

Abstract
The majority of malignant primary brain tumors are gliomas, derived from glial cells. Grade IV gliomas, Glioblastoma multiforme, are extremely invasive and the clinical prognosis for patients is dismal. Gliomas utilize a number of proteins and pathways to infiltrate the brain parenchyma including ion channels and calcium signaling pathways. In this study, we investigated the localization and functional relevance of transient receptor potential canonical (TRPC) channels in glioma migration. We show that gliomas are attracted in a chemotactic manner to epidermal growth factor (EGF). Stimulation with EGF results in TRPC1 channel localization to the leading edge of migrating D54MG glioma cells. Additionally, TRPC1 channels co-localize with the lipid raft proteins, caveolin-1 and β-cholera toxin, and biochemical assays show TRPC1 in the caveolar raft fraction of the membrane. Chemotaxis toward EGF was lost when TRPC channels were pharmacologically inhibited or by shRNA knockdown of TRPC1 channels, yet without affecting unstimulated cell motility. Moreover, lipid raft integrity was required for gliomas chemotaxis. Disruption of lipid rafts not only impaired chemotaxis but also impaired TRPC currents in whole cell recordings and decreased store-operated calcium entry as revealed by ratiomeric calcium imaging. These data indicated that TRPC1 channel association with lipid rafts is essential for glioma chemotaxis in response to stimuli, such as EGF.
Gliomas constitute 78% of all malignant brain tumors and are characterized by diffuse invasion into the brain parenchyma (Demuth and Berens, 2004; Schwartzbaum et al., 2006). This invasiveness contributes to the poor prognosis of patients since complete resection is typically impossible (Giese et al., 2003; Bellail et al., 2004). Patients diagnosed with the most invasive gliomas, Glioblastoma multiforme, a grade IV astrocytoma, have a median survival under 1 year (Huncharek and Muscat, 1998; Butowski et al., 2006).
Numerous studies have focused on gaining a better understanding of different molecular mechanisms exploited by invading tumor cells. For example, research has shown that gliomas secrete matrix metalloproteinases to degrade the extracellular matrix surrounding invading cells (Levicar et al., 2003). Several integrins have been associated with glioma migration including αvβ3, whose inhibition decreases glioma motility in transwell assays (Platten et al., 2000). Further, integrins such as αvβ1 are crucial for new blood vessel formation (Soldi et al., 1999), which act as scaffolds for glioma cell invasion (Farin et al., 2006).
Recently, ion channels have been implicated in glioma invasion as a means to regulate cell size (Sontheimer, 2008) or by participation in Ca2+ signaling within migratory cells (Weaver et al., 2006; Arcangeli et al., 2009). Indeed, it has been proposed that ion channels have a role in setting the intracellular cytoplasmic environment by controlling cell volume, which both affects cytoskeletal elements and is affected by the cytoskeleton (Schwab et al., 2007). In addition, cell migration depends upon ion channels as calcium channels are known to be important for intracellular calcium concentration (Komuro and Kumada, 2005b).It has been shown that migrating neurons (Komuro and Rakic, 1998) and glioma cells (Bordey et al., 2000) display oscillatory changes in intracellular Ca2+ which correlate with the velocity of cell migration. Consequently, channels that allow the influx of Ca2+ are considered to be particularly important for signaling in migration (Cole and Kohn, 1994). One family of ion channels that is particularly well suited to participate in Ca2+ signaling is the transient receptor potential canonical (TRPC) channel family (Louis et al., 2008).
TRPC channels are tetrameric, non-selective cation channels which are activated by the phospholipase C cascade and involved in agonist-induced calcium entry and store-operated calcium entry (SOCE) (Freichel et al., 2004; Beech, 2005; Worley et al., 2007; Tai et al., 2008). TRPC channels have been linked to migration and chemotaxis in a number of cell types. For example, in skeletal myoblasts, TRPC1 inhibition decreases calcium signals and migration while activation enhances migration (Louis et al., 2008). Likewise, in renal epithelial cells, decreased TRPC1 channel expression resulted in decreased migration (Fabian et al., 2008). Possibly the best example of TRPC involvement in migration stems from chemotactic studies on axonal growth cone guidance. Here, pharmacological and siRNA studies show a role for TRPC1 channels in netrin-1 mediated axonal guidance (Wang and Poo, 2005). Additionally, TRPC-3 and -6 are involved in axonal growth cone guidance by brain-derived neurotrophic factor (BDNF) as shown by dominant negative studies and siRNA directed against TRPC3 (Li et al., 2005).
In order for cells to recognize concentration gradients, receptor and channel expression must be polarized. There has been evidence to suggest that TRPC channels and other calcium signaling components important for migration are localized to lipid rafts (McFerrin and Sontheimer, 2005; Manes and Viola, 2006; Pani and Singh, 2009), specialized membrane domains enriched in cholesterol and sphingolipids (Brown and London, 2000). The cholesterol composition of lipid rafts attracts proteins with specific post-translational modifications such as glycosylphosphatidlinositol (GPI)-anchors (Simons and Ikonen, 1997) or acylation (Brown and London, 2000), which direct proteins to the endoplasmic reticulum for trafficking. The spatial constraints of lipid rafts allows for localized signaling to occur between these proteins in processes such as SOCE, a process which TRPC channels are thought to be involved (Beech, 2005; Allen et al., 2007; Worley et al., 2007).
In a recent study, we described expression and function of TRPC channels in human malignant gliomas and show their involvement in SOCE (Bomben and Sontheimer, 2010). In the present study, we investigate the role of TRPC channels in glioma migration and chemotaxis. We find that TRPC1 channels are localized to lipid raft domains at the leading edge of migrating cells and that TRPC channel function is essential for glioma chemotaxis toward epidermal growth factor (EGF). Using inducible shRNA plasmids to suppress TRPC1 channel expression, we were able to attenuate glioma chemotaxis toward EGF without affecting non-directed migration; hence, specifically implicating TRPC1 channels in glioma chemotaxis.
Materials and Methods
Cell culture
Experiments were done using a human grade IV glioma cell line D54MG, a gift by Dr. D. Bigner (Duke University, Durham, NC, obtained 2001). Cells were maintained as described in (Bomben and Sontheimer, 2008) in DMEM/F12 with 7% fetal bovine serum. D54MG cells were also used for transfections of shRNA plasmids (Open Biosystems, Huntsville, AL) and clones of the inducible plasmids were generated using puromycin (Sigma–Aldrich, St. Louis, MO) resistance.
