

AAA+ ATPase molecular machines perform mechanical work in all organisms. A key question is how rings of identical subunits interact with asymmetric target macromolecules. This study by Nixon and colleagues elucidates the structure of AAA+ ATPase bacterial transcriptional activator NtrC1, providing mechanistic insight into transcription initiation. Partial ATP occupancy causes the heptameric closed ring of NtrC1 to rearrange into a hexameric split ring of striking asymmetry. Furthermore, the similarity between the structure of NtrC1 and those of distantly related helicases reveals a general mechanism for homomeric ATPase function.
Keywords: AAA+ ATPase, mechanochemical ATPases, multimeric ATPases, bacterial enhancer-binding protein (bEBP), σ54-dependent transcription, σ54-dependent transcription activators
Abstract
It is largely unknown how the typical homomeric ring geometry of ATPases associated with various cellular activities enables them to perform mechanical work. Small-angle solution X-ray scattering, crystallography, and electron microscopy (EM) reconstructions revealed that partial ATP occupancy caused the heptameric closed ring of the bacterial enhancer-binding protein (bEBP) NtrC1 to rearrange into a hexameric split ring of striking asymmetry. The highly conserved and functionally crucial GAFTGA loops responsible for interacting with σ54–RNA polymerase formed a spiral staircase. We propose that splitting of the ensemble directs ATP hydrolysis within the oligomer, and the ring's asymmetry guides interaction between ATPase and the complex of σ54 and promoter DNA. Similarity between the structure of the transcriptional activator NtrC1 and those of distantly related helicases Rho and E1 reveals a general mechanism in homomeric ATPases whereby complex allostery within the ring geometry forms asymmetric functional states that allow these biological motors to exert directional forces on their target macromolecules.
A long-standing, largely unanswered question about the functional mechanism of ATPases associated with various cellular activities (AAA+ ATPases) is: How do rings of chemically identical subunits interact with highly asymmetric target macromolecules? Answering this question is very important because AAA+ ATPase molecular machines perform mechanical work remodeling nearly every type of macromolecule in cells from all kingdoms of life.
In bacteria, a subfamily of AAA+ ATPases known as the bacterial enhancer-binding proteins (bEBPs) remodels the σ54 form of RNA polymerase (RNAP) (Eσ54) that is present in complexes with promoter DNA. The manipulation facilitates transformation of closed preinitiation complexes of Eσ54 and promoter DNA into opened, transcriptionally competent ones. This ability to remodel the Eσ54–promoter complex provides bacteria with adaptive expression of genes for nutrient acquisition, complex developmental programs, and virulence as pathogens. The functionally crucial surface “GAFTGA” (or L1) loop and supporting L2 loop are the hallmark features of bEBPs that distinguish them from other AAA+ ATPases, adapting the basic ATPase fold for direct interaction with Eσ54 (Lee et al. 2003).
Previously, an Eσ54 complex was shown to bind to a bEBP via contact with GAFTGA loops, but high-resolution details are lacking for that cryo-electron microscopy (cryo-EM) three-dimensional (3D) model (Bose et al. 2008). Several bEBPs have been crystallized as hexamers and heptamers, but no mechanism for hydrolysis has yet emerged from those structures (Lee et al. 2003; Rappas et al. 2005, 2006; Sallai and Tucker 2005; Batchelor et al. 2008; Chen et al. 2010). We showed previously that binding of ATP but not ADP causes large structural changes in the bEBP NtrC1 from the extreme hyperthermophile Aquifex aeolicus (Chen et al. 2007, 2010). Presumed symmetry required for modeling those small-angle solution X-ray scattering (SAXS) data and sevenfold symmetry in the ADP- and ATP-bound crystal structures suggested cooperative upward and downward motion of the crucial GAFTGA-containing loops that tracks the status of nucleotides in the hydrolysis cycle (Chen et al. 2010). The relevance of these symmetric structures needs to be further explored, as there is biochemical evidence from studies of the bEBP PspF that mixed nucleotide states and potential structural asymmetry are present in an optimally functioning bEBP (Schumacher et al. 2008; Joly and Buck 2011).
In the present study, we used isothermal calorimetry (ITC), time-resolved SAXS (TR-SAXS), equilibrium SAXS, crystallography, and 3D reconstruction from EM single particles to monitor the development and extent of conformational changes in NtrC1 that follow its binding to the metal fluoride ATP analogs ADP-BeFx or ADP-AlFx and its target, σ54, bound to promoter DNA. The results revealed that partial binding of ATP drives a dramatic reorganization of protomers from heptamer to hexamer rings in which target-binding GAFTGA loops are raised above the pore in a spiral staircase. A significant gap exists between two specific protomers (the ones with highest and lowest loops), forming a likely site for nucleotide exchange. This gap is uniquely oriented in complex with σ54 and promoter DNA, which appears to occur via four contacts. We propose that transcription-activating bEBPs hydrolyze ATP according to varied structural and functional identities that allostery imposes on different subunits as it builds a highly asymmetric ring. We call attention to the unexpected similarity of such an asymmetric ring structure to those of Rho and E1, two ATPases that function as RNA and DNA translocases, respectively. Finally, we discuss how this structural similarity among three distantly related asymmetric rings partially answers the introductory question: Heterogeneity within a hexamer of chemically identical subunits is available through the formation of quasisplit rings. Harnessing this heterogeneity in ring protomers to provide an asymmetric distribution of functional states allows these biological motors to exert directional forces on their targeted, asymmetric macromolecules.
Results
Complexity of ATP binding to NtrC1
As established earlier (Chen et al. 2007), adding 1–5 mM ATP and ATP analogs to the assembled ring form of apo NtrC1 ATPase causes it to undergo large-scale conformational changes, but adding ADP does not. It was also shown that beginning near 1 mM concentration, ATP inhibits ATPase activity of the σ54-dependent activators. For NtrC1, this inhibition is only partial (Chen et al. 2010); for PspF, it is more complete (Joly et al. 2006; Schumacher et al. 2008). Therefore, it appears that the activator is highly sensitive to the nucleotide concentration, and thus a more detailed study of structural changes induced by ATP is needed to understand how the bEBP motor functions.
Initially, we used isothermal titration calorimetry (ITC) to measure heat released upon binding of ADP and ATP analog to the NtrC1 activator. ADP binding released heat monotonically, pointing to the presence of a single type of binding site (Fig. 1A; Supplemental Table S1). In stark contrast, binding of ADP-BeFx was not monotonic, implying the presence of binding sites with different affinities for ATP (Fig. 1A; Supplemental Table S1). In fitting the ITC data to standard one- or two-site models, the total numbers of sites were fractional, ∼80% of the total available binding sites. Such substoichiometric occupancy could reflect the presence of prebound nucleotides in the purified protein samples or the presence of nonbinding ATPase subunits, and these could be either inactive or of low affinity. Following the ITC experiments, stopped-flow measurements of intrinsic fluorescence showed that conformational changes associated with binding ADP-BeFx were complex, associated with at least three different time trajectories, with the fastest phase of several milliseconds.
Figure 1.

Nucleotide binding to NtrC1 ATPase and its X-ray-scattering response. (A) ITC measurements for NtrC1 titrated with ADP (closed squares) and ADP-BeFx (open circles). (Solid lines) Fits to models with one or two binding sites, respectively (parameters are in Supplemental Table S1). (B) Time-resolved scattering profiles of NtrC1 after mixing with 1 mM ADP-BeFx. For clarity of presentation, only every third profile from 5 msec to 3 min is shown; errors are illustrated only in the inset. (Green) Profile for apo NtrC1; (red) profile for protein pre-equilibrated with nucleotide. (C) Scattering intensity integrated over the Q-vector range of 0.01 to 0.12 Å−1 (right axis; open circles) or radius of gyration (left axis; open rhombs) versus time after mixing NtrC1 with ADP-BeFx (final concentrations 327 μM and 1 mM, respectively). (D) Same as C, but for NtrC1 equilibrated with the indicated concentrations of ADP-BeFx before exposure to X-rays.
