

Abstract
The induction of corticostriatal long-term depression (LTD) in striatal spiny projection neurons (SPNs) requires coactivation of group I metabotropic glutamate receptors (mGluRs) and L-type Ca2+ channels. This combination leads to the postsynaptic production of endocannabinoids that act presynaptically to reduce glutamate release. Although the necessity of coactivation is agreed upon, why it is necessary in physiologically meaningful settings is not. The studies described here attempt to answer this question by using two-photon laser scanning microscopy and patch-clamp electrophysiology to interrogate the dendritic synapses of SPNs in ex vivo brain slices from transgenic mice. These experiments revealed that postsynaptic action potentials induce robust ryanodine receptor (RYR)-dependent Ca2+-induced-Ca2+ release (CICR) in SPN dendritic spines. Depolarization-induced opening of voltage-gated Ca2+ channels was necessary for CICR. CICR was more robust in indirect pathway SPNs than in direct pathway SPNs, particularly in distal dendrites. Although it did not increase intracellular Ca2+ concentration alone, group I mGluR activation enhanced CICR and slowed Ca2+ clearance, extending the activity-evoked intraspine transient. The mGluR modulation of CICR was sensitive to antagonism of inositol trisphosphate receptors, RYRs, src kinase, and Cav1.3 L-type Ca2+ channels. Uncaging glutamate at individual spines effectively activated mGluRs and facilitated CICR induced by back-propagating action potentials. Disrupting CICR by antagonizing RYRs prevented the induction of corticostriatal LTD with spike-timing protocols. In contrast, mGluRs had no effect on the induction of long-term potentiation. Taken together, these results make clearer how coactivation of mGluRs and L-type Ca2+ channels promotes the induction of activity-dependent LTD in SPNs.
Keywords: two-photon imaging, calcium, long-term depression, plasticity, striatum
the striatum plays an important role in learning context-appropriate actions (Gerfen and Surmeier 2011; Yin and Knowlton 2006). This learning is thought to depend upon long-term changes in the strength of corticostriatal synapses formed on principal striatal spiny projection neurons (SPNs) (Gerfen and Surmeier 2011; Lovinger 2010). These synapses are capable of both long-term potentiation (LTP) and depression (LTD) (Fino et al. 2005; Lovinger 2010; Pawlak and Kerr 2008; Shen et al. 2008).
LTD is the best studied of these forms of plasticity (Calabresi et al. 1992a; Lovinger et al. 2003; Surmeier et al. 2009). Endocannabinoid (eCB)-mediated corticostriatal LTD is induced postsynaptically and expressed presynaptically. Induction requires postsynaptic depolarization, coactivation of group I metabotropic glutamate receptors (mGluRs) and L-type Ca2+ channels with a Cav1.3 pore-forming subunit, and an elevation of cytosolic Ca2+ (Adermark and Lovinger 2007; Calabresi et al. 1994; Kreitzer and Malenka 2005; Lovinger 2010; Shindou et al. 2011). This combination leads to the production of eCBs that act presynaptically to reduce glutamate release (Lovinger 2010).
Although these processes are agreed upon, the nature of their interaction is not. Typically, LTD induction is achieved by combining strong somatic depolarization with high-frequency stimulation (HFS) of afferent fibers (Adermark and Lovinger 2007; Calabresi et al. 1992a, 1994; Kreitzer and Malenka 2005; Wang et al. 2006). While this combination is unlikely to be achieved normally in SPNs, it reflects the need for dendritic depolarization to achieve the conditions necessary for LTD induction. In vivo, this might be accomplished by convergent synaptic input that triggers state transitions in dendrites (Plotkin et al. 2011; Wilson and Kawaguchi 1996) or by the temporal convergence of synaptic input and back-propagating action potentials (bAPs). In SPNs, as in other neuron types (Christie et al. 1996; Magee and Johnston 1997; Nevian and Sakmann 2006), bAPs decrementally back-propagate into dendrites, providing sufficient depolarization to open voltage-dependent Ca2+ channels at synapses in the proximal portion of the SPN dendritic tree (Carter and Sabatini 2004; Day et al. 2008). When paired with trailing synaptic stimulation, repetition of short bAP bursts at theta frequencies induces LTD in SPNs (Shen et al. 2008). This spike timing-dependent plasticity (STDP)-LTD has all the pharmacological properties of conventional HFS-LTD, suggesting that the underlying mechanisms are the same (Kreitzer and Malenka 2008; Lovinger et al. 2003; Shen et al. 2008; Surmeier et al. 2009).
In pyramidal neurons, opening of voltage-dependent Ca2+ channels is thought to facilitate STDP-LTD induction by increasing mGluR-stimulation of phospholipase C (PLC) (Nevian and Sakmann 2006). However, it also is possible that by increasing cytosolic inositol trisphosphate (IP3) or activating src kinase mGluRs promote Ca2+-induced Ca2+ release (CICR) triggered by opening of voltage-dependent channels (Berridge 1998; Lerner and Kreitzer 2012; Nakamura et al. 1999, 2000; Nishiyama et al. 2000). In this scenario, mGluR signaling and activity-dependent Ca2+ entry through L-type Ca2+ channels would work in concert to elevate postsynaptic Ca2+ concentration into a range necessary to drive eCB generation. Because of its reciprocity, this model suggests that repetition, rather than precise timing of pre- and postsynaptic activity, is critical to LTD induction.
To explore these ideas, two-photon laser scanning microscopy (2PLSM) was used in conjunction with patch-clamp techniques in ex vivo brain slices from transgenic mice to monitor Ca2+ concentration in proximal and distal spines of direct pathway SPNs (dSPNs) and indirect pathway SPNs (iSPNs) in situations similar to those necessary for STDP-LTD.
MATERIALS AND METHODS
Brain slice preparation.
Parasagittal brain slices (275 μm) were obtained from 19- to 23-day-old male and female hemizygous BAC D1 or BAC D2 transgenic mice (Day et al. 2008; Gong et al. 2003) following procedures approved by the Northwestern University Animal Care and Use Committee. The mice were anesthetized with a mixture of ketamine (50 mg/kg) and xylazine (4.5 mg/kg) and perfused transcardially with 5–10 ml of ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM) 124 NaCl, 3 KCl, 1 CaCl2, 1.5 MgCl2, 26 NaHCO3, 1 NaH2PO4, and 16.66 glucose, continuously bubbled with carbogen (95% O2-5% CO2). The slices were then transferred to a holding chamber where they were incubated in ACSF containing (in mM) 2 CaCl2 and 1 MgCl2 at 35°C for 60 min, after which they were stored at room temperature until recording.
Electrophysiology.
Patch pipettes were pulled from thick-walled borosilicate glass on a Sutter P-97 puller. Pipette resistance was typically 3–4 MΩ when filled with recording solution. The internal recording solution contained (in mM) 135 KMeSO4, 5 KCl, 0.16 CaCl2, 10 HEPES, 2 ATP-Mg2+, 0.5 GTP-Na, 5 phosphocreatine-Tris, 5 phosphocreatine-Na, and 0.1 spermine; pH was adjusted to 7.25 with NaOH and osmolarity to 270–280 mosM. For some experiments the internal recording solution lacked CaCl2, as indicated in the text. For Ca2+ imaging experiments, the recording solution also contained 200 μM fluo-4 pentapotassium salt and 50 μM Alexa Fluor 568 hydrazide Na salt (Invitrogen), unless otherwise indicated. Slices were continuously perfused with carbogen-bubbled ACSF. For experiments in which cadmium was locally puffed onto dendrites, the puffer solution contained HEPES-buffered ACSF, 0 mM CaCl2, and 200 μM cadmium. Slices were transferred to a submersion-style recording chamber mounted on an Olympus BX51 upright, fixed-stage microscope. Electrophysiological recordings were obtained with a Multiclamp 700B amplifier. Stimulation and display were obtained as previously described (Day et al. 2008) with the custom-written shareware package WinFluor (John Dempster, Strathclyde University, Glasgow, UK), which automates and synchronizes the two-photon imaging and electrophysiological protocols. The amplifier bridge circuit was adjusted to compensate for serial resistance and continuously monitored during recordings.