Drugs and solutions
The TRPC inhibitors SKF96365, MRS-1845, and 2-aminoethoxydiphenylborane (2-APB) were obtained from Sigma–Aldrich. Puromycin, doxycycline, cyclopiazonic acid (CPA), and methyl-β-cyclodextrin (MβCD) were also obtained from Sigma. Electrophysiological recordings were done in a sulfate and phosphate free bath solution that consisted of the following (in mM): 130 NaCl, 5 KCl, 1 CaCl2, 10.5 d-glucose, 32.5 HEPES, pH adjusted to 7.4 with NaOH. For calcium imaging, bath solution consisted of the following (in mM): 125 NaCl, 5 KCl, 1.2 MgSO4, 1 CaCl2, 1.6 Na2HPO4, 0.4 NaH2PO4, 10.5 d-glucose, 32.5 HEPES, pH adjusted to 7.4 with NaOH. Calcium was omitted from the recipe when calcium-free solutions were necessary. Pipette solutions contained the following (in mM): 145 KCl, 1 MgCl2, 0.2 CaCl2, 10 EGTA, 10 HEPES sodium salt, pH adjusted to 7.2 with Tris-base. For all experiments using MβCD, treatment was carried out as follows. MβCD, at a concentration of 5 mg/ml, was resuspended in serum free media. Cells were washed twice with serum free media and then incubated with MβCD for 15–30 min. Cells were then immediately used for electrophysiology.
Immunocytochemistry
D54MG cells were seeded and stained as described in Bomben and Sontheimer (2008). For immunocytochemistry on transwell assays, fluorescence blocked transwell migration inserts were used, antibodies were incubated in the same way on the top and bottom of the filter. Following staining, the membrane was cut out of the filter and mounted in between two coverslips for imaging. Confocal images were obtained using a Hamatsu (Hamamatsu City, Japan) IEEE1392 CCD digital camera mounted on an Olympus (Melville, NY) IX81 motorized, inverted microscope containing a disk spinning unit (DSU). The microscope was controlled by Slidebook software (Intelligent Imaging Inovations, Inc., Denver, CO) and image stacks were obtained from scans spaced 0.5 μm apart using FITC, TRITC, and DAPI filter sets.
Lipid raft isolation
The entire process of isolation was carried out at 4°C. Following treatment with either SF media (control) or SF media with 5 mg/ml MβCD, confluent cultures of cells were washed two times in cold PBS. Cells were collected in TNE (5mm Tris–HCl, pH 7.6, 150mm NaCl, 5mm EDTA) buffer by scraping and centrifuged for 5 min at 12,000g. The supernatant was aspirated, and cells were resuspended in 500 μl of TNE and mechanically homogenized. Protein concentrations were determined using the DC protein assay kit (Bio-Rad, Hercules, CA) according to manufacturer's instructions. After determining protein concentration, all samples were diluted to the same concentration (typically 1 mg/ml) and the same volume to allow for comparison between samples. Lysates were then centrifuged for 1 h at 5,000 rpm, and the supernatant was transferred to another tube and labeled the “water-soluble” fraction. The remaining pellet was resuspended in cold lysis buffer (1% Triton X-100 in TNE buffer) and incubated on ice for 15 min with occasional vortexing. The water-insoluble fraction was then centrifuged at 5,000 rpm for 30 min. The supernatant was removed and labeled the “detergent-soluble” fraction. The pellet was then resuspended in a 40% Optiprep solution (60% stock Optiprep diluted in TNE) and placed in an ultracentrifuge tube. 3.5 ml of a 30% Optiprep solution were then layered on top of the 40% layer. Finally, 0.5 ml of a 5% Optiprep solution was layered on top. The samples were spun in a Beckman Ultracentrifuge using the SW-60 swinging bucket rotor at 36,000 rpm for 16 h at 4°C. Following centrifugation, 500-μl fractions were collected from the top to the bottom of each sample and numbered 1–9. Samples were then loaded onto a 4–20% gradient SDS–polyacrylamide gel and run according to the Western blotting protocol.
Western blots
Blots were run as described in Bomben and Sontheimer (2008). We obtained antibodies from Alomone Labs Ltd., Jerusalem, Israel (TRPC1), Abcam, Cambridge, MA (β-cholera toxin, EGFR), and Santa Cruz, Santa Cruz, CA (caveolin-1).
Transfections of shRNA and control plasmids
To knockdown TRPC1, we obtained pGIPZ-lentiviral shRNAmir vectors containing either non-silencing (NS) scrambled sequence or one of two hairpin sequences targeting TRPC1 (Open Biosystems). Plasmids were catalog numbers RHS4346 (NS), RHS4430-98486752 (shRNA1), RHS4430-99292249 (shRNA2). The pGIPZ vectors also expressed GFP, to identify transfected cells. For inducible knockdown, pTRIPZ-lentiviral vectors were obtained (catalog numbers RHS4743 and RHS4696-99683013) for NS and shRNA1 plasmids respectively and TurboRed® expression indicated induction of shRNA. Cells were transfected as described in Weaver et al. (2006). To generate stable lines, 1 μg/ml puromycin treatment began 96 h after transfection. After selection, cells were passaged (density: 0.5 cells/100 μl) into 96-well plates and scored for single colonies.
Calcium imaging
Cells were loaded with Fura-2-acetoxymethylester (5μmol/L, TEFLABS) reconstituted in 20% (w/v) pluronic acid in DMSO (Invitrogen, Carlsbad, CA). For SOCE, cells were in normal bath (containing calcium) and placed on microscope to equilibrate. Recordings were obtained with an Olympus DSU fluorescent imaging microscope where cells were alternately excited at 340 and 380 nm. Emitted light was collected at >520 nm. Slidebook software (Intelligent Imaging Innovations, Inc.) digitized images and obtained 340:380 nm ratios. After obtaining baselines, cells were rinsed twice and calcium-free bath was applied. Resuming imaging, we collected baseline data then added 10μM CPA (first peak). After 10 min, extracellular calcium was restored to 1mM (second peak) and imaging continued for 10 min.
Electrophysiology
Recordings of whole-cell currents were made using an Axopatch 200A amplifier (Axon Instruments, Foster City, CA) following standard recording techniques (Hamill et al., 1981) and described in Bomben and Sontheimer (2008).
Migration assay
One day prior to the experiment, a 70% confluent dish of cells was aspirated and supplied with serum-free media overnight. Cell culture inserts (BD Biosciences, San Diego, CA) with 8-μm pores were coated overnight with Vitronectin (BD Biosciences) at a concentration of 5μg/ml in PBS. The following day, inserts were blocked with 1% fatty acid-free bovine serum albumin for 1 h. Inserts were then washed two times in PBS, and 400μl of migration assay buffer (MAB, 0.1% fatty acid-free bovine serum albumin in serum-free media) was added to the bottom of each well. Cells were rinsed once in PBS and were lifted off the dish by the addition of 0.5mm EGTA for ∼20 min. Cells were rinsed twice by centrifugation and resuspended in MAB and counted. Forty thousand cells were plated on top of each filter and allowed to adhere for 30 min before drug was added. When EGF was added to the filters, it was only added to the bottom of the filter for chemotaxis as opposed to application to both sides of the filter. After addition of drug, cells were allowed to migrate for 5 h. Filters were then fixed and stained with Crystal Violet, and the tops were wiped clean of cells, and representative fields (five per filter) were counted with a Zeiss Axiovert 200M microscope with a 20× objective.