Distinct structural intermediates upon ATP binding to NtrC1
To analyze how the phases discovered in calorimetric and fluorescence experiments correlate with structural changes, we performed TR-SAXS experiments in which the ATP analog ADP-BeFx was mixed with apo NtrC1 subunits followed by recording the time trajectory of scattering profiles. Since the SAXS method is not sensitive enough to directly detect binding of nucleotides (∼0.5 kDa) to an NtrC1 oligomer (∼210 kDa), any observed changes in the scattering profiles must reflect conformational differences in the protein induced by interactions with nucleotides. We modified the existing TR-SAXS setup of the BioCAT beamline to minimize radiation damage and thus permit 10-msec sample exposures for monitoring changes in scattering over the time frame of milliseconds to several minutes. Since conventional fluorescence stopped-flow measurements showed that the fastest change in the intrinsic NtrC1 fluorescence happens in several milliseconds, we presumed that nucleotide binding occurred before the 10-msec X-ray exposures. Scattering curves acquired at different times after mixing NtrC1 ATPase with ATP analog are shown in Figure 1B. Inspecting the curves by monitoring the intensity summed over all Q vectors (integrated intensity) (Fig. 1C) and the radius of gyration (Rg) (Fig. 1C) revealed that the scattering profile changed in two time-dependent phases. The first phase occurred at Q vectors ranging from 0.007 Å−1 to 0.06 Å−1, reflecting a monomodal decrease in Rg from ∼46 Å to ∼42 Å. The second phase of change registered primarily at Q vectors ranging from 0.06 Å−1 to 0.10 Å−1, reflecting smaller-scale changes in the ATPase structure. We conclude that the apo NtrC1 oligomer undergoes a biphasic process of large- and then small-scale structural response to binding ADP-BeFx.
To establish whether the biphasic structural changes are correlated with nucleotide occupancy, titration experiments were conducted in which variable amounts of ATP analog were equilibrated with a constant amount of ATPase subunit prior to collecting SAXS data. A bimodal transition was observed in the titration experiments (Fig. 1D), with signals designating the two changes being distributed over similar Q-vector ranges as seen in the time-resolved experiment. Moreover, the changes of scattering intensities in the different Q-vector ranges showed distinct dependencies on ATP analog concentration, indicating that these changes reflect transitions between states that are occupied with nucleotides to different extents.
Crystal structure of the hexameric intermediate of ATP binding
The time-resolved and equilibrium SAXS experiments confirmed that the structure of the NtrC1 ensemble is extremely sensitive to the amount of bound nucleotide and that the ring transits through at least one intermediate conformation before reaching its high-occupancy state. At just below stoichiometric amounts of ADP-BeFx, we were able to grow diffracting crystals. One scattered X-rays isotropically to 3.6 Å, and one scattered X-rays anisotropically to 3.2 × 5.2 × 3.2 Å (crystals “S” and “Q” for Protein Data Bank [PDB] depositions 4LY6 and 4LZZ, respectively) (Table 1). In these crystals, four similar hexameric rings were packed into a primitive orthorhombic lattice; the symmetry was broken, however, by local features in each of the rings (Fig. 2A; Supplemental Fig. S1). Unlike the previously described symmetric heptamer rings of the NtrC1 ATPase domain, all of the newly discovered hexamers exhibited a striking asymmetry in subunit juxtaposition.
Table 1.
Data collection and refinement statistics
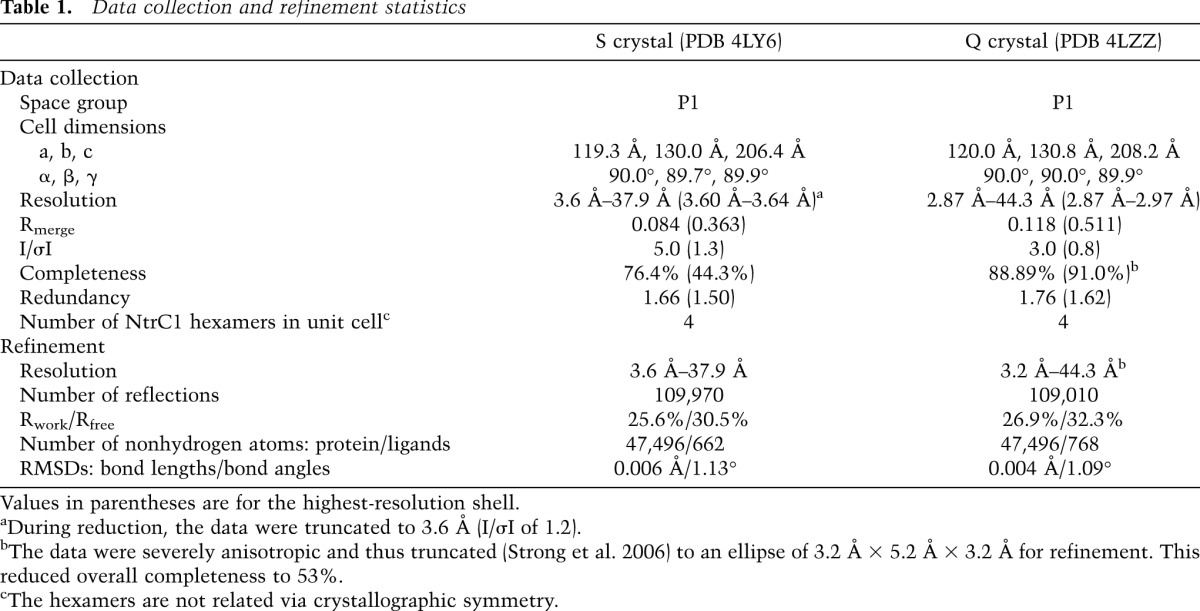
Figure 2.

Crystal structure of the hexameric intermediate of the NtrC1 ATPase domain. (A) Top and side views of the NtrC1 hexamer; boundaries are enhanced by variously colored subunits A–F. Contact surface areas between subunits (PISA) (Krissinel and Henrick 2007) are shown as colored circles scaled to the listed areas (see also Supplemental Table S2). See also Supplemental Figure S1 for details of the NtrC1 monomer and oligomer reorganization upon nucleotide binding. (B) Heptamer-to-hexamer transition of the NtrC1 ATPase. Solution-scattering profiles of NtrC1 in the presence of 0, 200, and 1000 mM ADP-BeFx (gray with black outline, solid gray, and solid black, respectively) onto the scattering profiles calculated by CRYSOL (Svergun et al. 1995) for the hexamer from the new crystals (black dashed line). This and other illustrations for the 3D structure were rendered using Pymol (The PyMOL Molecular Graphics System, version 1.5.0.4, Schrödinger, LLC.) unless indicated otherwise.
The new hexameric structures were used to calculate theoretical scattering in solution. The calculated profiles, essentially identical to one another, were compared with the measured scattering from NtrC1 under the presence of different amounts of ADP-BeFx (Fig. 2B); they matched best and well to the observed scattering from the ATPase in the presence of 1 mM ATP analog. This figure also illustrates the presence of an intermediate structure, showing how partial occupancy of 330 μM protomers at 200 μM nucleotide was sufficient to stabilize the first phase of conformational change at low Q vectors but not the second one at higher scattering angles.