Two-photon laser scanning microscopy and Ca2+ imaging.
D1 receptor-expressing dSPNs or D2 receptor-expressing iSPNs were identified by somatic enhanced green fluorescent protein (eGFP) two-photon excited fluorescence with an Ultima Laser Scanning Microscope system (Prairie Technologies). A DODT contrast detector system was used to provide a bright-field transmission image in registration with the fluorescent images. The green GFP signals (490–560 nm) were acquired with 810-nm excitation (Verdi/Mira laser). SPNs were patched with video microscopy with a Hitachi CCD camera and an Olympus ×60/0.9 NA lens. Alexa Fluor 568 fluorescence was used for visualization of cell bodies, dendrites, and spines. After patch rupture, the internal solution was allowed to equilibrate for 15–20 min before imaging. Whole cell maximum-projection images of the soma and dendrites were acquired with 0.36-μm2 pixels with 10-μs pixel dwell time; ∼80 images were taken with 1-μm focal steps. High-magnification maximum-projection images of dendrites were acquired with 0.072-μm2 pixels with 10-μs pixel dwell time; ∼20 images were taken with 0.5-μm focal steps.
Single bAPs were generated by injecting current pulses (2 nA, 2 ms) in a theta burst pattern: five bursts, each burst containing three bAPs with a 20-ms interevent interval (50 Hz); bursts were delivered at 5 Hz. Drugs were either bath applied by dissolving them in the external ACSF or focally applied with pressure ejection through a micropipette, unless otherwise stated. All Ca2+ imaging recordings were performed in a cocktail of synaptic blockers (in μM: 5 NBQX, 50 AP-5, 10 SR-95531, 1 CGP-55845, 1 MPEP, and 50 CPCCOET, all from Tocris) to isolate postsynaptic components of the mechanisms being examined. For experiments involving (S)-3,5-dihydroxyphenylglycine (DHPG) application, MPEP and CPCCOET were omitted. Experiments testing the involvement of protein kinase C, PLA2, and phosphatidylinositol 4,5-bisphosphate (PIP2) depletion were performed by loading the cell with 1 μM calphostin, 20 μM N-(p-amylcinnamoyl)anthranilic acid (ACA) or 200 nM PIP2, respectively. Dendritic changes in Ca2+ were measured with fluo-4 as previously described (Day et al. 2008). Ca2+ transients were expressed as a ratio of green to red (G/R) fluorescence. As this measure is sensitive to photomultiplier tube settings, only data collected under identical settings were compared. In some instances, where drugs were locally puffed, transients are expressed as change in fluorescence (ΔF/Fo). This is because the puffer solution contained Alexa Fluor 568, to confirm the local application of drug, and thus altered the red fluorescence channel. Locally puffing drugs had two benefits: 1) Alterations of the somatic waveform and bAP propagation to the dendritic point of measurement were minimal, and 2) before and after Ca2+ measurements could be performed in the same continuous line scan. Green fluorescence line-scan signals were acquired at 6 ms per line and 512 pixels per line with 0.08-μm pixels and 10-μs pixel dwell time. The laser-scanned images were acquired with 810-nm light pulsed at 90 MHz (∼250-fs pulse duration). Power attenuation was achieved with two Pockels cells (electro-optic modulators; models 350-80 and 350-50, Con Optics, Danbury, CT). The two cells were aligned in series to provide an enhanced modulation range for fine control of the excitation dose (0.1% steps over 4 decades). The line scan was started 200 ms before the stimulation protocol and continued 4 s after the stimulation to obtain the background fluorescence and to record the decay of the optical signal after stimulation. To reduce photo-damage and photo-bleaching, the laser was fully attenuated with the second Pockels cell at all times during the scan except for the period directly flanking the bAP burst.
Glutamate uncaging.
Two-photon uncaging (2PU) of MNI-glutamate was performed simultaneously with Ca2+ imaging using a Chameleon-XR laser system (Coherent Laser Group, Santa Clara, CA) as previously described (Plotkin et al. 2011). MNI-glutamate (Tocris Cookson, Ellisville, MO) was superfused at 5 mM with a system of syringe pumps (WPI, Sarasota, FL) and a multibarreled perfusion manifold fitted with a small-volume mixing tip that allowed rapid switching between solutions (Cell MicroControls, Norfolk, VA). MNI-glutamate was uncaged with 1-ms pulses of 720-nm light independently controlled by a third Pockels cell modulator (model 302, Con Optics). Experiments involved uncaging on a single spine to evoke a somatic excitatory postsynaptic potential (EPSP) measuring 0.8–4 mV. The uncaging pulses were typically ∼10 mW in strength measured at the sample plane. Photolysis power was adjusted to closely mimic spontaneously occurring EPSPs and tuned to achieve the predetermined somatic EPSP amplitude. Simultaneous photolysis and line scan images were made from shafts/spines. As described above, the custom-written software package WinFluor automated and synchronized the Ca2+ imaging with the electrophysiological stimulation and the photolysis.
Synaptic plasticity.
STDP was induced as previously described (Shen et al. 2008). Briefly, iSPNs were recorded in current-clamp mode with perforated-patch recordings. The internal recording solution contained (in mM) 126 KMeSO4, 14 KCl, 3 MgCl2, 0.5 CaCl2, 5 EGTA, and 10 HEPES, with 180 μg/ml amphotericin B. The external recording solution contained (in mM) 126 NaCl, 3 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 26 NaHCO3, and 14 glucose, bubbled with 95% O2-5% CO2. A theta glass stimulating electrode was placed near a dendrite visualized under IR-DIC optics ∼100 μm from the soma. STDP was induced by pairing a theta burst pattern of somatically induced APs (as described above) with timed synaptically evoked EPSPs. For the negative timing protocol, EPSPs followed each AP burst by 10 ms. For the positive timing protocol, EPSPs preceded every AP by 5 ms. Stimulus intensity was adjusted to evoke baseline single-component EPSPs. Postsynaptic cells were depolarized to −70 mV, and GABAA receptors were blocked with gabazine (10 μM). The induction protocol was repeated 15 times at 0.1 Hz. Capacitance current was continuously monitored during perforation by applying a 5- to 10-mV pulse from a holding potential of −70 mV, and input resistance was continuously monitored during recordings. For whole cell current-clamp LTD induction, the internal recording solution contained (in mM) 135 KMeSO4, 5 KCl, 5 HEPES, 0.25 EGTA, 10 phosphocreatine-Na, 2 ATP-Mg, and 0.5 GTP-Na. Recordings were performed at 30–32°C and constantly perfused with 10 μM SR 95531 hydrobromide and 10 μM (R)-CPP. A concentric bipolar stimulating electrode was placed ∼100 μm from the soma and was calibrated to evoke ∼10-mV somatic EPSPs every 10 s. Baseline was recorded for 10 min before induction. The induction protocol consisted of a 3-s, 100-Hz train of electrical stimulation, with APs (evoked by 2-ms, 1-nA current injections) preceding each stimulation by 4 ms. The induction protocol was repeated at 20-s intervals three times. Baseline was calculated as the average amplitude for the 5 min preceding LTD induction, and LTD amplitude was measured as the average of the responses 20–25 min after induction.
Fluorescence-assisted cell sorting and gene expression profiling.