Data analysis
Results were analyzed using Origin (v.6.0, MicroCal Software, Northhampton, MA). Significance was determined by one-way ANOVA or Student's t-tests, as appropriate, since all data showed normal distribution. All data reported are mean±SEM and *denotes P<0.05 unless otherwise stated.
Results
TRPC channels co-localize with lipid raft proteins at the leading edge of glioma cells
As channels that are potentially important for calcium entry, TRPC channels have been shown, in some cells, to localize to particular microdomains, called lipid rafts, in the plasma membrane (Beech, 2005). The two types of lipid rafts described are planar rafts and caveolae (Nicolau et al., 2006). Because of the cholesterol composition of the rafts, marker proteins that often segregate to lipid rafts are caveolin-1 and β-cholera toxin (Brown and London, 2000; Allen et al., 2007). To assess whether TRPC1 channels localize to lipid rafts, we performed immunocytochemistry on D54MG glioma cells, derived from a grade IV glioblastoma multiforme patient. Using confocal microscopy, image stacks in Figure 1A revealed TRPC1 channels co-localized to regions of the plasma membrane with caveolin-1. To look more closely at regions of co-localization, a single 0.5μm z-plane is shown in Figure 1B, where we utilized the three-dimensional image, at this plane, of D54 cells stained with caveolin and TRPC1 channel antibodies (white arrows in side views point out leading edge co-localization). We also see that another lipid raft marker, β-cholera toxin, co-localized with TRPC1 channels at a protruding edge of glioma cells (Fig. 1C).
Fig. 1.

TRPC1 channels localize to lipid rafts of glioma cells. A: Immunocytochemistry on D54MG cells revealed co-localization with TRPC1 channels (red) and caveolin-1 (green) on protruding edges of glioma cells. B: Three-view image of D54MG cells stained with TRPC1 channels (red) and caveolin-1 (green). White arrows within the merged image show the line scans of the side views demonstrated within this 0.5 μm z-plane. White arrows within the side views of the image point out co-localization of TRPC1 andcaveolin-1 attheedge of the glioma cell. C:β-Choleratoxin (green) and TRPC1 channels (red) were also co-localized on the protruding edge of glioma cells. D: In D54MG cells grown in serum conditions, TRPC1 channels were located in the lipid raft fraction of detergent insoluble proteins fractionated by density gradient. Since lipid rafts are buoyant between the first two fractions, the proteins associated with these rafts (i.e., caveolin-1) fractionated to the second fraction. A small proportion of caveolin-1 was also located within the detergent soluble fraction. E: In chronic serum starvation conditions, TRPC1 channels were located in the detergent soluble fraction of the biochemical dissociation and the majority proportion of caveolin-1 became localized to the detergent soluble fraction.
To confirm that TRPC1 channels localize to lipid rafts, we turned to a well characterized biochemical separation of lipid raft components (Rujoi et al., 2003). The protocol separates whole cell lysates into water-soluble, detergent-soluble, and detergent-insoluble fractions at 4°C as determined by their relative solubilities in aqueous solutions with or without detergent. Cytosolic constituents were isolated within the water-soluble fraction leaving behind membrane and cytoskeletal elements. These elements were further dissociated into both a detergent-soluble fraction that was solubilized in Triton X-100, containing membrane-associated proteins and detergent-insoluble proteins. The detergent-insoluble fraction consists of cytoskeletal proteins as well as proteins associated with regions of the membrane exhibiting concentrated amounts of lipid which renders them insoluble in detergent at 4°C. The insoluble fraction was further separated into nine fractions by discontinuous gradient centrifugation. All 11 isolated fractions were run on an SDS–polyacrylamide gel and then Western blotted and probed for both lipid raft markers and TRPC1 channels. As shown in Figure 1D, caveolin-1, a common lipid raft-associated protein used to biochemically define the lipid raft containing gradient fractions, was found in fraction 2 of the gradient. Since lipid rafts are buoyant in the density gradient, this lane is referred to as the lipid raft fraction. A smaller proportion of caveolin-1 was also found in the detergent-soluble lane, which likely consisted of partial solubilization of some lipid rafts. Intriguingly, the lipid raft fraction was where the majority of TRPC1 channels were seen (Fig. 1D), which implies that TRPC1 channels localize to lipid raft microdomains when glioma cells are maintained in normal growth conditions.
One mechanism of disrupting the formation of lipid rafts is by maintaining cells chronically in serum-free conditions. This depletes the rafts of cholesterol (Weaver et al., 2007). When D54MG cells were chronically serum deprived for 3 days, we observed that a greater proportion of caveolin-1 localized to the detergent soluble fraction indicating that lipid rafts were indeed disrupted. Importantly, we also observed that TRPC1 channels no longer localized to the lipid raft fraction (Fig. 1E). In fact, following serum depletion, TRPC1 channels associated with the detergent soluble fraction of the biochemical isolation. Importantly, as shown in Supplemental Figure 1A, serum starvation does not alter the amount of TRPC1 channels localized to the plasma membrane as revealed by biotinylations of surface proteins and western blot analysis. This implies that the localization of TRPC1 channels to the detergent soluble fraction when lipid rafts were disrupted did not remove these channels from the plasma membrane.
Disruption of lipid rafts impairs TRPC channel function
Since TRPC1 channels localized to lipid rafts, we next wanted to determine whether this localization affected their function. We have previously reported that TRPC channels mediate small, linear currents in D54MG cells that are inhibited by pharmacological TRPC inhibitors such as SKF96365 and 2-APB (Bomben and Sontheimer, 2008). Patch clamp recordings were obtained, under conditions where large conductance K+ channels were inhibited with paxilline (Basrai et al., 2002), and by applying 100 ms voltage steps ranging from −80 to 140mV from a holding potential of −40 mV. The subtraction of currents before and after drug application allowed for the isolation of drug sensitive current. By whole-cell patch clamp electrophysiology, D54MG cells grown under control conditions displayed small, linear currents using this voltage step protocol. Figure 2A illustrates mean data from electrophysiological recordings from 17 cells in which the SKF-sensitive current was determined and normalized to the membrane capacitance for each cell and plotted as a function of voltage. In contrast, D54MG cells that were chronically deprived of serum for 3 days displayed no SKF-sensitive current (n=16), which implicated TRPC1 lipid raft localization as important for TRPC channel function. To further investigate TRPC1 channel function in relation to localization within lipid rafts, we utilized MβCD, a member of the cyclodextrin family of oligosaccharides, which is used to solubilize cholesterol in aqueous solutions (Hartel et al., 1998). Additionally, MβCD has been shown to remove and sequester cholesterol from the plasma membrane (Christian et al., 1997). We have previously demonstrated that MβCD disrupts lipid raft in glioma cells (Weaver et al., 2007). Treatment with MβCD involved dissolving the compound in serum free media; however, acute treatment in serum free media was not sufficient to affect SKF-sensitive currents (n=25). When D54MG cells were treated with MβCD for 15–30 min (n=24), the SKF-sensitive current no longer displayed linear voltage-independent properties as seen in control conditions. This indicated that disruption of lipid rafts, by acute or chronic cholesterol depletion, interfered with TRPC channels function. This loss of SKF-sensitive current indicated that TRPC1 channels were functionally associated with lipid rafts.