Asymmetry of the NtrC1 hexamer
The roots of the striking ring asymmetry were evident upon examining the subunits' shapes, binding pockets, and intersubunit interfaces. While five of the six subunits of each ring were similar to one another and were found in molecular replacement using the ATP-saturated subunit in the structure 3M0E (Chen et al. 2010), the sixth subunit (labeled chains F, L, R, and X) had to be built using subdomains of the ATPase. None of the ADP-bound subunits of the NtrC1 structures 1NY5 and 1NY6 (Lee et al. 2003) gave solutions in molecular replacement. In closing the rings, the interfaces between subunits 1 and 6 were dramatically less tight than for the rest of the subunit pairs (Supplemental Fig. S1). This observation was confirmed by quantifying contact surface areas (Fig. 2A; Supplemental Table S2; Krissinel and Henrick 2007). We henceforth refer to this as the “gap interface” (between protomers A/F, G/L, M/R, and S/X in the PDB files). The intersubunit contact areas are seen to differ by 2.7-fold to 4.2-fold around the NtrC1 hexamers, ranging from the highest of 1600–1700 Å2 for the subunit D/E type to the lowest of 400–600 Å2 for the subunit pair A/F. Interestingly, electron densities for the A and F subunits were weaker than for the rest of the ring subunits. Consequently, B factors of the respective residues were higher. The distribution of B factors around the hexameric ring is shown in Figure 3A. The plot reveals that the A subunit and the α subdomain of the F subunit are the most flexible or mobile components of the ensemble.
Figure 3.
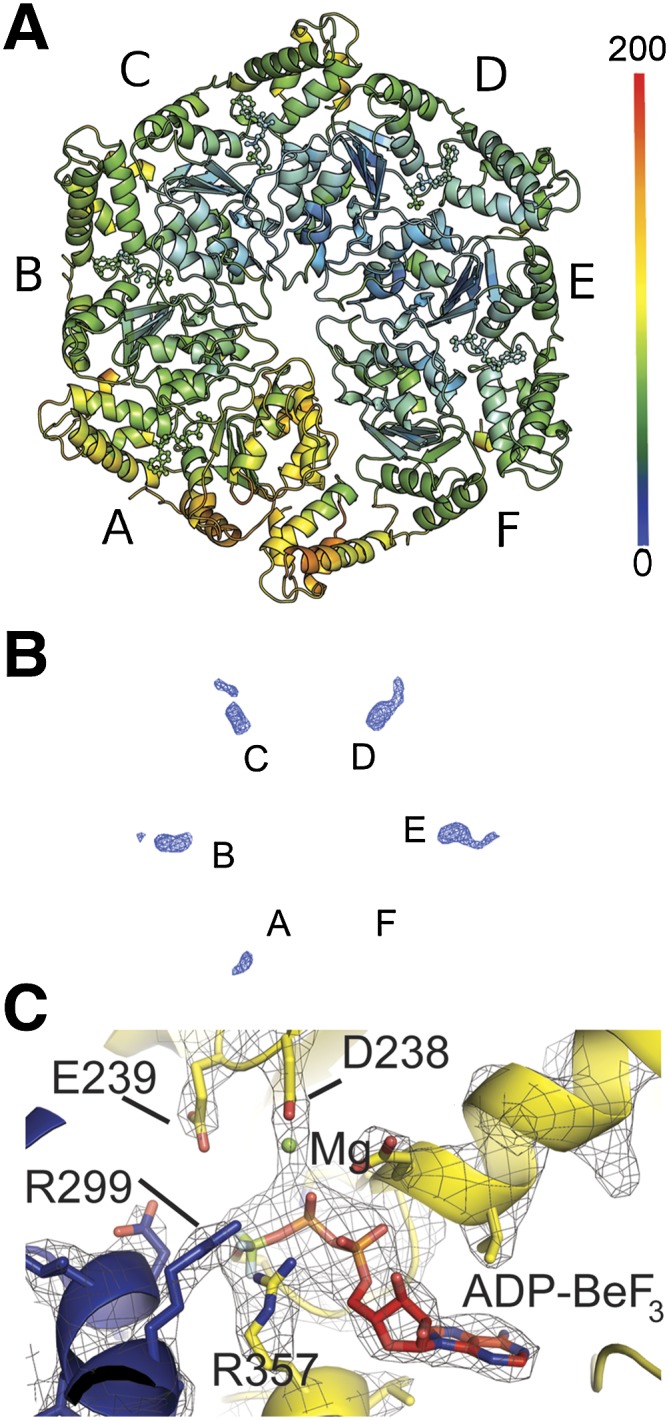
Asymmetry of the NtrC1 hexamer. (A) NtrC1 hexamer is colored according to B factors. (B) An example an omit map (S crystal, subunits A–F) showing ligand densities in the active sites of the NtrC1 hexamer that were used for placing nucleotides. See also Supplemental Figure S2. (C) Active site of subunit E showing modeled ADP-BeF3 ligand and its interaction with P-loop, arginine finger R299, and Walker B residues.
Omit maps in Figure 3B (2Fo–1Fc; 0.125 e/Å3 or 1.7 RMSD) and Supplemental Figure S3A (2Fo–1Fc; 0.177 e/Å3 or 2.4 RMSD) summarize the nucleotide densities present in the four hexameric rings of both crystals. Supplemental Figure S3A shows the major differences seen in the presence of ligand densities in the active sites between the eight hexamer rings. Some densities are well modeled by the presence of ADP-BeF3 (e.g., see Fig. 3C), others suggest ADP, and some appear to be weakly occupied or exist in an apo state. The poorly occupied subunits contain a relocated P loop. According to the strength of electron density for nucleotides present in the subunits that flank the gap, we classified the hexamer rings as type 1 (S crystal, with subunits F or R lacking density), type 2 (S crystal, with subunits L or X having moderate density), type 3 (Q crystal, with subunits A and M having moderate density), and type 4 (Q crystal, with subunits G and S lacking density). Note that the subunits lacking density for nucleotides were always adjacent to the gap in the ring but abutted opposite sides of the gap in the S and Q crystals. Likewise, the B-factor values suggest that the tightness of nucleotide binding varies from one interface to another and may reflect whether the gap interface lacks nucleotides (as in type 1 and type 4 rings) or is bound by ADP or ATP analog (as in type 2 and type 3 rings). Beyond their sensor 2 helixes, the backbone trajectories of the α-helical domains correlated with the presence or absence of nucleotides: Different paths were observed between nucleotide-bound subunits and those of chains F, L, R, and X in the S crystal. This observation may reveal a rigid body conformational change within the α domains that is sensitive to nucleotide binding.
Comparison of the refined structures with the previously seen ADP-bound (1NY6) and ATP-bound (3M0E) structures revealed two significant differences. First, protomers were packed as hexamers versus heptamers via a small shift in their orientation relative to the ring's plane (Supplemental Figs. S1, S2). The sequential progression of this shift caused the L1 and L2 stem loops to cascade in a downward spiral from subunit A to subunit F (Fig. 4A,B; Supplemental Fig. S1B). Second, analysis of protomer structures with RAPIDO and DynDom revealed rigid body movement that is illustrated in Figure 4D (Hayward et al. 1997; Hayward and Berendsen 1998; Mosca and Schneider 2008; Mosca et al. 2008). This movement is very similar to the rigid body roll observed upon comparison of the ADP- and ATP-bound heptamers (Chen et al. 2010) and includes rotation of the functional stem–loops (Fig. 4C,D) in subunit F with respect to subunits A–E (or their equivalents in other rings). The extent of rotation of rigid body 2 away from rigid body 1 is less extreme for subunits L and X of the S crystal than for subunits F and R of the S crystal and F, L, R, and X of the Q crystal. The first five subunits of each ring share similar conformations, with the root-mean-square deviation (RMSD) for all-atom overlays varying from 0.49 Å to 0.94 Å (Supplemental Table S3). These conformations are also very similar to that of the ATP-bound subunits present in the heptamer ring of the Walker B mutant E239A (Supplemental Table S3).