Quantitative polymerase chain reaction (qPCR) was used to quantify transcripts of interest with procedures similar to those described previously (Chan et al. 2012). In brief, striata were microdissected, dSPNs and iSPNs were separated with fluorescence-assisted cell sorting (FACS) based on GFP expression, and total mRNA was isolated with the RNeasy Micro Kit (Qiagen). cDNA was synthesized with qScript cDNA Supermix (Quanta Biosciences). Real-time PCR was performed using Fast SYBR Mastermix (Applied Biosystems) on a StepOnePlus thermocycler (Applied Biosystems). The thermal cycling conditions comprised an initial denaturing step at 95°C for 20 s and 40 cycles at 95°C for 3 s, 60°C for 30 s. The PCR cycle threshold (CT) values were measured within the exponential phase of the PCR reaction with StepOnePlus software version 2.1 (Applied Biosystems). A correction was performed with a passive reference dye (Rox) present in the PCR master mix. Reactions with any evidence of nonspecificity (i.e., low melting temperatures or multiple peaks in melting point analysis) were excluded from the analysis. A relative quantification method (ΔΔCT method) was used to quantify differences in gene expression level. To increase accuracy of gene expression analysis, a panel of reference genes (Atp5b, Cyc1, Gapdh, H2afz, Hmbs, Uchl1) was included. Weighted CTs based on the stability of each reference gene were calculated. Experiments for each gene of interest were run in triplicate. Desalted primers were custom synthesized (Invitrogen) and intron-spanning whenever possible. No-template and no-reverse transcriptase control assays produced negligible signals, suggesting that primer dimer formation and genomic DNA contamination effects were small. The mRNA levels in each subgroup of samples were characterized by their median values. Results are presented as fold differences between cell types.
Statistical analysis.
Differences in dendritic spines or shafts were examined with the Mann-Whitney U-nonparametric test of significance in most cases. In cases in which comparisons were made between the same spines/shafts before and after experimental manipulation, statistical significance was tested with the Wilcoxon signed-rank test (a nonparametric test). Differences were considered statistically significant if P < 0.05.
RESULTS
CICR contributes to bAP-induced Ca2+ transients.
Recent work has pointed to the involvement of ryanodine receptors (RYRs) and CICR in corticostriatal LTD induction (Lerner and Kreitzer 2012), but whether or not APs engage dendritically localized RYRs (Martone et al. 1997; Verkhratsky 2002), where LTD induction occurs, is unknown. To answer this question, SPNs were studied in brain slices with patch-clamp techniques and 2PLSM Ca2+ imaging. SPNs were identified in slices from transgenic mice expressing eGFP under control of either the D1 dopamine receptor promoter (for dSPNs) or the D2 dopamine receptor promoter (for iSPNs) (Fig. 1A). Ca2+ transients evoked by somatically generated AP theta bursts [APs were evoked by 2-nA, 2-ms current pulses, 3 pulses per burst (50 Hz), bursts delivered at 5 Hz, 5 bursts total; Fig. 1] were measured in proximal (40–60 μm from soma) and distal (100–120 μm from soma) dendritic spines. The theta burst bAP train evoked reliable elevations in intraspine Ca2+, as measured with the high-affinity Ca2+ dye fluo-4, at both proximal and distal sites, although the magnitude of the Ca2+ transient at distal sites was consistently smaller. Because bAPs produce progressively smaller fluorescent signals as they propagate from the soma, a relatively high concentration of fluo-4 (200 μM) was used to ensure reliable detection of Ca2+ in distal dendrites (Fig. 1B), with minimal effects on decay kinetics (Fig. 1C). This protocol induced dendritic Ca2+ signals well below dye saturation, as confirmed by enhancing bAP invasion into dendrites with millimolar concentrations of the Kv1/4 K+ channel antagonist 4-aminopyridine (4-AP) (Fig. 1D). The Ca2+ transients rose rapidly but decayed slowly (rise τ = 24.9 ± 3.2 ms, decay τ = 192.5 ± 22.7 ms; 11 spines), leading to summation of intracellular Ca2+ when bAP bursts were separated by 200 ms.
Fig. 1.

Intracellular Ca2+ stores contribute to back-propagating action potential (bAP)-induced dendritic Ca2+ transients in direct pathway (dSPNs) and indirect pathway (iSPNs) spiny projection neurons. A: maximum-intensity projection images of an iSPN (left) and a dSPN (right). Patch electrodes are shaded gray. B: comparison of Ca2+ transients in distal (100–120 μm from soma) iSPN dendritic spines loaded with either 100 μM (n = 4 cells, 28 spines) or 200 μM (n = 5 cells, 11 spines) fluo-4, in response to a theta burst pattern of bAPs. Average Ca2+ transients are fit with summating biexponentials. The larger-amplitude transients afforded by higher fluo-4 concentration allowed for reliable quantification of pharmacologically decreased events. C: Ca2+ transient decay kinetics (measured from peak of 5th burst in a theta burst train of bAPs) were not significantly (ns) altered by 200 μM (n = 5 cells, 11 spines) vs. 100 μM (n = 4 cells, 28 spines) fluo-4. D: average dendritic spine Ca2+ transients from distal (100–120 μm from soma) iSPNs in response to a theta burst pattern of bAPs in the presence (N = 8 spines, 3 cells) or absence (ACSF; N = 9 spines, 3 cells) of 1 mM 4-aminopyridine (4-AP). G/R, ratio of green to red fluorescence. E: average dendritic spine Ca2+ transients from proximal (40–60 μm from soma) and distal (100–120 μm from soma) dendritic spines loaded with or without (control) 75 μM ryanodine. Example images of spines from which line scans were taken are shown (line scan is represented by yellow line). Transients are in response to somatically generated theta bursts of bAPs. Shaded areas indicate SE; solid lines are summating biexponential fits of the average G/R transients. Dashed horizontal lines indicate G/R = 0. Ca2+ transient areas corresponding to each burst are shown at bottom (temporal boundaries indicated by dashed lines; dots are averages, bars are SE). Ca2+ transient areas are fit with a single exponential. Average somatic voltage traces and current pulses used for AP initiation are shown in register below in the presence and absence of ryanodine. Ca2+ transient magnitudes were decreased by ryanodine, indicating a significant contribution of intracellular Ca2+ release to bAP-evoked dendritic Ca2+ transients (proximal: control n = 5 cells, 26 spines, ryanodine n = 5 cells, 20 spines; distal: control n = 5 cells, 13 spines, ryanodine n = 5 cells, 13 spines). F: as in E, for dSPNs (proximal: control n = 4 cells, 24 spines, ryanodine n = 4 cells, 32 spines; distal: control n = 4 cells, 27 spines, ryanodine n = 4 cells, 36 spines). Ca2+ transient magnitudes were decreased by ryanodine in distal dSPN spines. *P < 0.05, Mann-Whitney nonparametric test.
In the presence of a high concentration (75 μM) of ryanodine in the patch pipette to antagonize RYRs, the theta burst train generated significantly smaller Ca2+ transients in proximal spines of both SPN populations (Fig. 1, E and F; Fig. 2A). Ryanodine had no discernible effect on the somatic AP waveform, suggesting that the effect was mediated by an action at the spine (Fig. 1, E and F). Ryanodine also reduced bAP-induced Ca2+ transients in distal dendritic spines of iSPNs but not dSPNs (Fig. 1, E and F; Fig. 2A). The lack of ryanodine action in distal dSPN spines was not due to the higher concentration of Ca2+ indicator used. Rather, it is likely attributable to the more rapid attenuation of bAP amplitude in dSPN dendrites (Day et al. 2008), although differential RYR subunit expression cannot be ruled out (see Fig. 3). RYR-mediated CICR required repetitive activity, as it was not evoked in iSPN dendritic spines by a single bAP (Fig. 2B). The higher Ca2+ indicator concentration used to enhance signal detection did not interfere with the processes examined here, as the effect of ryanodine was robustly observed with a lower fluo-4 concentration (Fig. 2C).
Fig. 2.

