiotti A, Berthelsen J, Bosotti R, Ciavolella A, Cirla A, Cristiani C, D’Alessio R, Forte B, et al. 2008. Cdc7 kinase inhibitors: Pyrrolopyridinones as potential antitumor agents. 1. Synthesis and structure-activity relationships. J Med Chem 51: 487–501 [DOI] [PubMed] [Google Scholar]
- Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M 2004. Rad18 guides polη to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J 23: 3886–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich M, Stillman B 1999. Cdc7p–Dbf4p kinase binds to chromatin during S phase and is regulated by both the APC and the RAD53 checkpoint pathway. EMBO J 18: 5334–5346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SA, Longerich S, Sung P, Vaziri C, Kupfer GM 2011. The E3 ubiquitin ligase RAD18 regulates ubiquitylation and chromatin loading of FANCD2 and FANCI. Blood 117: 5078–5087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Lee H 2002. Human Dbf4/ASK promoter is activated through the Sp1 and MluI cell-cycle box (MCB) transcription elements. Oncogene 21: 7786–7796 [DOI] [PubMed] [Google Scholar]
- Xie K, Doles J, Hemann MT, Walker GC 2010. Error-prone translesion synthesis mediates acquired chemoresistance. Proc Natl Acad Sci 107: 20792–20797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Sato N, Taniyama C, Ohtani K, Arai K, Masai H 2002. A 63-base pair DNA segment containing an Sp1 site but not a canonical E2F site can confer growth-dependent and E2F-mediated transcriptional stimulation of the human ASK gene encoding the regulatory subunit for human Cdc7-related kinase. J Biol Chem 277: 27668–27681 [DOI] [PubMed] [Google Scholar]
- Yamashita YM, Okada T, Matsusaka T, Sonoda E, Zhao GY, Araki K, Tateishi S, Yamaizumi M, Takeda S 2002. RAD18 and RAD54 cooperatively contribute to maintenance of genomic stability in vertebrate cells. EMBO J 21: 5558–5566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita N, Kim JM, Koiwai O, Arai K, Masai H 2005. Functional analyses of mouse ASK, an activation subunit for Cdc7 kinase, using conditional ASK knockout ES cells. Genes Cells 10: 551–563 [DOI] [PubMed] [Google Scholar]
- Zachariae W, Schwab M, Nasmyth K, Seufert W 1998. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science 282: 1721–1724 [DOI] [PubMed] [Google Scholar]
- Zegerman P, Diffley JF 2010. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 467: 474–478 [DOI] [PMC free article] [PubMed] [Google Scholar]