Fig. 2.

Lipid raft disruption impairs TRPC channel function. A:Whole-cell patch clamp recordings were used to isolate currents sensitive to 25 μM SKF96365, a TRPC inhibitor. The drug sensitive current was plotted as a function of applied voltage. In normal serum conditions, D54MG cells displayed small, linear SKF-sensitive currents (n=17). When lipid rafts were disrupted with either chronic serum depletion (n=16) or MβCD (n=24), D54MG cells did not display SKF-sensitive currents. Acute serum conditions (n=25) were used for MβCD treatment, but did not themselves affect SKF-sensitive currents. B: Plots of Fura-2 imaging using the SOCE protocol (see Materials and Methods Section). SOCE was significantly decreased (P<0.05) with chronic serum depletion disruption of lipid rafts (n=335) as compared to cell grown in normal serum conditions (n=341). C: Quantification of SOCE peak revealed that chronic serum depletion to disrupt lipid rafts decreased SOCE by 41±1% (P < 0.05).
Lipid raft disruption inhibits store-operated calcium entry in gliomas
TRPC channels play a role in the process of SOCE (Liao et al., 2009) whereby depletion of endoplasmic reticulum Ca2+ stores triggers the influx of Ca2+ across the cell membrane presumably via TRPC channels. We have recently shown that TRPC channels, particularly TRPC1, are involved in SOCE in glioma cells (Bomben and Sontheimer, 2010). In order to assess whether TRPC1 channel lipid raft association also affected SOCE, we used quantitative intracellular Ca2+ imaging with the ratiometric dye FURA-2. To visualize extracellular calcium entry through store-operated channels (SOC), the experimental protocol first required that the endoplasmic reticulum stores be emptied using 10 μM CPA in an extracellular bath free of calcium. This store depletion then signals SOC on the plasma membrane to open. Once stores were emptied for 10 min, 1mM extracellular calcium was applied and calcium entry from the extracellular space was observed. The experiments utilizing this protocol are demonstrated in Figure 2B. The initial increase in [Ca2+]i following store depletion with CPA (first peak) was followed by the rapid entry of Ca2+ upon application of 1mM extracellular Ca2+ (second peak) and was prominent in control cells grown in normal serum conditions (n=341). This second peak was much attenuated when D54MG cells had been chronically starved of serum for 3 days (n=335). Analysis of peak data indicated that SOCE was decreased significantly (P<0.05) by 41±1% (Fig. 2C). This implies that lipid rafts are important for SOCE in gliomas most likely by affecting TRPC channel localization.
Epidermal growth factor stimulation increase glioma chemotaxis, TRPC1 channel localization to the leading edge, and TRPC currents
TRPC channels have been shown to be involved in both migration and chemotactic movement. In fact, TRPC-3, and -6 channels have been shown to be required for axonal growth cone turning toward netrin-1 and BDNF (Wang and Poo, 2005). In glioma cells, a receptor that is often chronically amplified is the EGF receptor (EGFR) also known as ErbB-1. EGF stimulation affects both TRPC channel trafficking (Bezzerides et al., 2004) and activation (Beech, 2005; Liu et al., 2009). We show here that EGF acts as a chemoattractant for D54MG glioma cells (Fig. 3A) without increasing non-directed migration (n=5). Glioma migration was increased almost fivefold (to 473%; P<0.05) only when 10 ng/ml EGF was applied to the bottom of a transwell filter and not when EGF was applied to both sides. This indicated directional specific migration or chemotaxis. Western blot analysis and immunocytochemistry (Fig. 3B) revealed that D54MG cells expressed the EGFR. In Supplemental Figure 1B, we show that like many growth factors, EGF affects glioma cells in a dose-dependent manner and we further utilized 10 ng/ml EGF as an optimal dose for maximal stimulation. Further, we determined that application of EGF to the bottom of a transwell filter increased expression of TRPC1 channels at the leading edge of glioma cells. When fluorescence-blocked transwell filters were stained with antibodies for the EGF receptor and TRPC1 channels, the leading edge of the glioma cells was demarcated by TRPC1 staining (Fig. 3C). These staining were done using confocal microscopy after only 3 h of migration to catch populations of cells that have completely moved through the filters and those that were still in the process of migration. In contrast, the top of the transwell filter had non-migrating cells that revealed more ubiquitous staining of the EGF receptor (green) and TRPC1 channels (red) (Fig. 3D). Additionally, Figure 3E shows TRPC1 staining on the bottom of the filter under unstimulated migration conditions. In cases where EGF was not used to stimulate glioma cells, the localization of TRPC1 channels to the leading edge of glioma cells is not apparent by immunocytochemistry.
Fig. 3.

EGF stimulates glioma chemotaxis. A: Epidermal growth factor stimulated glioma migration in a chemotactic manner up to 473% (P<0.05), but not chemokinesis since EGF application to both sides of the filter did not enhance migration. B: Western blot and immunocytochemistry revealed EGFR expression in D54MG cells. Immunocytochemistry revealed diffuse localization of the EGFR (green) and TRPC1 channels (red) in D54MG cells. Scale bar is 20 μm. C: Immunocytochemistry on the bottom of fluorescence blocked transwell filters revealed increased TRPC1 channel expression (red) on the leading edge of glioma cells when stimulated in a chemotactic manner with EGF. Scale bar is 20 μm. D: Immunocytochemistry on the top of fluorescence blocked transwell filters revealed ubiquitous expression of the EGF receptor (green) and TRPC1 channels (red). Scale bar is 20 μm. E: Immunocytochemistry on the bottom of a fluorescence blocked transwell migration filter without EGF stimulation did not appear to increase TRPC1 channel expression (red) on the leading edge of gliomacells. Scale bar is 20 μm. F: Transwell migration inserts were used to assay glioma migration (n = 5). TRPC inhibitors 100 μM 2-APB, 100 μM MRS1845, and 25 μM SKF reduced glioma migration significantly by 98±1%, 47±5%, and 58±6%, respectively (P<0.05) when compared to control (0.1% DMSO). All drugs were applied to MAB (migration assay buffer). G: Whole-cell patch clamp recordings were used to isolate currents sensitive to 100 μM 2-APB, another TRPC inhibitor. The drug sensitive current was plotted as a function of applied voltage. In control conditions, 0.2% BSA treated D54MG cells displayed small, linear SKF-sensitive currents (n =11). In comparison, when 10ng/ml EGF was applied to glioma cells prior to 2-APB application, there was a significantly greater conductance block at voltages below −30 mV.