bAP-mediated intracellular Ca2+ release requires repetitive activity. A: box plots showing % block of the entire calcium transient (sum of bursts 1–5) by ryanodine in proximal and distal iSPNs and dSPNs (from Fig. 1, E and F). No effect of ryanodine was seen in distal dSPN spines with a lower concentration (100 μM) of fluo-4 (control n = 3 cells, 32 spines, ryanodine n = 3 cells, 27 spines), although the variance was large because of the small Ca2+ signal. B: average Ca2+ transients in distal iSPN spines induced by a single bAP in the presence (n = 5 cells, 30 spines) and absence (control; n = 3 cells, 19 spines) of ryanodine. C: comparison of Ca2+ transients in proximal (40–60 μm from soma) iSPN dendritic spines loaded with 100 μM fluo-4 with (n = 4 cells, 32 spines) and without (n = 4 cells, 26 spines) 75 μM ryanodine in the electrode. Areas of Ca2+ transients corresponding to each burst are shown at bottom. Ca2+ transient areas are fit with a single exponential. Ryanodine significantly reduces bAP-evoked Ca2+ transients in lower (100 μM fluo-4) Ca2+ dye concentration conditions. *P < 0.05, Mann-Whitney nonparametric test.
Fig. 3.

Activation of group I metabotropic glutamate receptors (mGluRs) enhances bAP-evoked Ca2+ transients in iSPN and dSPN dendritic spines. A: box plots showing basal intracellular Ca2+ concentrations in distal (100–120 μm from soma) spines (sp) and shafts (sh) of iSPNs and dSPNs, before and after bath application of (S)-3,5-dihydroxyphenylglycine (DHPG, 50 μM; n = 5 cells, 8–10 spines, 7–9 shafts). DHPG itself had no significant effect on dendritic basal Ca2+. B: average theta burst bAP-evoked Ca2+ transients in iSPN dendritic spines before and after bath application of DHPG (50 μM). Shaded areas indicate SE; solid lines are summating biexponential fits of the average G/R transients. Dashed horizontal lines indicate G/R = 0. Ca2+ transient areas corresponding to each burst are shown at bottom (temporal boundaries indicated by dashed lines; dots are averages, bars are SE). Ca2+ transient areas are fit with a single exponential. DHPG significantly increased Ca2+ transients in iSPN spines (proximal: n = 5 cells, 10 spines; distal: n = 5 cells, 11 spines). C: as in B, for dSPNs (proximal: n = 4 cells, 6 spines; distal: n = 5 cells, 9 spines). DHPG significantly increased Ca2+ transients in distal dSPN spines. D: average theta burst bAP-evoked Ca2+ transient areas in distal dendritic spines of 2-mo-old iSPNs (n = 4 cells, 24 spines). DHPG significantly increased Ca2+ transients in mature iSPN spines. *P < 0.05, Wilcoxon signed-rank test. E: relative mRNA expression in FACS-pooled dSPNs and iSPNs (n = 4–17), showing cell-specific differential regulation of membrane-, scaffold-, and endoplasmic reticulum (er)-associated genes. VGCC, voltage-gated Ca2+ channel. #P < 0.05, 2-tailed t-test.
Group I mGluR activation enhances bAP-induced dendritic Ca2+ transients.
In hippocampal pyramidal neurons, bAP-induced dendritic CICR is enhanced by activation of group I mGluRs (Nakamura et al. 1999, 2000). To determine whether similar mechanisms are engaged by SPNs, the group I mGluR agonist DHPG (50 μM) was rapidly applied to distal SPN dendrites with a puffer pipette while basal dendritic Ca2+ concentration was measured. In the absence of somatic APs, DHPG had no detectable effect on intraspine Ca2+ concentration in either type of SPN, regardless of holding potential (Fig. 3A). In contrast, bath application of DHPG significantly increased both the proximal and distal iSPN intraspine Ca2+ signal evoked by a theta burst bAP train (Fig. 3, B and C). In dSPNs, DHPG only significantly enhanced the bAP Ca2+ signal in distal dendrites (Fig. 3C). As with ryanodine, DHPG had no discernible effect on somatic APs (data not shown), suggesting that the effect was dendritically mediated. These studies were performed in SPNs from 3-wk-old mice to allow ready imaging of dendrites. However, this reliance raises the possibility that the phenomenon is developmentally regulated, as these mice have not fully matured (Plotkin et al. 2005; Tepper et al. 1998; Uryu et al. 1999). To test this possibility, the effect of DHPG was examined in ex vivo slices from 2-mo-old mice. The effect at this age was the same as that seen in the younger neurons (Fig. 3D), suggesting that CICR was not just a feature of immature SPNs.
To better understand the differences between dSPNs and iSPNs in the response to DHPG, qPCR approaches were used to characterize the expression of CICR-related genes. SPNs were separated by FACS of cells acutely dissociated from brain slices taken from D1 and D2 receptor-labeled BAC transgenic mice. The relative abundance of mRNAs encoding mGluR1 and mGluR5, as well as those encoding RYR2, RYR3, Cav1.2, and Cav2.3, was higher in iSPNs than dSPNs (Fig. 3E). Expression of mRNA for IP3 receptors (IP3Rs) and Homer 1 and 3 proteins was similar in the two SPN types (Fig. 3E). These differences in gene expression provide a potential explanation for the more robust CICR in iSPNs and the ability of mGluR activation to enhance this response.
As expected, dialysis with ryanodine (75 μM) blocked the enhancement of CICR by DHPG (Fig. 4A). Dialysis with the IP3R antagonist xestospongin C (5 μM) also blocked the DHPG effect (Fig. 4B). Recent work has suggested that mGluR activation of src kinase promotes RYR-dependent CICR in the induction of corticostriatal LTD (Lerner and Kreitzer 2012). To test this hypothesis, the effect of DHPG on bAP-evoked Ca2+ transients was examined in the presence of the src inhibitor PP-2 (10 μM in bath). Indeed, PP-2 completely blocked the DHPG-induced enhancement of bAP-evoked Ca2+ transients (Fig. 4C). Although group I mGluR activation enhances SPN dendritic CICR via both IP3Rs and RYRs, enhanced RYR engagement may be sufficient for LTD induced by HFS induction protocols (Lerner and Kreitzer 2012), as xestospongin C (IP3R antagonist) did not prevent HFS-induced LTD in iSPNs (3 × 3-s, 100-Hz trains of activity; depression = 64.5 ± 11.6% in control vs. 62.9 ± 7.6% in 5 μM xestospongin C; n = 4).
Fig. 4.