Figure 4.

Spiraling arrangement of the functional pore loops of the NtrC1 activator. (A) The L1 (GAFTGA) and L2 loops surrounding the pore of the NtrC1 hexamer; the coloring is the same as in A. Electron density is contoured at 1.5σ. (B) Left-handed spiral staircase of loops L1 and L2, with labels for subunits A and F illustrating the change in elevation. (C) GAFTGA loops of subunits F and A. (D) Overlay of subunit A with F, L, R, and X of the S-crystal structure, showing rigid body roll of stem–loops L1 and L2. Subunit A (essentially identical to subunits B–E in all four rings) follows the ATP-bound state of 3M0E, while subunits F and R follow the ADP-bound state of 1NY6 subunits L and X, which are poised between the two extremes. In the Q-crystal structure, the first five subunits of each ring are in the ATP-like state, and subunits F, L, R, and X are like F and R in the S crystal.
Ring asymmetry guides interaction with σ54 and promoter DNA
The good fit between the scattering profiles of NtrC1 calculated from the novel crystal structures and those experimentally measured for the protein partially occupied with ATP analog suggests the presence of asymmetric hexamer rings in solution. To determine whether the asymmetry in the hexameric NtrC1 ring is important for interaction with σ54 and promoter DNA, we derived a 3D reconstruction from transmission electron microscopy and single-particle analysis of the negative-stained ternary complex of NtrC1 ATPase, σ54, and promoter DNA (MW ∼256 kDa) that was stabilized by ADP-AlFx. Visual inspection of the class averages and the reconstruction at a resolution of 24 Å shows a gap in the ATPase ring (Fig. 5; Supplemental Movie S1). Additional density can be observed above the ATPase, presumably for σ54 and promoter DNA. Agreement between reprojections of the 3D reconstruction and the class averages confirms that a large number of unique orientations are present among the particles, validating the model. Importantly, the gap in the ring could also be observed in the reference-free class averages (Fig. 5D).
Figure 5.

Subunit A/F gap of the ATPase crystal structure persists in 3D EM reconstruction of a complex of ATPase, σ54, and promoter DNA. (A) 3D EM reconstruction of the complex of ATPase, σ54, and promoter DNA. Density for ATPase (pink) is distinguished from density for σ54 and promoter DNA. The hexamer crystal structure of the ATPase (colors as in Fig. 2) was fit to its EM density; the arrowhead points to the aligned gap in both structures. (B) EM density contoured at a threshold showing only the density corresponding to the ATPase. (C) Four points of contact between the ATPase and σ54 promoter DNA. Contact 1 is magnified to show the GAFTGA loop of subunit A (red ribbon) and σ54/promoter DNA. (D) Selected forward projections (top row) of the complex and averages of the corresponding aligned particles (middle row) are above similar looking reference-free class averages (bottom row); yellow dots locate the gap in the ATPase ring. See also Supplemental Figure S3 and the Supplemental Movies.
The split hexamer ring of NtrC1 was fit into the ring-shaped density of the reconstruction based on the highest cross-correlation of the ATPase's molecular envelope with the electron density map. The gap region of the NtrC1 hexamer also lacked density in the EM map (subunits A and F) (Fig. 5B; Supplemental Movie S2). The observed correlation of the SAXS, EM, and crystal data is consistent with some regions being flexible in both solution and the crystal lattice. Most interestingly, the EM reconstruction reveals four potential regions of ATPase that form two types of contact with σ54/promoter DNA (Fig. 5A,C; Supplemental Movie S1): with the functional GAFTGA-containing loops and with interfaces of other subunits. For the loops, these include the GAFTGA loop of protomer A and above the loop region of protomer E. While the GAFTGA-containing loop of the activator subunit A is the most poorly defined in the crystal structures, it seems to be stabilized by the presence of the interacting partners σ54/promoter. A second type of contact appears at the interface between the α-helical and α/β subdomains of protomers C and D and of protomers E and F. Combined, the contacts both bridge the A/F gap and anchor distant parts of the target with subunits across the ring from the gap. While clearly present, the density for the σ54/promoter part of the complex is less well defined than that of the ATPase. This may be caused by the absence of the rest of RNAP in this complex.
Discussion
The degree of saturation with ATP dictates the high-order structure of the NtrC1 ATPase activator
Our prior biochemical and structural analyses suggested two things: (1) that apo and ADP-saturated rings form similar heptamer rings (SAXS data in Chen et al. 2007) and (2) that strong clashes predicted for the interface between neighboring subunits that are alternately bound to ATP or ADP can be avoided by highly cooperative nucleotide binding and hydrolysis (Chen et al. 2010). In this study, we see that the symmetry in those ring structures is broken as binding of ATP analogs gradually drives the heptamers to form split-ring hexamers. The bimodal curve for heat release upon interaction of the apo NtrC1 ATPase domain and ATP analog is in fact consistent with a strong negative cooperativity for ATP binding. Perhaps a set of high-affinity binding events drives the ring to adopt a conformation that imposes negative cooperativity on binding beyond about two-thirds saturation. Similar observations were made for DnaB of Escherichia coli, another multimeric ATPase (Bujalowski and Klonowska 1993), and mass spectroscopy data were recently published for the EBP PspF that are quite consistent with heptamer EBP rings converting to hexameric ones upon partial saturation with nucleotides (Zhang et al. 2013). The SAXS data for NtrC1 binding of metal fluoride ATP analogs suggest that consecutive binding events generate a series of structural rearrangements, including changes in both the stoichiometry of the oligomer and subdomain movements. The results are consistent with the hypothesis that the EBP ATPase oligomer behaves as an integrated ensemble of subunits whose relative occupancies with nucleotides strongly influence their neighbors, as suggested earlier for other multimeric ATPases (Sanders et al. 2007; Glynn et al. 2012).
Since σ54 binding to the activator was observed only upon binding of NtrC1 with an ATP analog and not ADP, we hypothesized that structural changes mediating cooperativity are mainly induced by the presence of the γ-phosphate (Chen et al. 2007, 2010). This phosphate was previously shown to bridge between the ATP-binding cleft on one subunit and the arginine finger (R299 in NtrC1) of its adjacent partner in the ring (Chen et al. 2010). Although the electron density for the gapped hexamer ring that we show here is from low-resolution diffraction data, it is consistent with the proposed contact between subunits (Fig. 3C). It follows that any ATP-binding event will break the symmetry of the apo heptamer ring. The asymmetry would in turn influence properties of the remaining unoccupied binding sites via cooperative nucleotide binding. While higher-resolution data are required for confirmation, inspecting the active sites for electron density around the γ-BeF3, β-phosphate, arginine finger, sensors 1 and 2, and Walker glutamates (R299, N280, R357, E238, and E239 of NtrC1) suggests unique features for each subunit, consistent with them having unique functional roles (Supplemental Fig. S3B). The presence of heptamers may be an artifact of having truncated the NtrC1 protein to yield the ATPase domain alone. In full-length NtrC1, the equilibrium between heptamer and hexamer rings may be influenced by the presence of the N-terminal two-component receiver and C-terminal domains. Mass spectroscopy data showed that hexamer is favored over heptamer in full-length constructs of NtrC4, an EBP related to NtrC1 (Batchelor et al. 2009). The receiver domain forms stable homodimers, and the C-terminal DNA-binding domain directs assembly of four protomers in a tandem array on “enhancers” (Doucleff et al. 2005; Vidangos et al. 2013). However, this potential artifact underscores the stabilization of split hexamer rings of EBP and does not diminish the evidence for γ-phosphate-directed asymmetry in this structural state.