To further examine whether EGF stimulation affects TRPC channel functions, we again utilized voltage steps in whole-cell patch clamp recordings. We have previously shown that 2-aminoethoxyphenylborane (2-APB) blocks small, linear currents similar to SKF96365 TRPC channel inhibitors (Bomben and Sontheimer, 2008). Applying 10 ng/ml EGF to glioma cells (n=10) before the TRPC channel inhibitor 2-APB, resulted in a change in the TRPC inhibitor sensitive current (Fig. 1G) from control conditions (n=11). When EGF was applied to glioma cells, a significantly greater conductance was blocked by 2-APB at voltages below −30mV in comparison to control conditions. These are voltages that glioma cells typically rest which indicates that application of EGF can alter TRPC channel function.
TRPC channel inhibition decreases glioma transwell migration
Calcium signaling, lipid raft components, and TRPC channels have all been implicated in cellular migration (Wang and Poo, 2005; Manes and Viola, 2006; Zheng and Poo, 2007; Louis et al., 2008). In light of this evidence and the knowledge that TRPC1 is localized to lipid rafts, we investigated whether pharmacological inhibitors to TRPC channels would decrease glioma migration. To assay this, we used three pharmacological inhibitors of TRPC channels, namely 25μM SKF96365, 100μM 2-APB, and 100μM MRS1845, which are concentrations we have previously determined inhibit TRPC channels as demonstrated by whole cell patch clamp electrophysiology and SOCE experiments (Bomben and Sontheimer, 2010). D54MG cells were harvested, treated with drug, and plated on transwell migration assay inserts (n=5). The glioma cells were then allowed to migrate for 5 h and cells in the presence or absence of drug. Cells were counted as migrated if their nuclei were present on the bottom of the filter. We found that in the presence of TRPC channel inhibitors, glioma migration was reduced by 98±1%, 47±5%, and 58±6% with 2-APB, MRS1845, and SKF treatment, respectively (Fig. 3F). Though none of these inhibitors is completely specific, SKF and MRS1845 have both been reported to block calcium entry through TRPC channels (Malarkey et al., 2008). The third inhibitor, 2-APB, also inhibits the inositol triphosphate receptor (IP3R) (Weaver et al., 2007), which likely accounts for increased block of migration compared to the other inhibitors.
TRPC1 channel inhibition does not impact basal glioma migration
Since TRPC1 channels have specifically been shown to be involved in migration of cells (Fabian et al., 2008; Louis et al., 2008) and we saw increased leading edge expression, we wanted to test whether glioma cells with decreased TRPC1 channel expression would migrate as well as control glioma cells. We previously stably transfected D54MG cells with an inducible shRNA plasmid directed against TRPC1 (Bomben and Sontheimer, 2010). In these cells, when doxycycline is present, the shRNA is transcribed along with a TurboRed® protein. We previously reported that this shRNA decreases TRPC1 protein expression, SKF-sensitive currents, SOCE, and glioma proliferation (Bomben and Sontheimer, 2010). Morphologically, the leading edge staining that we observed with TRPC1 channels (green) was decreased with doxycycline treatment (Fig. 4B) as compared to control cells and non-induced TRPC1 shRNA cells (Fig. 4A). However, when plated on transwell migration inserts with equal cell numbers after 5 days of treatment with doxycycline (n=5), we observed no difference in unstimulated glioma motility (Fig. 4C). This indicated that TRPC1 channels are not involved in the non-directed transwell migration of glioma cells.
Fig. 4.

TRPC1 channels are not involved in basal glioma migration. A: Immunocytochemistry on D54MG cells transfected with inducible shRNA plasmids and stained with TRPC1 antibodies (green). Example staining of non induced control cells, induced control cells (red from TurboRed®) expression, and non-induced TRPC1 shRNA cells. B: Example staining of induced TRPC1 shRNA cells displayed markedly different morphology with TRPC1 channel staining (green) and TurboRed® expression. C: Basal migration was not affected by TRPC1 shRNA being induced. There was no significant difference in transwell migration with either control cells in the presence or absence of doxycycline or in TRPC1 shRNA cells in the presence or absence of doxycycline.
Lipid raft integrity and TRPC channels are essential for EGF chemotaxis
TRPC channels have been associated with chemotactic movement toward growth factors. Since the localization of TRPC1 channels to lipid rafts was shown to be important for TRPC channel function, we first investigated whether lipid raft disruption impaired chemotaxis toward EGF. In Figure 5A, we show that MβCD treatment (n=5) did not significantly affect basal glioma transwell migration (87±8%) of control (P>0.05) while chemotactic migration (217±17%) was attenuated in comparison to control (366±21%) (P<0.05). This indicated that lipid raft integrity is important for chemotactic movement.
Fig. 5.
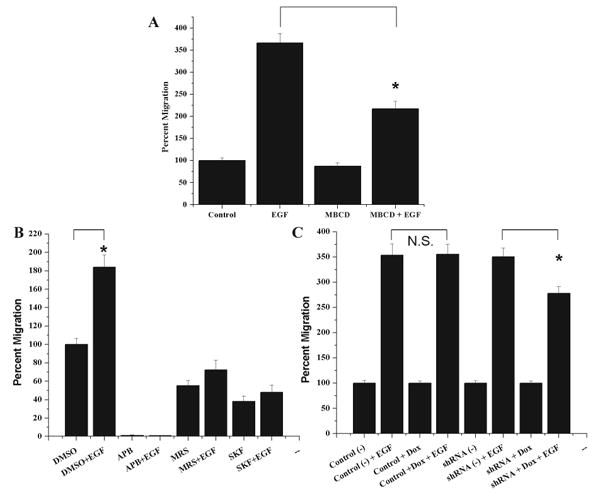
Lipid raft disruption or TRPC channel inhibition decreases glioma chemotaxis toward EGF. A: Treatment with the lipid raft disrupter MβCD (n=5) did not significantly decrease the amount of unstimulated glioma migration (87±8%) of control (P > 0.05) while chemotactic migration in the presence of MβCD (217±17%) was significantly attenuated in comparison to control chemotaxis (366±21%).B:In the presence of pharmacological inhibitors 2-APB, MRS1845, and SKF96365, EGF was not able to stimulate glioma chemotaxis and cells migrated only 1±0.1%, 73±10%, and 48±8% in comparison to control (DMSO), which were stimulated 184±13%. C: D54MG cells transfected with inducible shRNA plasmids were stimulated in a chemotactic manner with EGF. Non-induced control, induced control, and non-induced TRPC1 shRNA cells were stimulated 354±22%, 356±20%, and 351±17%. Cells with induced TRPC1 shRNA had an attenuated response to EGF and were stimulated to 278±14% which was significantly (P < 0.05) less chemotaxis than their sister cells.