DHPG enhances bAP-evoked dendritic Ca2+ transients via Ca2+-induced-Ca2+ release (CICR) and non-CICR mechanisms. A–C: average theta burst bAP-evoked Ca2+ transients in distal iSPN dendritic spines in the presence or absence of DHPG (50 μM). Traces are from iSPNs loaded with the ryanodine receptor (RYR) antagonist ryanodine (75 μM, n = 6 cells, 20 spines; A), loaded with the inositol trisphosphate receptor (IP3R) antagonist xestospongin C (XC, 5 μM, n = 5 cells, 14 spines; B), or incubated in PP-2 (10 μM, bath, n = 5 cells, 25 spines; C). Example images of spines from which line scans were taken are shown. Shaded areas are SE, and solid lines are fits of the average G/R Ca2+ transients. Ca2+ transient areas corresponding to each burst are shown in register at bottom. Ca2+ transient areas are fit with a single exponential. Ryanodine, xestospongin C, and PP-2 all prevented the DHPG-induced enhancement of Ca2+ transients in dendritic spines. D: normalized Ca2+ transient amplitudes corresponding to the decaying portion (starting from peak of last burst) of the transient. DHPG significantly slowed the decay in iSPNs but not dSPNs in the presence of ryanodine, xestospongin C, and PP-2. Graphs produced from same data shown in A–C and Fig. 3, B and C. The effect of DHPG on decay kinetics is preserved with lower Ca2+ indicator concentration (n = 4 cells, 26 spines). E: relative mRNA expression of Na+/Ca2+ exchanger (NCX)1–3 in FACS-pooled dSPNs and iSPNs (n = 4–8). F: averaged fit Ca2+ transients in distal iSPN spines in response to the NCX pump antagonist SN-6 (n = 5 cells, 11 spines). Fits are normalized to the peak of the first burst. Normalized Ca2+ transient amplitudes corresponding to the decaying portion of the transient are shown at bottom. SN-6 significantly slowed the decay kinetics. G: normalized Ca2+ transient amplitudes corresponding to the decaying portion of the transient before and after incubation in DHPG in distal dendritic spines of iSPNs in the presence of SN-6 (N = 22 spines, 4 cells). H: box plots showing % DHPG-induced slowing of the Ca2+ transient decay, in the presence of SN-6, taken from data in G. Blockade of NCX pumps partially occluded the DHPG-induced slowing of the Ca2+ decay. I: correlation of DHPG-induced slowing and DHPG-induced changes in Ca2+ transients in distal spines of iSPNs and dSPNs, taken from data in D. Lines are linear fits of data points; dashed portion shows extrapolated fit. *P < 0.05, Wilcoxon signed-rank test.
DHPG also slowed the decay of intraspine Ca2+ concentration following the bAP-evoked transient in iSPNs but not dSPNs (Fig. 4D). Because the effects of mGluR stimulation were most robust in iSPNs, subsequent studies focused on them. The decay kinetics reflect a variety of processes, including diffusion and intracellular sequestration, but plasma membrane Ca2+ pumps are thought to be the principal determinants of Ca2+ clearance in dendritic spines (Sabatini et al. 2002). A prominently expressed Ca2+ pump in the striatum is the Na+/Ca2+ exchanger (NCX) (Canitano et al. 2002). Profiling SPNs with qPCR revealed that mRNAs for NCX1–3 were all detectable, with NCX1 being the most abundant in iSPNs (Fig. 4E). In agreement with the inferred role of NCXs, the NCX antagonist SN-6 (10 μM) slowed the decay of the bAP-evoked Ca2+ transient, much like the slowing produced by DHPG (Fig. 4F). Antagonizing NCX pumps with SN-6 diminished but did not fully occlude the DHPG effect on the Ca2+ decay kinetics (Fig. 4, G and H), suggesting that the effects of mGluR stimulation might be mediated in part by modulating NCX pumps. How mGluR signaling might be affecting NCX kinetics and Ca2+ clearance is unclear. NCX has been shown to be modulated by signaling through Gq-linked G protein-coupled receptors (GPCRs), like group I mGluRs (Annunziato et al. 2004; Katanosaka et al. 2005). However, inhibition of the obvious intermediaries (protein kinase C, PLA2, or membrane PIP2; see materials and methods) had no effect on the DHPG-induced slowing of decay kinetics (Table 1). Another possibility is that there is a direct interaction between NCX and mGluRs (Kim et al. 2007), but this was not tested. The observation that DHPG increased dendritic spine Ca2+ amplitude in both iSPNs and dSPNs, but only significantly slowed decay kinetics in iSPNs, argues that group I mGluRs enhanced CICR and slowed Ca2+ decay kinetics through two independent, dissociable mechanisms.
Table 1.
DHPG-mediated slowing of calcium decay kinetics is not mediated by three common PLC-linked pathways
| ACSF | DHPG | |
|---|---|---|
| Calphostin | 0.72 ± 0.01 | 0.78 ± 0.02* |
| PIP2 | 0.75 ± 0.01 | 0.83 ± 0.01* |
| ACA | 0.77 ± 0.01 | 0.86 ± 0.01* |
Values are mean ± SE normalized calcium transient decay amplitudes (normalized to peak Ca2+ amplitude of last back-propagating action potential burst) in response to 50 μM (S)-3,5-dihydroxyphenylglycine (DHPG) in the presence and absence of 1 μM calphostin (n = 4 cells, 22 spines), 200 μM phosphatidylinositol 4,5-bisphosphate (PIP2; n = 4 cells, 24 spines), or 20 μM N-(p-amylcinnamoyl)anthranilic acid (ACA; n = 4 cells, 16 spines), all in the presence of 75 μM ryanodine. Comparisons made at 300 ms.
P < 0.05, Wilcoxon signed-rank test.
Cav1.3 L-type Ca2+ channels are necessary for mGluR effects.
In skeletal muscle, strong depolarization of the plasma membrane is capable of inducing ER Ca2+ release without Ca2+ entry from the extracellular space by bringing about a conformational change in L-type channels that are physically coupled to RYRs (Dulhunty et al. 2002). However, in SPNs, Ca2+ influx was necessary for CICR, as blocking plasma membrane Ca2+ channels by locally puffing on Cd2+ virtually eliminated the dendritic Ca2+ elevation triggered by bAP bursts (Fig. 5, A and B). Moreover, in the presence of Cd2+, DHPG had no effect on the bAP-evoked Ca2+ signal (Fig. 5, B and C).
Fig. 5.

DHPG enhancement of bAP-evoked dendritic Ca2+ transients requires Cav1.3-mediated CICR. A: maximum-intensity projection image of an iSPN loaded with 50 μM Alexa Fluor 568. Inset: high-magnification image of region identified by dashed box. Puffer electrode used to locally deliver cadmium to the distal dendritic region is shaded pink. B: average theta burst bAP-induced Ca2+ transients in distal iSPN dendritic spines. Recordings were made in the presence of a local dendritic cadmium puff in the absence of DHPG (n = 5 cells, 15 spines) or a local dendritic cadmium puff (puffer contained 200 μM cadmium + 50 μM DHPG; n = 5 cells, 14 spines) after bath application of DHPG (50 μM). Shaded areas are SE, and solid lines are fits of the average ΔF/Fo Ca2+ transients. Puffer solution was supplemented with 25 μM Alexa Fluor 568 to visually confirm drug delivery. Ca2+ transient areas corresponding to each bAP burst are shown at bottom. Ca2+ transient areas are fit with a single exponential. Extracellular Ca2+ was required for DHPG enhancement of bAP-evoked Ca2+ transients. C: the magnitude of the bAP-evoked Ca2+ transient component due to bath application of DHPG in the presence of cadmium was calculated by subtraction. DHPG produced no additional Ca2+ component in the presence of cadmium. For reference, the magnitude of the Ca2+ transient due to DHPG in the absence of cadmium is shown in black (calculated from data presented in Fig. 1). D: average theta burst bAP-evoked Ca2+ transients in distal iSPN dendritic spines in the presence of isradipine (5 μM) ± 50 μM DHPG (n = 3 cells, 15 spines). L-type VGCCs were necessary for the DHPG-induced enhancement of bAP-evoked Ca2+ transients. E: average theta burst bAP-evoked Ca2+ transients in distal iSPN dendritic spines in the presence or absence of 50 μM DHPG in Cav1.3-null mice (Cav1.3 KO; n = 5 cells, 11 spines). Cav1.3 L-type VGCCs were necessary for the DHPG-induced enhancement of bAP-evoked Ca2+ transients. F: average theta burst bAP-induced Ca2+ transients in distal iSPN dendritic spines in the presence or absence of the L-type VGCC positive modulator BAY K8644 (5 μM, bath applied; n = 5 cells, 13 spines). Recordings are from wild-type D2 BAC mice. Increasing L-type VGCC contribution produced a decrementing enhancement of theta burst bAP-evoked dendritic Ca2+ transients. G: % change in theta burst bAP-evoked dendritic Ca2+ transients caused by application of DHPG in control or Cav1.3-null mice or application of BAY K in control mice. Data for the wild-type (WT) DHPG application group were calculated from Fig. 1 and plotted for comparison. While both DHPG and BAY K enhanced bAP-evoked dendritic Ca2+ transients, the kinetics of the enhancement were opposite. *P < 0.05, Wilcoxon signed-rank test. H: total Ca2+ transient areas (sum of bursts 1–5) in response to theta burst bAP trains evoked from resting membrane potential (hyperpolarized) or subthreshold membrane potential (∼approximately −60 mV; depolarized) in ACSF or 20 μM BPN 4689, loaded with 75 μM ryanodine, or loaded with 75 μM ryanodine + 20 μM BPN 4689 (n = 2–5 cells, 9–25 spines). In the absence of DHPG, non L-type Ca2+ channels also contribute to bAP-mediated CICR. *P < 0.05, Mann-Whitney nonparametric test.