Unexpected similarity of the NtrC1 activator and hexameric helicases
Strikingly, the staircase-like arrangement of the pore loops has been found in several other ring-shaped ATPases from different families: the Rho termination factor helicase from E. coli, the E1 helicase of bovine papiloma virus, and the DnaB helicase from Geobacillus stearothermophilus (Enemark and Joshua-Tor 2006; Thomsen and Berger 2009; Itsathitphaisarn et al. 2012). The presence of this similar architectural feature among these diverse classes of ATPases is particularly interesting because of the obvious differences in their topologies giving rise to different origins of the pore loops (Fig. 6A). This suggests convergent evolution to common structural features that sustain dramatically different functions; bEBPs such as NtrC1 remodel the σ54–RNAP holoenzyme bound in a closed complex with promoter DNA, while E1 and DnaB helicases unwind DNA, and Rho factor translocates along an RNA strand.
Figure 6.

Structural similarity in the arrangement of functional loops in diverse homomeric ATPases with different topologies. (A) The functional loops are shown for each ATPase in both the 3D models and the cartoon depicting structural folds. Loop L1 (embellished H2 insertion) of NtrC1 contains at its tip the “GAFTGA” motif for binding σ54. Structures used: 2GXA for E1 helicase and 3ICE for Rho factor (Enemark and Joshua-Tor 2006; Thomsen and Berger 2009) (B) A hypothetical model of an α helix (orange) interacting with the pore of NtrC1 through the T217 residues facing the central channel. See also Supplemental Figure S4.
The unifying features of the NtrC1, E1, and Rho structures include a staircased arrangement of pore loops, overall planarity of the hexameric ring, one semi-open protomer/protomer interface, and an asymmetric distribution of B factors of protein and nucleotides around the ATPase ring. The DnaB helicase shows most of these features, except that its subunits are organized with a helical shift, breaking the mostly planar arrangement seen for subunits of the other ATPases. The MCM helicase and dynein ATPase domain also clearly show a loose or open interface between neighboring subunits (Carter et al. 2011; Lyubimov et al. 2012). A more distant and extreme version of this open and asymmetric architecture of multimeric ATPases is present in clamp loader pentamers (Kelch et al. 2011). All of these ATPases are also shown to contain binding sites with different nucleotide affinities and functionality.
Topologically open hexamer as a general trend?
Is there a general mechanism for multimeric ATPases that invokes the opening of the topologically closed ring? Recent work by the Sauer group (Glynn et al. 2012) showed that cross-linking of the ClpX ATPase subunits with “disulfide staples” between the neighboring subunits does not dramatically disrupt the functionality of the enzyme. However, double stapling of the subunits with two disulfide bonds reduced the processivity of the ClpX substrate unfolding. Using a similar cross-linking approach, the Tsai group (Biter et al. 2012) showed that ClpB ATPase does not need to “disaggregate” for its refolding activity. Such evidence can be interpreted in two ways. Glynn et al. (2012) proposed that properties of the linker between α/β and α subdomains of ClpX permits conformational variation that is crucial for function but that such variation can take place in a covalently closed ring. While the retention of partial function seen in the double-staple ClpX experiments could reflect no need for opening the ring, it could also reflect a reduction but not total loss of subunit–subunit engagement (i.e., “ring opening”) that can take place in the covalently closed topology. The equivalent linker is important for EBP function, as amino acid substitutions in it can uncouple ATP hydrolysis from transcription activation (Xu et al. 2004). It is true that topologically open Clp-type ATPases have not been observed in crystals (except those due to continuous helical packing in which any given hexamer can be considered “topologically open”). Until this study, the same was true for rings of NtrC1; they have been seen as closed, flat rings. Only in the presence of subsaturating amounts of ADP-BeFx has the split-ring form been observed. Perhaps the single snapshots of structure that crystallography provides have yet to reveal important intermediate structures for all AAA+ motors. It thus seems possible but not certain that helicases and bEBP activators may function using the open or helical states, while the protease- and chaperone-associated ATPases prefer closed topologies with lower than sixfold symmetry (for example, see Glynn et al. 2009). What does appear certain is that all of the multimeric ATPases need to break rotational symmetry or lower it in order to function efficiently. That requirement may explain inhibition of the ATPase activity by saturating amounts of ATP in the bEBPs PspF and NtrC1 (Schumacher et al. 2008; Chen et al. 2010).
Extensive data for many ATPases accumulated to date show that each of the oligomeric ATPases may exist in a variety of states. It is possible that different ATPases use different sets of states for functioning, but it is also plausible that each ATPase may invoke most of the geometries in a general multistep mechanism that is yet to be elucidated.
The gap interface of the NtrC1 ATPase as a nucleotide release site
The results reported here begin to reveal how cooperative nucleotide binding allosterically manipulates the bEBP ATPase oligomer structure. An important consequence of pronounced ring cooperativity could be the assignment of unique propensities for ATP hydrolysis among active sites. We suggest that the varied distribution of B factors for proteins and nucleotides around the ring is evidence for such position-dependent roles. The uniquely loose binding pocket formed by subunits A and F may serve as an exit and entrance site for the nucleotide product and reactant. For the EBP, the binding affinity between subunits A and B also appears weak and may play a role in ATPase/nucleotide dynamics. A similar model was previously proposed for Rho and E1 helicases based on the nucleotide-binding pocket structures around the hexameric rings (Enemark and Joshua-Tor 2006; Thomsen and Berger 2009). Another consideration arises from several biochemical studies of various AAA ATPases (Donmez and Patel 2008; Wang et al. 2009; Adams and Reid 2012). These studies showed that the rate-limiting step for ATP hydrolysis is phosphate release, suggesting that the product state entails tight association between ATPase and phosphate anion rather than ADP. Establishing a high-resolution 3D structure of the open hexamer and other possible intermediates of the bEBP activators is needed to clarify this aspect of the mechanism.
Nucleotide status is sufficient to stabilize staircased GAFTGA loops
The split-ring nature of the NtrC1 activator structure is determined solely by the nucleotide status of the ring without needing any additional interaction with its target substrates, σ54 and promoter DNA. This observation is consistent with solution scattering by NtrC1, and thus one can exclude possible artifacts arising from crystal packing. The only other example of a homomeric ATPase with a staircase-like arrangement of pore loops forming in the absence of the macromolecular substrate is the apo crystal structure of bovine papilloma virus helicase E1 (Sanders et al. 2007). It was crystallized in the presence of phosphate and magnesium ions that are found in the binding pockets of the E1 hexamers in the crystal lattice. The investigators attributed the apparent E1 asymmetry to the intrinsic property of the apo homo-oligomer, but it is possible that the asymmetry was induced by the phosphate and Mg2+ ions being bound to the active sites. If so, nucleotide binding may commonly present γ-phosphate to induce large-scale changes to significantly distort rotational symmetry in homomeric, ring-like ATPases. This hypothesis does not exclude the possibility that binding of the ATPase to its macromolecular substrate or accessory factor may enhance, stabilize, or eliminate such a nucleotide-induced asymmetry.