Since EGF stimulated TRPC1 channel localization to the leading edge of migrating gliomas, we wanted to determine whether TRPC channels, TRPC1 channels in particular, contributed to chemotactic movement toward EGF. To ascertain whether TRPC channels are important for EGF induced migration, we again utilized transwell migration assay inserts, harvested cells and pharmacologically inhibited TRPC channels with either control (0.1% DMSO), 100 μM 2-APB, 100μM MRS1845, or 25 μM SKF96365. While TRPC channels were inhibited, we stimulated glioma cells with EGF on the bottom of the transwell filter and allowed them to migrate for 5h (n=5). When TRPC channels were inhibited, EGF did not increase migration either in comparison to control conditions or in comparison to drug conditions without EGF stimulation (Fig. 5B). Migration remained at just 1±0.1%, 73±10%, and 48±8% for 2-APB, MRS1845, and SKF, respectively, when also stimulated with EGF. This suggests that calcium dynamics through TRPC channels play an important role in directed glioma migration.
To further ascertain whether TRPC1 channels specifically play a role in chemotaxis toward EGF, control and TRPC1 shRNA cells were treated with or without doxycycline for 4–5 days. Following treatment, equal numbers of cells were plated on transwell filters and half of each condition stimulated to migrate with EGF. While EGF significantly enhanced glioma migration in all conditions, the inducible TRPC1 shRNA cells migration was not enhanced to the same extent as the other conditions (n=6). As shown in Figure 5C, control cells stimulated with EGF were enhanced 354±22% in the absence of doxycycline and 356±20% in the presence of doxycycline. TRPC1 shRNA stimulated with EGF were enhanced 351±17% in the absence of doxycycline and 278±14% in the presence of doxycycline. Since the EGF chemotactic effect was significantly attenuated compared to control cells and non-induced TRPC1 shRNA cells, TRPC1 channels appear to impact glioma chemotaxis. These results also implied that there are either other TRPC channels involved in basal migration and chemotaxis or that the level of TRPC1 knockdown achieved was insufficient to affect basal migration.
Discussion
Glioma invasion is a much studied but complex and poorly understood phenomenon. Here we show that glioma cells migrate along an EGF gradient being chemotactically attracted to EGF but without any effect of EGF on cell motility per se since glioma migration was increased only when EGF was applied in a directed manner. This chemotactic migration was lost when TRPC channels were inhibited pharmacologically and reduced when the expression of TRPC1 was compromised through shRNA knockdown. Interestingly, TRPC1 channels localize to the leading edge of migrating glioma cells where they co-localize with markers of caveolar lipid rafts. This raft association appears important since disruption of lipid rafts by depletion of cholesterol impaired TRPC1 channels, channel-mediated Ca2+ entry and EGF mediated chemotaxis.
While pharmacological inhibitors of TRPC channels, which lack specificity for individual members of the TRPC family, showed an almost complete loss of chemotaxis, a shRNA mediated knockdown of TRPC1 significantly reduced but did not eliminate chemotaxis. This suggests that, while TRPC1 channels are involved in EGF chemotaxis, they are likely not the sole TRPC channel involved in the process. We have previously reported TRPC-1, -3, -4, -5 expression in glioma cells (Bomben and Sontheimer, 2008) and, in a recent study, have shown that shRNA inhibition of TRPC1 channels decreases, but does not completely inhibit, both TRPC1 channel expression and SOCE (Bomben and Sontheimer, 2010). This incomplete reduction of SOCE in comparison to pharmacological inhibitors implies that there is still calcium entry possible through remaining TRPC1 channels or other TRPC subunits.
Calcium signaling has been shown to play important roles in glioma cell invasion (Komuro and Kumada, 2005a). Particularly, the localization of the calcium signal in both space and time has important consequences for the downstream physiological response (Gomez et al., 2001; Manes and Viola, 2006). Additionally, lipid raft integrity is important for chemotaxis in many cell types including breast cancer cells and lymphocytes (Feske, 2007; El Hiani et al., 2009). Following trafficking to the plasma membrane, proteins that are important for signal transduction sequester to lipid rafts, in part, because of post-translational modifications that confer an affinity for cholesterol-enriched domains (Simons and Ikonen, 1997; Brown and London, 2000). The EGF receptor has been shown to localize to lipid rafts (Takebayashi et al., 2004) and, in human breast cancer cells, this localization is essential for chemotaxis (El Hiani et al., 2009). Additional signal transduction components such as TRPC channels, particularly TRPC1, have been shown to localize to lipid rafts (Remillard and Yuan, 2006). Further, signaling components for SOCE have been linked to caveolar lipid rafts (Pani and Singh, 2009). TRPC1 channels, in particular, have been shown to directly interact with caveolin and this interaction was shown to have a role in calcium entry (Beech, 2005; Remillard and Yuan, 2006). Stromal interacting molecule-1 (STIM 1), a key component of SOCE, has also been shown to scaffold with lipid raft domains upon stimulation in HEK293 cells (Liao et al., 2008; Liao et al., 2009) and it has been proposed that the localization of TRPC1 and other components to lipid rafts is what confers SOCE ability (Jardin et al., 2008; Pani and Singh, 2009). We now show that TRPC1 channels localize to lipid rafts and that this localization is important for SOCE in glioma cells. As channels localize to lipid rafts and that this localization is important for SOCE in glioma cells. As signaling components, including TRPC channels, to lipid rafts that enhances glioma migration and chemotaxis.
Since glioma cells can restructure the extracellular matrix and respond to extracellular cues, individual cells invade diffusely throughout the brain (Bellail et al., 2004; Johnson et al., 2009) and this invasion is a primary reason gliomas remain difficult to resect (Giese et al., 2003). The mature brain is typically not a permissive environment for differentiated glial cells to migrate; therefore, mechanisms that glioma cells utilize to invade must be investigated thoroughly (Cayre et al., 2009). An important regulator of glioma signaling is the EGFR as it has been shown to be mutated or amplified in many glioblastoma patients (Bryant et al., 2004). Here, we find another important role for the receptor as its ligand is important for glioma chemotactic movement. Further, we have shown that TRPC channels mediate the chemotactic migration of glioma cells toward EGF and future studies on the role of TRPC channels in normal and malignant glial migration would be essential. Specific pharmacological inhibitors for TRPC channels may have therapeutic value in controlling glioma invasion and future studies should address this question.
Supplementary Material
Acknowledgments
The authors of this manuscript have no competing financial interests. This work was supported by National Institutes of Health grants F31-NS063688, RO1-NS031234, and RO1-NS036692.
Contract grant sponsor: National Institutes of Health grants; Contract grant numbers: F31-NS063688, RO1-NS031234, RO1-NS036692.
Footnotes
Supporting information may be found in the online version of this article.