A variety of voltage-dependent Ca2+ channels contribute to bAP-evoked transients in SPN dendrites, including L-type Ca2+ channels with a Cav1.3 pore-forming subunit (Carter and Sabatini 2004; Day et al. 2008; Plotkin et al. 2011). In cardiac muscle and cerebellar granule cells, membrane depolarization and Ca2+ entry specifically through L-type channels triggers CICR (Chavis et al. 1996; Dulhunty et al. 2002). The effect of DHPG on bAP-evoked Ca2+ transients in distal iSPN dendritic spines was examined in the presence of the L-type Ca2+ channel antagonist isradipine (5 μM). Isradipine fully blocked the effect of DHPG (Fig. 5D). To determine which subtype of L-type channels mediates this effect, mice lacking the Cacna1d gene, which codes for the Cav1.3 subunit, were examined (Platzer et al. 2000). In iSPNs from these mice, bAPs evoked an elevation in dendritic Ca2+ concentration, as expected from previous work showing the engagement of other Ca2+ channels by bAPs. However, DHPG had no effect on the dendritic Ca2+ transients in Cacna1d−/− mice (Fig. 5E).
One of the ways in which deletion of Cacna1d might have blunted the effects of DHPG is if mGluR signaling facilitated Cav1.3 channel opening (Lerner and Kreitzer 2012; Topolnik et al. 2009). To address this possibility, Cav1.3 channel opening was enhanced with the dihydropyridine agonist BAY K8644 (Adermark and Lovinger 2007) in wild-type mice and the bAP-evoked Ca2+ transient measured. BAY K8644 increased the amplitude of the Ca2+ transient. However, unlike the effect of DHPG, the response to the first burst of APs was enhanced the most, diminishing with repetition (Fig. 5, F and G). These results are not consistent with the proposition that mGluRs positively modulate Cav1.3 channel opening. Rather, these results suggest that mGluRs enhance CICR, which is triggered by repetitive opening of Cav1.3 channels.
Although mGluR-mediated enhancement of CICR is dependent upon Cav1.3 Ca2+ channels, CICR itself was not dependent upon these channels. Antagonism of Cav1.3 L-type Ca2+ channels with BPN 4689 [1-(3-chlorophenethyl)-3-cyclopentylpyrimidine-2,4,6-trione, also referred to as compound 8] at saturating concentrations (20 μM) (Kang et al. 2012) reduced, but did not fully eliminate, the effect of ryanodine on distal iSPN dendritic spines (Fig. 5H). The effects of ryanodine on CICR were significant in the presence of BPN 4689, even at depolarized membrane potentials where Cav3 channels should be largely inactivated (Fig. 5H), suggesting that Cav1.2 and/or Cav2.3 Ca2+ channels also were capable of promoting CICR in response to bAP bursts (Carter and Sabatini 2004; Plotkin et al. 2011).
In the experiments described thus far, mGluRs were stimulated by bath application of DHPG. To determine whether precisely timed transient activation of mGluRs would engage the same CICR-mediated pathways, glutamate was uncaged at distal iSPN dendritic spines 10 ms after each bAP burst in a train (Fig. 6, A–C). The intensity of the uncaging laser pulse was adjusted 1) to produce an uncaging EPSP (uEPSP) that was similar in amplitude to spontaneously occurring EPSPs and 2) to produce a dendritic Ca2+ transient that was restricted to the targeted spine (Fig. 6, A–C). Uncaging glutamate significantly increased the maximum Ca2+ transient evoked by bAP bursts (Fig. 6, C and D). The group I mGluR antagonists MPEP (1 μM) + CPCCOET (50 μM) significantly attenuated the Ca2+ responses to paired and unpaired bAP trains, but not uncaging alone, as did the combined antagonism of RYRs and IP3Rs with ryanodine and xestospongin C (Fig. 6D). Somatic EPSP amplitudes were unaffected by antagonizing mGluRs or disrupting CICR (data not shown). Normalization revealed that post-pre pairing led to a progressive enhancement of the bAP-evoked Ca2+ transient, which was similar to what was observed with DHPG. This progressive enhancement was prevented by MPEP + CPCCOEt and by the combination of ryanodine and xestospongin C (Fig. 6E).
Fig. 6.
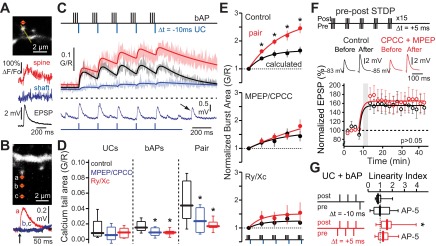
Paired pre- and postsynaptic activity can modulate CICR in iSPNs. A: example of 2-photon uncaging (2PU) of glutamate on a distal SPN spine (top), showing a Ca2+ transient restricted to the targeted spine (middle) and the corresponding somatic uncaging excitatory postsynaptic potential (uEPSP; bottom). B: example of the spatial restriction of 2PU-induced glutamate spread at a distal SPN spine (top), as determined by the uEPSP (bottom) C, top: schematic showing the negative timing of presynaptic glutamate uncaging events (UCs) and postsynaptically triggered bAPs in distal iSPNs. Middle: average Ca2+ transients in distal iSPN dendritic spines in response to uncaging events only (pre; blue), bAPs only (post; black), or negatively timed uncaging events + bAPs (paired; red). Shaded areas are SEs, and solid lines are fits of the average G/R Ca2+ transients. Bottom: example somatic voltage recordings in response to uncaging events, in register with Ca2+ transients above. Arrow indicates a spontaneous depolarization. D: box plots showing the integrated areas under the final 400 ms of the measured Ca2+ transients triggered by glutamate uncaging alone (UCs), theta burst bAPs alone (bAPs), or glutamate uncaging + theta burst bAPs (Pair). Paired and unpaired UC and theta burst bAP recordings were made under control conditions (n = cells, 14 spines), + 2.5 μM MPEP and 125 μM CPCCOEt (CPCC, bath applied; n = 4 cells, 8 spines), or +75 μM ryanodine and 5 μM xestospongin C (loaded via electrode; n = 4 cells, 7 spines). Both mGluRs and intracellular Ca2+ stores were activated by this transient stimulation protocol. E: areas corresponding to each bAP burst of the paired protocol are compared to the Ca2+ transients calculated by the arithmetic sum of the UC and bAP components. Plots are normalized to the first burst for comparison. Measured sequential bursts of paired theta burst trains are significantly higher than arithmetically predicted; this observation is prevented by MPEP + CPCCOET or ryanodine + xestospongin C. *P < 0.05, Wilcoxon signed-rank test. F: iSPNs were held in current-clamp mode under perforated-patch conditions, and a positively timed spike timing-dependent plasticity (STDP) protocol known to induce long-term potentiation (LTP) was delivered in the presence or absence of bath-applied CPCCOEt + MPEP (n = 4) with intrastriatal electrical stimulation. Circles are means, error bars are SEs. Blockade of group I mGluRs had no significant effect on STDP-LTP induction. *P < 0.05, Mann-Whitney nonparametric test. G: distal dendritic spine Ca2+ transients were measured in iSPNs in response to paired bAPs (3 bAPs, 50 Hz) and glutamate uncaging either in a post-pre configuration or a pre-post configuration in the presence (N = 15–17 spines, 5–9 cells) or absence (N = 9–12 spines, 3–9 cells) of 100 μM AP-5. The linearity index was calculated by dividing the response to paired stimuli by the arithmetic sum of the individual stimuli. This brief pairing scenario induced NMDA receptor-mediated supralinear Ca2+ summation in the pre-post but not post-pre scenario. *P < 0.05, Wilcoxon signed-rank test.