Asymmetry of the NtrC1 hexamer guides its interaction with the target σ54 protein
Interaction of the helicases with nucleic acids is believed to be assisted by the match between their staircased loops and the helical nature of the substrates. Opening of the ring was hypothesized also to serve as an entry point for the DNA or RNA strands to initiate translocation of the substrates by the ATPase motor. In the case of bEBPs, it is not believed that the ATPase acts like a helicase to unwind promoter DNA; rather, the ATPase is thought to act on the σ54–RNAP holoenzyme via remodeling the σ54 factor (Wedel et al. 1990; Wang et al. 1995; Wedel and Kustu 1995). One of the major questions in the field is how the bEBP homo-oligomer performs this mechanical work on the σ54 protein–RNAP complex. It is well established that intact GAFTGA loops are absolutely required for ATPase to bind the σ54 protein and for subsequent steps in open complex formation. Moreover, the Buck and Zhang groups (Joly and Buck 2011; Zhang et al. 2012) provided compelling results that more than one GAFTGA loop is involved in the interaction.
The ring asymmetry shown here indicates that each of the staircased GAFTGA loops can be considered a unique binding environment. A straightforward hypothesis is that the uniqueness of these ring features guides interaction with σ54. Our EM data show that is indeed the case, revealing a particular geometry between the ATPase, σ54, and promoter DNA. In this geometry, the σ54/promoter complex forms asymmetrical lateral contacts with some exposed GAFTGA loops but also to some intersubunit interfaces (Fig. 5). Although it was observed earlier that σ54 protein can bind with other oligomeric states of the bEBPs (Rappas et al. 2005; Chen et al. 2010), we propose that the asymmetric hexamer provides a unique configuration for guiding the protein–protein interaction. Our preliminary EM data for the NtrC1 ATPase in complex with σ54–promoter–RNAP suggest the presence of a similarly asymmetric NtrC1 hexamer (S Chowdhury, S DeCarlo, and BT Nixon, unpubl.). A pronounced “notch” in the ATPase activator is also notable in the previously reported EM structure of σ54–RNAP in complex with another bEBP protein, PspF (EMDataBank EMD 1566) (Supplemental Fig. S5; Bose et al. 2008).
In the prior and more recent PspF studies, the investigators suggest two points of contact between the σ54–RNAP complex and the activator (Rappas et al. 2005; Zhang et al. 2012). It is interesting to wonder whether there is a second site of specific interaction with the bEBP created by the gap in the ring. The gap interface also creates a new available binding interface. Recent crystal structure of a nonplanar hexameric state of the DnaB helicase shows that its ring opening results in extensive contacts of the N-terminal DnaB domain with the ATPase subunit adjacent to the gap; such interactions are absent for the nongap subunits (Itsathitphaisarn et al. 2012). It is also possible that the bEBP ensemble undergoes significant rearrangements during transcription initiation.
Roles of the GAFTGA loops and the central pore
As mentioned above, the novel structure for the NtrC1 transcription activator resembles structures of helicases that thread ssDNA or RNA substrates through their respective central pores. Also, other homomultimeric ATPases like ClpX thread polypeptide substrates through their pores. It is thus possible that bEBP activators use the GAFTGA loops to thread a portion of σ54 or promoter DNA through the central pore.
Extensive mutagenesis studies suggest that bEBPs remodel σ54 protein via interactions with region I, the protein's N-terminal domain (Gonzalez et al. 1998; Bordes et al. 2004; Dago et al. 2007; Schumacher et al. 2007; Zhang et al. 2009). Region I spans ∼50 amino acids that are predicted to form an α helix; it possesses two short motifs that are suggested to make direct contact with the GAFTGA motifs of the ATPase (Merrick 1993; Zhang et al. 2012). The dimensions of the NtrC1 pore in the hexameric ensemble are close to the diameter of a protein helix and thus is large enough to pass region I through the channel (Fig. 6B). Nuclear magnetic resonance (NMR) studies of pieces of σ54 from A. aeolicus were interpreted by the Wemmer laboratory (Hong et al. 2009) to suggest that pulling region I could trigger a refolding of a portion of σ54 that binds the core of RNAP. Prior studies by Zhang and Buck (Bose et al. 2008) were interpreted as evidence for this core-binding portion of σ54 physically blocking the active site of RNAP. Nevertheless, in the EM model presented in this study and the EM data published earlier (Rappas et al. 2005; Chen et al. 2010), there is no observable density obstructing the pore of the bEBP ring. The new data imply that such pulling would begin with binding to specific portions of the asymmetric surface of the split ring. Subsequent threading could take place, or pulling could occur from the surface. Precedent for lateral as opposed to threading interaction exists for the dynein ATPase. Its ring is thought to apply force on the stalk portion of the dynein molecule with the help of the tail domain that is positioned on one side of the dynein ring, interacting with two or three ATPase domains of the ring (Carter et al. 2011). This lateral interaction between the protein substrate and tail differs dramatically from the centrally threaded polymer models proposed for the helicases and the ClpX and ClpB unfoldases. To distinguish among these possibilities, higher-resolution structures need to be established for a bEBP activator in complex with the σ54 substrate.
In summary, our studies of the NtrC1 ATPase domain underscore the flexibility in packing stoichiometry and interface angles inherent to AAA+ ATPases. The work suggests how allosteric pathways sensitive to the presence or absence of the γ-phosphate of ATP can provide homomeric ATPase rings with an ordered asymmetry—an asymmetry that imparts unique identity and thus unique function to each subunit that is harnessed to deliver mechanical work to asymmetric target macromolecules.
Materials and methods
Details of sample preparations and experiments are given in the Supplemental Material.
ITC
Binding of nucleotides to NtrC1 at 25°C was assayed using a Microcal VP-ITC calorimeter (GE Healthcare). After degassing, 200 μM NtrC1 solution in base buffer (20 mM TrisHCl at pH 7.9, 5% [w/v] glycerol, 200 mM KCl) was loaded into the cell, and the syringe was filled with the nucleotide solution (5 mM).
Static SAXS measurements
X-ray scattering experiments were performed at the BioCAT beamline (ID18) (Fischetti et al. 2004) of the Advanced Photon Source. Solutions were passed through the capillary and exposed to the X-ray beam. For titrations, varied amounts of ADP were placed in the presence of constant concentrations of the NtrC1 protein (330 μM protein in base buffer supplemented with 5 mM TCEP) and salts BeCl2 (1 mM), NaF (8 mM), and MgCl2 (5 mM).
Stopped-flow SAXS
A Biolog stopped-flow device was inserted in the beam path so that X-rays passed through the reaction cuvette contained an aging sample. Scattering was recorded with an Aviex CCD camera or Pilatus detector built with CMOS hybrid-pixel technology.
Crystallization, data collection, and refinement
NtrC1 protein was screened for crystallization conditions in the presence of ADP-BeFx and Mg2+ under oil using Crystal screens I/II (Hampton Research). Optimal crystals were obtained in 20% ethylene glycol with 300–500 μM ADP-BeFx in the initial protein solution (20 g/L). The data were collected at beamline 8.3.1 of the Advanced Light Source and X29 at the National Synchrotron Light Source. Data were processed with CrystalClear software (Rigaku). For both crystals, initial phase estimates were found by molecular replacement with a subunit from the ATP-saturated structure (PDB 3M0E) as a search model (Phaser) (Mccoy et al. 2007). Refinement was done first in CNS (Brunger 2007; Schroder et al. 2010) and then with reference model restraints in Phenix (Adams et al. 2010) and Coot (Emsley and Cowtan 2004). Details of handling the metal fluoride analogs, crystallization, and initial processing of the diffraction data are described elsewhere (Sysoeva et al. 2013).