Literature Cited
- Allen JA, Halverson-Tamboli RA, Rasenick MM. Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci. 2007;8:128–140. doi: 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- Arcangeli A, Crociani O, Lastraioli E, Masi A, Pillozzi S, Becchetti A. Targeting ion channels in cancer: A novel frontier in antineoplastic therapy. Curr Med Chem. 2009;16:66–93. doi: 10.2174/092986709787002835. [DOI] [PubMed] [Google Scholar]
- Basrai D, Kraft R, Bollensdorff C, Liebmann L, Benndorf K, Patt S. BK channel blockers inhibit potassium-induced proliferation of human astrocytoma cells. Neuroreport. 2002;13:403–407. doi: 10.1097/00001756-200203250-00008. [DOI] [PubMed] [Google Scholar]
- Beech DJ. TRPC1: Store-operated channel and more. Pflugers Arch. 2005;451:53–60. doi: 10.1007/s00424-005-1441-3. [DOI] [PubMed] [Google Scholar]
- Bellail AC, Hunter SB, Brat DJ, Tan C, Van Meir EG. Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int J Biochem Cell Biol. 2004;36:1046–1069. doi: 10.1016/j.biocel.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Bezzerides VJ, Ramsey IS, Kotecha S, Greka A, Clapham DE. Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol. 2004;6:709–720. doi: 10.1038/ncb1150. [DOI] [PubMed] [Google Scholar]
- Bomben VC, Sontheimer HW. Inhibition of transient receptor potential canonical channels impairs cytokinesis in human malignant gliomas. Cell Prolif. 2008;41:98–121. doi: 10.1111/j.1365-2184.2007.00504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomben VC, Sontheimer H. Disruption of transient receptor potential canonical channel 1 causes incomplete cytokinesis and slows the growth of human malignant gliomas. Glia. 2010;58:1145–1156. doi: 10.1002/glia.20994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H, Trouslard J. Muscarinic activation of BK channels induces membrane oscillations in glioma cells and leads to inhibition of cell migration. J Membr Biol. 2000;176:31–40. doi: 10.1007/s00232001073. [DOI] [PubMed] [Google Scholar]
- Brown DA, London E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem. 2000;275:17221–17224. doi: 10.1074/jbc.R000005200. [DOI] [PubMed] [Google Scholar]
- Bryant JA, Finn RS, Slamon DJ, Cloughesy TF, Charles AC. EGF activates intracellular and intercellular calcium signaling by distinct pathways in tumor cells. Cancer Biol Ther. 2004;3:1243–1249. doi: 10.4161/cbt.3.12.1233. [DOI] [PubMed] [Google Scholar]
- Butowski NA, Sneed PK, Chang SM. Diagnosis and treatment of recurrent high-grade astrocytoma. J Clin Oncol. 2006;24:1273–1280. doi: 10.1200/JCO.2005.04.7522. [DOI] [PubMed] [Google Scholar]
- Cayre M, Canoll P, Goldman JE. Cell migration in the normal and pathological postnatal mammalian brain. Prog Neurobiol. 2009;88:41–63. doi: 10.1016/j.pneurobio.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian AE, Haynes MP, Phillips MC, Rothblat GH. Use of cyclodextrins for manipulating cellular cholesterol content. J Lipid Res. 1997;38:2264–2272. [PubMed] [Google Scholar]
- Cole K, Kohn E. Calcium-mediated signal transduction: Biology, biochemistry, and therapy. Cancer Metastasis Rev. 1994;13:31–44. doi: 10.1007/BF00690417. [DOI] [PubMed] [Google Scholar]
- Demuth T, Berens ME. Molecular mechanisms of glioma cell migration and invasion. J Neurooncol. 2004;70:217–228. doi: 10.1007/s11060-004-2751-6. [DOI] [PubMed] [Google Scholar]
- El Hiani Y, Lehen'kyi V, Ouadid-Ahidouch H, Ahidouch A. Activation of the calcium-sensing receptor by high calcium induced breast cancer cell proliferation and TRPC1 cation channel over-expression potentially through EGFR pathways. Arch Biochem Biophys. 2009;486:58–63. doi: 10.1016/j.abb.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Fabian A, Fortmann T, Dieterich P, Riethmuller C, Schon P, Mally S, Nilius B, Schwab A. TRPC1 channels regulate directionality of migrating cells. Pflugers Arch. 2008;457:475–484. doi: 10.1007/s00424-008-0515-4. [DOI] [PubMed] [Google Scholar]
- Farin A, Suzuki SO, Weiker M, Goldman JE, Bruce JN, Canoll P. Transplanted glioma cells migrate and proliferate on host brain vasculature: A dynamic analysis. Glia. 2006;53:799–808. doi: 10.1002/glia.20334. [DOI] [PubMed] [Google Scholar]
- Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7:690–702. doi: 10.1038/nri2152. [DOI] [PubMed] [Google Scholar]
- Freichel M, Philipp S, Cavalie A, Flockerzi V. TRPC4 and TRPC4-deficient mice. Novartis Found Symp. 2004;258:189–199. [PubMed] [Google Scholar]
- Giese A, Bjerkvig R, Berens ME, Westphal M. Cost of migration: Invasion of malignant gliomas and implications for treatment. J Clin Oncol. 2003;21:1624–1636. doi: 10.1200/JCO.2003.05.063. [DOI] [PubMed] [Google Scholar]
- Gomez TM, Robles E, Poo M, Spitzer NC. Filopodial calcium transients promote substrate-dependent growth cone turning. Science. 2001;291:1983–1987. doi: 10.1126/science.1056490. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hartel S, Diehl HA, Ojeda F. Methyl-beta-cyclodextrins and liposomes as water-soluble carriers for cholesterol incorporation into membranes and its evaluation by a microenzymatic fluorescence assay and membrane fluidity-sensitive dyes. Anal Biochem. 1998;258:277–284. doi: 10.1006/abio.1998.2594. [DOI] [PubMed] [Google Scholar]
- Huncharek M, Muscat J. Treatment of recurrent high grade astrocytoma; results of a systematic review of 1,415 patients. Anticancer Res. 1998;18:1303–1311. [PubMed] [Google Scholar]
- Jardin I, Salido GM, Rosado JA. Role of lipid rafts in the interaction between hTRPC1, Orai1 and STIM1. Channels (Austin) 2008;2:401–403. doi: 10.4161/chan.2.6.7055. [DOI] [PubMed] [Google Scholar]
- Johnson JK, Nowicki MO, Lee CH, Chiocca EA, Viapiano MS, Lawler SE, Lannutti J. Quantitative analysis of glioma cell migration on electrospun polycaprolactone using time-lapse microscopy. Tissue Eng Part C Methods. 2009;15:534–540. doi: 10.1089/ten.tec.2008.0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro H, Kumada T. Ca2+ transients control CNS neuronal migration. Cell Calcium. 2005a;37:387–393. doi: 10.1016/j.ceca.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Komuro H, Kumada T. Ca2+ transients control CNS neuronal migration. Cell Calcium. 2005b;37:387–393. doi: 10.1016/j.ceca.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Komuro H, Rakic P. Orchestration of neuronal migration by activity of ion channels, neurotransmitter receptors, and intracellular Ca2+ fluctuations. J Neurobiol. 1998;37:110–130. [PubMed] [Google Scholar]