As both LTD and LTP depend upon an elevation in postsynaptic Ca2+ concentration (Lovinger 2010), activation of group I mGluRs might promote both forms of plasticity. To address this question, LTP was induced by pairing the bAP theta burst with interleaved positively timed (+5 ms between synaptic stimulation and bAP) presynaptic electrical stimulation to induce STDP-LTP (Shen et al. 2008). Surprisingly, antagonizing group I mGluRs had no effect on the magnitude of LTP (Fig. 6F). Examination of postsynaptic spine Ca2+ transients induced by STDP-LTD (post-pre) and -LTP (pre-post) pairing protocols offered a clue about how this result should be interpreted. Brief post-pre pairing (1 burst, below the threshold necessary for mGluR-enhancement of CICR) induced linear cytosolic Ca2+ summation (calculated as the response to paired stimuli divided by the arithmetic means of pre- and postsynaptic stimuli), whereas pre-post pairing, consistent with previous reports (Carter and Sabatini 2004), produced supralinear Ca2+ summation (Fig. 6G). Antagonizing NMDA receptors (NMDARs) with AP-5 eliminated the supralinearity (Fig. 6G). These results suggest that Ca2+ influx through NMDARs, not simply an elevation in intraspine Ca2+, is critical to LTP induction in SPNs and that mGluRs are not necessary for this to occur.
STDP-LTD is dependent upon CICR.
Both HFS-LTD and STDP-LTD are dependent upon group I mGluR stimulation and an elevation in cytosolic Ca2+ (Adermark and Lovinger 2007; Kreitzer and Malenka 2008; Shen et al. 2008; Surmeier et al. 2009). Recent work by Kreitzer and colleagues (Lerner and Kreitzer 2012) has shown that HFS-LTD is dependent upon RYRs, clearly implicating CICR in the phenomenon. To determine whether CICR also plays a central role in STDP-LTD, iSPNs were subjected to a post-pre timing protocol that has been shown to induce LTD (Shen et al. 2008) in the presence or absence of ryanodine (75 μM). Blocking RYR-dependent CICR significantly diminished STDP-LTD (Fig. 7A), suggesting that both HFS- and STDP-LTD engaged similar mechanisms.
Fig. 7.

Corticostriatal long-term depression (LTD) requires CICR in iSPNs. A: iSPNs were held in current-clamp mode under perforated-patch conditions, and the negatively timed STDP protocol was delivered in the presence or absence of bath-applied ryanodine (75 μm, preincubated for 1 h; control: n = 6, ryanodine: n = 5) with local intrastriatal electrical stimulation. Blockade of RYRs significantly reduced STDP-LTD (P < 0.05, Mann-Whitney nonparametric test). B: proposed model of corticostriatal LTD induction, based on Lerner et al. (Lerner and Kreitzer 2012). PLD, phospholipase D; AEA, anandamide.
DISCUSSION
The data presented demonstrate four key features of activity-dependent regulation of intracellular Ca2+ concentration in the dendrites of SPNs. First, the opening of low-threshold, voltage-dependent Ca2+ channels by bAPs triggered RYR-dependent CICR in dendritic spines in both iSPNs and dSPNs. Second, activation of group I mGluRs enhanced bAP-evoked dendritic CICR; this modulation required Ca2+ entry through Cav1.3 L-type channels and both src kinase and IP3Rs/RYRs. Third, mGluR activation slowed Ca2+ clearance from dendritic spines, broadening the activity-dependent Ca2+ signal. Fourth, CICR was necessary for the induction of STDP-LTD. These studies provide a mechanistic footing for the interaction between postsynaptic spiking and presynaptic glutamate release in the induction of synaptic plasticity.
bAPs evoked CICR in SPNs.
A heterogeneous group of voltage-dependent Ca2+ channels contribute to bAP-evoked cytosolic Ca2+ transients in SPN dendrites (Carter and Sabatini 2004; Day et al. 2008; Plotkin et al. 2011). However, this Ca2+ transient is not solely attributable to extracellular Ca2+ entering the cytoplasm through these channels. Antagonism of RYRs significantly diminished the dendritic Ca2+ transient evoked by short trains of bAPs, demonstrating that a part of it was attributable to release of Ca2+ from intracellular stores triggered by opening of plasma membrane Ca2+ channels—so-called CICR (Armisén et al. 1996; Berridge 1998; Verkhratsky 2002). Our finding that both iSPNs and dSPNs express mRNA for all three RYRs (RYR1–3) and previous work showing RYR protein in SPN dendritic spines (Martone et al. 1997; Verkhratsky 2002) buttress the conclusion.
Somatic CICR in neurons is well documented (Alford et al. 1993; Berridge 1998; Cohen et al. 1997; Jacobs and Meyer 1997; Lipscombe et al. 1988; Richter et al. 2005; Shmigol et al. 1995; Tully and Treistman 2004; Usachev and Thayer 1997). However, there are only a handful of examples of dendritic CICR (Carter et al. 2002; Emptage et al. 1999; Goussakov et al. 2010; Nakamura et al. 1999, 2000; Rose and Konnerth 2001). In our experiments, bAPs evoked CICR in the proximal dendritic spines of both iSPNs and dSPNs; bAPs did not evoke CICR in distal dendrites of dSPNs, in contrast to iSPNs. Although this difference could be attributable in part to the more robust expression of mGluR1/5s or RYR2/3s in iSPNs, it also could be due to the relatively poor invasion of distal dendrites by bAPs in dSPNs (in comparison to iSPNs) (Day et al. 2008). This feature of dSPNs would limit bAP opening of voltage-dependent Ca2+ channels necessary to trigger CICR. As basal dopamine levels are minimal in superfused ex vivo brain slices (Day et al. 2008), mGluR1/5 enhancement of dendritic CICR occurred in the absence of either D1 or D2 receptor signaling. Thus the D2 receptor dependence of LTD induction lies elsewhere, as shown by recent work implicating regulation of RGS4 (Lerner and Kreitzer 2012).
In SPNs, RYR-dependent CICR can be triggered by Ca2+ entry through both Cav1 (L type) and non-Cav1 channels. This lack of specificity is similar to that found in hippocampal pyramidal neurons (Nakamura et al. 2000) and dissimilar to that reported in other cell types where Cav1 channels play a dominant role (Chavis et al. 1996; Dulhunty et al. 2002). This promiscuous coupling should allow CICR to be engaged in both up-states, where depolarization-evoked Ca2+ transients are dominated by Cav1 Ca2+ channels, and down-states, where Cav3 Ca2+ channels make a larger contribution (Carter and Sabatini 2004; Plotkin et al. 2011).
mGluR activation enhanced CICR.