EM
The ternary complex contained the ATPase domain of NtrC1 (A. aeolicus), σ54 (Klebsiella pneumoniae), and the 36-base-pair (bp) promoter DNA (Sinorhizobium meliloti nifH). Components were mixed in base buffer supplemented with 1 mM ADP-AlFx, and the complex was purified by size exclusion chromatography (Superdex200, GE Healthcare). Purified complex was absorbed on glow-discharged, carbon-coated grids and stained with uranyl formate. Low-dose images were obtained on a JEOL2100F transmission electron microscope. Particle picking, maximum likelihood-based two-dimensional classification, and initial model building using random conical tilt were performed using XMIPP (Marabini et al. 1996). The ISAC program of SPARX (Yang et al. 2012) provided reference-free classification of untilted data. The initial RCT reconstruction was refined to 24 Å resolution (Fourier shell correlation of 0.5) by iterative projection matching and back projection using EMAN2 and SPARX libraries (Hohn et al. 2007). Models were analyzed with University of California at San Francisco Chimera (Pettersen et al. 2004).
Accession Numbers
Atomic coordinates and structure factors have been deposited in PDB with ID codes 4LY6 (S crystal) and 4LZZ (Q crystal), and the EM model of the ternary complex has been deposited in the EMDataBank with accession code EMD 5698.
Acknowledgments
We thank Mark Signs at the fermentation facility of the Huck Institute of the Pennsylvania State University for helping develop high-yield fermentations conditions for NtrC1 and σ54 purifications; Marc Allaire of National Synchrotron Light Source at Brookhaven National Laboratory and Neela Yennawar of the X-ray Crystallography Facility of The Huck Institutes for Life Sciences at Pennsylvania State University for assistance with the diffraction data collection; Andrea Berger and Ra Hel of the Statistical Consulting Services of the Eberly College of Science at Pennsylvania State University; Sacha De Carlo, Ruben Diaz, and William Rice for help with EM data collection and model building; Michael Fenn of the Research Computing and CyberInfrastructure at Pennsylvania State University and Borries Demeler of The University of Texas Health Science Center at San Antonio for access to the computational resources for crystal structure refinement and model building from SAXS and EM data. Use of the Advanced Photon Source, the Advanced Light Source, and the National Synchrotron Light Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract numbers W-31-109-ENG-38, DE-AC02-05CH11231, and DEAC02-98CH10886. The BioCAT is a National Institutes of Health-supported Research Center RR-08630. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Center for Research Resources or the National Institutes of Health.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.229385.113.
References
- Adams NB, Reid JD 2012. Nonequilibrium isotope exchange reveals a catalytically significant enzyme-phosphate complex in the ATP hydrolysis pathway of the AAA+ ATPase magnesium chelatase. Biochemistry 51: 2029–2031 [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor JD, Doucleff M, Lee CJ, Matsubara K, De Carlo S, Heideker J, Lamers MH, Pelton JG, Wemmer DE 2008. Structure and regulatory mechanism of Aquifex aeolicus NtrC4: Variability and evolution in bacterial transcriptional regulation. J Mol Biol 384: 1058–1075 [DOI] [PubMed] [Google Scholar]
- Batchelor JD, Sterling HJ, Hong E, Williams ER, Wemmer DE 2009. Receiver domains control the active-state stoichiometry of Aquifex aeolicus σ54 activator NtrC4, as revealed by electrospray ionization mass spectrometry. J Mol Biol 393: 634–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biter AB, Lee S, Sung N, Tsai FT 2012. Structural basis for intersubunit signaling in a protein disaggregating machine. Proc Natl Acad Sci 109: 12515–12520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordes P, Wigneshweraraj SR, Chaney M, Dago AE, Morett E, Buck M 2004. Communication between Eσ(54), promoter DNA and the conserved threonine residue in the GAFTGA motif of the PspF σ-dependent activator during transcription activation. Mol Microbiol 54: 489–506 [DOI] [PubMed] [Google Scholar]
- Bose D, Pape T, Burrows PC, Rappas M, Wigneshweraraj SR, Buck M, Zhang X 2008. Organization of an activator-bound RNA polymerase holoenzyme. Mol Cell 32: 337–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT 2007. Version 1.2 of the crystallography and NMR system. Nat Protoc 2: 2728–2733 [DOI] [PubMed] [Google Scholar]
- Bujalowski W, Klonowska MM 1993. Negative cooperativity in the binding of nucleotides to Escherichia coli replicative helicase DnaB protein. Interactions with fluorescent nucleotide analogs. Biochemistry 32: 5888–5900 [DOI] [PubMed] [Google Scholar]
- Carter AP, Cho C, Jin L, Vale RD 2011. Crystal structure of the dynein motor domain. Science 331: 1159–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Doucleff M, Wemmer DE, De Carlo S, Huang HH, Nogales E, Hoover TR, Kondrashkina E, Guo L, Nixon BT 2007. ATP ground- and transition states of bacterial enhancer binding AAA+ ATPases support complex formation with their target protein, σ54. Structure 15: 429–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Sysoeva TA, Chowdhury S, Guo L, De Carlo S, Hanson JA, Yang H, Nixon BT 2010. Engagement of arginine finger to ATP triggers large conformational changes in NtrC1 AAA+ ATPase for remodeling bacterial RNA polymerase. Structure 18: 1420–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dago AE, Wigneshweraraj SR, Buck M, Morett E 2007. A role for the conserved GAFTGA motif of AAA+ transcription activators in sensing promoter DNA conformation. J Biol Chem 282: 1087–1097 [DOI] [PubMed] [Google Scholar]
- Donmez I, Patel SS 2008. Coupling of DNA unwinding to nucleotide hydrolysis in a ring-shaped helicase. EMBO J 27: 1718–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucleff M, Chen B, Maris AE, Wemmer DE, Kondrashkina E, Nixon BT 2005. Negative regulation of AAA+ ATPase assembly by two component receiver domains: A transcription activation mechanism that is conserved in mesophilic and extremely hyperthermophilic bacteria. J Mol Biol 353: 242–255 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K 2004. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Enemark EJ, Joshua-Tor L 2006. Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442: 270–275 [DOI] [PubMed] [Google Scholar]
- Fischetti R, Stepanov S, Rosenbaum G, Barrea R, Black E, Gore D, Heurich R, Kondrashkina E, Kropf AJ, Wang S, et al. 2004. The BioCAT undulator beamline 18ID: A facility for biological non-crystalline diffraction and X-ray absorption spectroscopy at the Advanced Photon Source. J Synchrotron Radiat 11: 399–405 [DOI] [PubMed] [Google Scholar]
- Glynn SE, Martin A, Nager AR, Baker TA, Sauer RT 2009. Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell 139: 744–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn SE, Nager AR, Baker TA, Sauer RT 2012. Dynamic and static components power unfolding in topologically closed rings of a AAA+ proteolytic machine. Nat Struct Mol Biol 19: 616–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez V, Olvera L, Soberon X, Morett E 1998. In vivo studies on the positive control function of NifA: A conserved hydrophobic amino acid patch at the central domain involved in transcriptional activation. Mol Microbiol 28: 55–67 [DOI] [PubMed] [Google Scholar]
- Hayward S, Berendsen HJ 1998. Systematic analysis of domain motions in proteins from conformational change: New results on citrate synthase and T4 lysozyme. Proteins 30: 144–154 [PubMed] [Google Scholar]