- Levicar N, Nuttall RK, Lah TT. Proteases in brain tumour progression. Acta Neurochir (Wien) 2003;145:825–838. doi: 10.1007/s00701-003-0097-z. [DOI] [PubMed] [Google Scholar]
- Li Y, Jia YC, Cui K, Li N, Zheng ZY, Wang YZ, Yuan XB. Essential role of TRPC channels in the guidance of nerve growth cones by brain-derived neurotrophic factor. Nature. 2005;434:894–898. doi: 10.1038/nature03477. [DOI] [PubMed] [Google Scholar]
- Liao Y, Erxleben C, Abramowitz J, Flockerzi V, Zhu MX, Armstrong DL, Birnbaumer L. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci USA. 2008;105:2895–2900. doi: 10.1073/pnas.0712288105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Plummer NW, George MD, Abramowitz J, Zhu MX, Birnbaumer L. A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc Natl Acad Sci USA. 2009;106:3202–3206. doi: 10.1073/pnas.0813346106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Garg P, Yang S, Gong P, Pallero MA, Annis DS, Liu Y, Passaniti A, Mann D, Mosher DF, Murphy-Ullrich JE, Goldblum SE. Epidermal growth factor-like repeats of thrombospondins activate phospholipase Cgamma and increase epithelial cell migration through indirect epidermal growth factor receptor activation. J Biol Chem. 2009;284:6389–6402. doi: 10.1074/jbc.M809198200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis M, Zanou N, Van Schoor M, Gailly P. TRPC1 regulates skeletal myoblast migration and differentiation. J Cell Sci. 2008;121:23–29. doi: 10.1242/jcs.037218. [DOI] [PubMed] [Google Scholar]
- Malarkey EB, Ni Y, Parpura V. Ca(2+) entry through TRPC1 channels contributes to intracellular Ca(2+) dynamics and consequent glutamate release from rat astrocytes. Glia. 2008;56:821–835. doi: 10.1002/glia.20656. [DOI] [PubMed] [Google Scholar]
- Manes S, Viola A. Lipid rafts in lymphocyte activation and migration. Mol Membr Biol. 2006;23:59–69. doi: 10.1080/09687860500430069. [DOI] [PubMed] [Google Scholar]
- McFerrin MB, Sontheimer H. A role for ion channels in glioma cell invasion. Neuron Glia Biol. 2005;2:39–49. doi: 10.1017/S17440925X06000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolau DV, Jr, Burrage K, Parton RG, Hancock JF. Identifying optimal lipid raft characteristics required to promote nanoscale protein-protein interactions on the plasma membrane. Mol Cell Biol. 2006;26:313–323. doi: 10.1128/MCB.26.1.313-323.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pani B, Singh BB. Lipid rafts/caveolae as microdomains of calcium signaling. Cell Calcium. 2009;45:625–633. doi: 10.1016/j.ceca.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platten M, Wick W, Wild-Bode C, Aulwurm S, Dichgans J, Weller M. Transforming growth factors beta(1) (TGF-beta(1)) and TGF-beta(2) promote glioma cell migration via Up-regulation of alpha(V)beta(3) integrin expression. Biochem Biophys Res Commun. 2000;268:607–611. doi: 10.1006/bbrc.2000.2176. [DOI] [PubMed] [Google Scholar]
- Remillard CV, Yuan JX. Transient receptor potential channels and caveolin-1: Good friends in tight spaces. Mol Pharmacol. 2006;70:1151–1154. doi: 10.1124/mol.106.029280. [DOI] [PubMed] [Google Scholar]
- Rujoi M, Jin J, Borchman D, Tang D, Yappert MC. Isolation and lipid characterization of cholesterol-enriched fractions in cortical and nuclear human lens fibers. Invest Ophthalmol Vis Sci. 2003;44:1634–1642. doi: 10.1167/iovs.02-0786. [DOI] [PubMed] [Google Scholar]
- Schwab A, Nechyporuk-Zloy V, Fabian A, Stock C. Cells move when ions and water flow. Pflugers Arch. 2007;453:421–432. doi: 10.1007/s00424-006-0138-6. [DOI] [PubMed] [Google Scholar]
- Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol. 2006;2:494–503. doi: 10.1038/ncpneuro0289. [DOI] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Soldi R, Mitola S, Strasly M, Defilippi P, Tarone G, Bussolino F. Role of alphavbeta3 integrin in the activation of vascular endothelial growth factor receptor-2. EMBOJ. 1999;18:882–892. doi: 10.1093/emboj/18.4.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontheimer HW. An unexpected role for ion channels in brain tumor metastasis. Exp Biol Med. 2008;233:779–791. doi: 10.3181/0711-MR-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai K, Hamaide MC, Debaix H, Gailly P, Wibo M, Morel N. Agonist-evoked calcium entry in vascular smooth muscle cells requires IP3 receptor-mediated activation of TRPC1. Eur J Pharmacol. 2008;583:135–147. doi: 10.1016/j.ejphar.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Takebayashi M, Hayashi T, Su TP. Sigma-1 receptors potentiate epidermal growth factor signaling towards neuritogenesis in PC12 cells: Potential relation to lipid raft reconstitution. Synapse. 2004;53:90–103. doi: 10.1002/syn.20041. [DOI] [PubMed] [Google Scholar]
- Wang GX, Poo MM. Requirement of TRPC channels in netrin-1-induced chemotropic turning of nerve growth cones. Nature. 2005;434:898–904. doi: 10.1038/nature03478. [DOI] [PubMed] [Google Scholar]
- Weaver AK, Bomben VC, Sontheimer H. Expression and function of calcium-activated potassium channels in human glioma cells. Glia. 2006;54:223–233. doi: 10.1002/glia.20364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver AK, Olsen ML, McFerrin MB, Sontheimer H. BK channels are linked to IP3-receptors via lipid rafts: A novel mechanism for coupling [CA2+]1 to ion channel activation. J Biol Chem. 2007;282:31558–31568. doi: 10.1074/jbc.M702866200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley PF, Zeng W, Huang GN, Yuan JP, Kim JY, Lee MG, Muallem S. TRPC channels as STIM1-regulated store-operated channels. Cell Calcium. 2007;42:205–211. doi: 10.1016/j.ceca.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JQ, Poo MM. Calcium signaling in neuronal motility. Annu Rev Cell Dev Biol. 2007;23:375–404. doi: 10.1146/annurev.cellbio.23.090506.123221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.