- Hayward S, Kitao A, Berendsen HJ 1997. Model-free methods of analyzing domain motions in proteins from simulation: A comparison of normal mode analysis and molecular dynamics simulation of lysozyme. Proteins 27: 425–437 [DOI] [PubMed] [Google Scholar]
- Hohn M, Tang G, Goodyear G, Baldwin PR, Huang Z, Penczek PA, Yang C, Glaeser RM, Adams PD, Ludtke SJ 2007. SPARX, a new environment for Cryo-EM image processing. J Struct Biol 157: 47–55 [DOI] [PubMed] [Google Scholar]
- Hong E, Doucleff M, Wemmer DE 2009. Structure of the RNA polymerase core-binding domain of σ(54) reveals a likely conformational fracture point. J Mol Biol 390: 70–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itsathitphaisarn O, Wing RA, Eliason WK, Wang J, Steitz TA 2012. The hexameric helicase DnaB adopts a nonplanar conformation during translocation. Cell 151: 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly N, Buck M 2011. Single chain forms of the enhancer binding protein PspF provide insights into geometric requirements for gene activation. J Biol Chem 286: 12734–12742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly N, Schumacher J, Buck M 2006. Heterogeneous nucleotide occupancy stimulates functionality of phage shock protein F, an AAA+ transcriptional activator. J Biol Chem 281: 34997–35007 [DOI] [PubMed] [Google Scholar]
- Kelch BA, Makino DL, O'Donnell M, Kuriyan J 2011. How a DNA polymerase clamp loader opens a sliding clamp. Science 334: 1675–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K 2007. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774–797 [DOI] [PubMed] [Google Scholar]
- Lee SY, De La Torre A, Yan D, Kustu S, Nixon BT, Wemmer DE 2003. Regulation of the transcriptional activator NtrC1: Structural studies of the regulatory and AAA+ ATPase domains. Genes Dev 17: 2552–2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyubimov AY, Costa A, Bleichert F, Botchan MR, Berger JM 2012. ATP-dependent conformational dynamics underlie the functional asymmetry of the replicative helicase from a minimalist eukaryote. Proc Natl Acad Sci 109: 11999–12004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabini R, Masegosa IM, San Martin MC, Marco S, Fernandez JJ, de la Fraga LG, Vaquerizo C, Carazo JM 1996. Xmipp: An image processing package for electron microscopy. J Struct Biol 116: 237–240 [DOI] [PubMed] [Google Scholar]
- Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ 2007. Phaser crystallographic software. J Appl Crystallogr 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick MJ 1993. In a class of its own–the RNA polymerase σ factor σ54 (σN). Mol Microbiol 10: 903–909 [DOI] [PubMed] [Google Scholar]
- Mosca R, Schneider TR 2008. RAPIDO: A Web server for the alignment of protein structures in the presence of conformational changes. Nucleic Acids Res 36: W42-W46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosca R, Brannetti B, Schneider TR 2008. Alignment of protein structures in the presence of domain motions. BMC Bioinformatics 9: 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Rappas M, Schumacher J, Beuron F, Niwa H, Bordes P, Wigneshweraraj S, Keetch CA, Robinson CV, Buck M, Zhang X 2005. Structural insights into the activity of enhancer-binding proteins. Science 307: 1972–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappas M, Schumacher J, Niwa H, Buck M, Zhang X 2006. Structural basis of the nucleotide driven conformational changes in the AAA+ domain of transcription activator PspF. J Mol Biol 357: 481–492 [DOI] [PubMed] [Google Scholar]
- Sallai L, Tucker PA 2005. Crystal structure of the central and C-terminal domain of the σ(54)-activator ZraR. J Struct Biol 151: 160–170 [DOI] [PubMed] [Google Scholar]
- Sanders CM, Kovalevskiy OV, Sizov D, Lebedev AA, Isupov MN, Antson AA 2007. Papillomavirus E1 helicase assembly maintains an asymmetric state in the absence of DNA and nucleotide cofactors. Nucleic Acids Res 35: 6451–6457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder GF, Levitt M, Brunger AT 2010. Super-resolution biomolecular crystallography with low-resolution data. Nature 464: 1218–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher J, Joly N, Rappas M, Bradley D, Wigneshweraraj SR, Zhang X, Buck M 2007. Sensor I threonine of the AAA+ ATPase transcriptional activator PspF is involved in coupling nucleotide triphosphate hydrolysis to the restructuring of σ 54-RNA polymerase. J Biol Chem 282: 9825–9833 [DOI] [PubMed] [Google Scholar]
- Schumacher J, Joly N, Claeys-Bouuaert IL, Aziz SA, Rappas M, Zhang X, Buck M 2008. Mechanism of homotropic control to coordinate hydrolysis in a hexameric AAA+ ring ATPase. J Mol Biol 381: 1–12 [DOI] [PubMed] [Google Scholar]
- Strong M, Sawaya MR, Wang S, Phillips M, Cascio D, Eisenberg D 2006. Toward the structural genomics of complexes: Crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci 103: 8060–8065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D, Barberato C, Koch MHJ 1995. CRYSOL—a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr 28: 768–773 [Google Scholar]
- Sysoeva TA, Yennawar N, Allaire M, Nixon BT 2013. Crystallization and preliminary X-ray analysis of the ATPase domain of the σ54-dependent transcription activator NtrC1 from Aquifex aeolicus bound to ATP analog ADP-BeFx. Acta Crystallogr Sect F Struct Biol Cryst Commun doi: 10.1107/S174430911302976X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen ND, Berger JM 2009. Running in reverse: The structural basis for translocation polarity in hexameric helicases. Cell 139: 523–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidangos N, Maris AE, Young A, Hong E, Pelton JG, Batchelor JD, Wemmer DE 2013. Structure, function, and tethering of DNA-binding domains in σ transcriptional activators. Biopolymers 99: 1082–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Syed A, Hsieh M, Gralla JD 1995. Converting Escherichia coli RNA polymerase into an enhancer-responsive enzyme: Role of an NH2-terminal leucine patch in σ54. Science 270: 992–994 [DOI] [PubMed] [Google Scholar]
- Wang Q, Arnold JJ, Uchida A, Raney KD, Cameron CE 2009. Phosphate release contributes to the rate-limiting step for unwinding by an RNA helicase. Nucleic Acids Res 38: 1312–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedel A, Kustu S 1995. The bacterial enhancer-binding protein NTRC is a molecular machine: ATP hydrolysis is coupled to transcriptional activation. Genes Dev 9: 2042–2052 [DOI] [PubMed] [Google Scholar]
- Wedel A, Weiss DS, Popham D, Droge P, Kustu S 1990. A bacterial enhancer functions to tether a transcriptional activator near a promoter. Science 248: 486–490 [DOI] [PubMed] [Google Scholar]
- Xu H, Kelly MT, Nixon BT, Hoover TR 2004. Novel substitutions in the σ54-dependent activator DctD that increase dependence on upstream activation sequences or uncouple ATP hydrolysis from transcriptional activation. Mol Microbiol 54: 32–44 [DOI] [PubMed] [Google Scholar]
- Yang Z, Fang J, Chittuluru J, Asturias FJ, Penczek PA 2012. Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure 20: 237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Joly N, Burrows PC, Jovanovic M, Wigneshweraraj SR, Buck M 2009. The role of the conserved phenylalanine in the σ54-interacting GAFTGA motif of bacterial enhancer binding proteins. Nucleic Acids Res 37: 5981–5992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Joly N, Buck M 2012. A common feature from different subunits of a homomeric AAA+ protein contacts three spatially distinct transcription elements. Nucleic Acids Res 40: 9139–9152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Gordivenko Y, Joly N, Lawton E, Robinson CV, Buck M 2013. Subunit dynamics and nucleotide-dependent asymmetry of an AAA+ transcription complex. J Mol Biol. doi: 10.1016/j.jmb.2013.08.018 [DOI] [PubMed]