Both pharmacological and synaptic activation of group I mGluRs led to a progressive enhancement in dendritic CICR evoked by bAPs. This enhancement was observed in both dSPNs and iSPNs, albeit more robust in iSPNs. Consistent with this observation, mRNA for group I mGluRs (mGluR1, mGluR5) was found in both types of SPN, in agreement with inferences drawn from previous studies (Testa et al. 1994). In hippocampal pyramidal neurons, pharmacological activation of mGluRs also enhances bAP-evoked CICR by engaging IP3Rs (Nakamura et al. 1999, 2000). IP3Rs also appeared to contribute to the enhancement of CICR in SPNs. Both types of SPN robustly expressed IP3Rs, and the IP3R antagonist xestospongin C reduced the mGluR effect on CICR. However, inhibition of src kinase completely eliminated the effects of mGluR stimulation. Because mGluR stimulation alone had no measurable effect on intracellular Ca2+ concentration, our interpretation of this result is that mGluR activation initiates two parallel signaling cascades: one that is anchored by src kinase, which leads to enhanced RYR Ca2+ sensitivity (Fagni et al. 2000; Lerner and Kreitzer 2012), and another that involves PLC, which leads to an elevation in the sensitivity of IP3Rs to Ca2+ (Nakamura et al. 1999, 2000; Nevian and Sakmann 2006).
The enhancement of RYR-dependent CICR by IP3R signaling creates a context for understanding discrepancies in the literature about the requirements for LTD induction in SPNs. First, our results are consistent with the recent report that RYRs are necessary for LTD induction (Lerner and Kreitzer 2012). With a robust induction protocol involving HFS and postsynaptic depolarization that is likely to strongly elevate postsynaptic Ca2+ concentration, there was no dependence on IP3Rs. With a less robust STDP protocol, the added engagement of IP3R signaling appears to be necessary to reach the threshold needed for LTD induction (Fino et al. 2010).
The coordinated facilitation of CICR by mGluR signaling was most prominent in distal dendrites (>100 μm from the soma). The most likely explanation for this regional difference is a “ceiling” effect. In SPN dendrites, bAPs decrementally propagate, leading to progressively weaker activation of voltage-dependent Ca2+ channels and smaller CICR. In the proximal dendrites, where the bAP amplitude is the greatest, the robust opening of voltage-dependent Ca2+ channels might effectively maximize CICR. In the distal dendrites, where bAP amplitudes are considerably smaller and the opening of voltage-dependent channels more restricted, CICR should be submaximal and the mGluR enhancement of CICR easier to see.
The mGluR-mediated enhancement of CICR was dependent upon Cav1.3 L-type Ca2+ channels. The basis for this exclusivity (in contrast to CICR per se) is not entirely clear but might depend on scaffolding of mGluRs and Cav1.3 channels into microdomains near synapses (Olson et al. 2005; Zhang et al. 2005). In hippocampal interneurons, mGluR stimulation of protein kinase C appears to potentiate the opening of L-type Ca2+ channels, rather than CICR per se (Topolnik et al. 2009). The most compelling evidence against this interpretation of our results is that direct enhancement of L-type channel opening with BAY K8644 led to a very different pattern of changes in postsynaptic Ca2+ concentration with repetitive stimulation than mGluR stimulation. It is possible that this difference underlies the contrasting effects of mGluR stimulation in SPNs, where it induces LTD, and in hippocampal interneurons, where it induces LTP (Sung et al. 2001; Topolnik et al. 2009).
CICR was necessary for STDP-LTD induction.
Our studies show that, like HFS-LTD (Lerner and Kreitzer 2012), STDP-LTD also depended upon CICR in SPNs, establishing another mechanistic link between the two induction protocols. Previous work (Lerner and Kreitzer 2012) also demonstrated that the mGluR effect on LTD induction was dependent upon src kinase. Again, our results are in agreement with these results and extend them by showing that this dependence is likely to be mediated by regulation of RYRs, whose Ca2+ sensitivity is increased by src kinase phosphorylation (Zhang et al. 2004). Our studies also provide a partial explanation for the long-standing observation that while corticostriatal LTD can be induced at glutamatergic synapses onto both SPN populations (Shen et al. 2008; Wang et al. 2006), it is more readily inducible in iSPNs (Kreitzer and Malenka 2007). Clearly, CICR and its enhancement by mGluRs were more robust in iSPNs than dSPNs. The involvement of CICR in LTD induction creates a reliance upon activity in the recent past, as intracellular Ca2+ stores are “leaky integrators” of past activity (Berridge 1998). This dependence could be homeostatic, helping to ensure that quiescent SPNs do not undergo changes in synaptic strength that could further lower their activity below a set point.
Our studies also provide insight into the roles of timing and repetition in the induction of STDP. Previous work (Fino et al. 2010; Pawlak and Kerr 2008; Shen et al. 2008) has shown that in the presence of GABAA receptor antagonists repeated pairing of postsynaptic spikes with trailing synaptic stimulation (post-pre pairing) leads to LTD in SPNs, whereas reversing the order of stimulation (pre-post pairing) induces LTP in conditions permissive for second messenger signaling (e.g., perforated-patch recording). Our work suggests that the timing dependence of STDP-LTP is largely a consequence of the ability of pre-post protocols to effectively engage NMDARs, leading to supralinear elevations in postsynaptic Ca2+ concentration. NMDARs are also necessary for HFS-LTP (Calabresi et al. 1992b; Lovinger 2010; Paillé et al. 2010). In the absence of NMDAR engagement, the timing dependence of plasticity should be less stringent. Both pre-post and post-pre pairing should be effective means of producing CICR and STDP-LTD as long as the repetition rate of the induction protocol allows for summation of mGluR-mediated signaling and intracellular Ca2+. This is certainly the case in HFS protocols.
This also implies that neuromodulators that regulate SPN NMDARs could shape the timing dependence of STDP. D2 dopamine receptors, for example, are likely to promote LTD not only through intracellular signaling cascades that enhance eCB synthesis (Lerner and Kreitzer 2012; Lovinger 2010) but also by suppressing Ca2+ entry through NMDARs (Higley and Sabatini 2010) and weakening timing dependence. M1 muscarinic receptors also shape LTD induction, as might GABAergic signaling (Calabresi et al. 1998, 1999; Fino et al. 2010; Wang et al. 2006). These factors could contribute to discrepancies in the literature about the timing dependence of STDP-LTD (Fino et al. 2010; Pawlak and Kerr 2008; Shen et al. 2008; Shindou et al. 2011). Unraveling these factors will require experimental models in which the timing of neuromodulatory and GABAergic input to SPNs is tightly controlled, something that is now achievable with optogenetic approaches.
Summary.
Our results show that CICR is a significant component of dendritic integration in SPNs and critical to corticostriatal STDP-LTD induction. In particular, our studies elucidate the relationship between two obligatory participants in the most common form of corticostriatal LTD: group I mGluRs and Cav1.3 L-type voltage-dependent Ca2+ channels (Lovinger 2010). Activation of group I mGluRs sensitized IP3Rs and RYRs to Ca2+ entry through Cav1.3 channels in SPN dendrites, promoting CICR. CICR was necessary for the induction of corticostriatal LTD, explaining the requirement for coactivation of mGluRs and Cav1.3 channels. In addition, mGluR signaling slowed the extrusion of Ca2+, broadening the duration of the postsynaptic Ca2+ signaling resulting from CICR.
GRANTS
This work was supported by National Institute of Neurological Disorders and Stroke Grant NS-34696 and CHDI.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.L.P., W.S., I.R., L.E.S., M.D., and D.J.S. conception and design of research; J.L.P., W.S., I.R., L.E.S., and C.S.C. performed experiments; J.L.P., W.S., I.R., L.E.S., and C.S.C. analyzed data; J.L.P., W.S., I.R., L.E.S., M.D., C.S.C., and D.J.S. interpreted results of experiments; J.L.P. prepared figures; J.L.P. and D.J.S. drafted manuscript; J.L.P. and D.J.S. edited and revised manuscript; J.L.P. and D.J.S. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank S. Ulrich and K. Saporito for their technical assistance.
REFERENCES
- Adermark L, Lovinger DM. Combined activation of L-type Ca2+ channels and synaptic transmission is sufficient to induce striatal long-term depression. J Neurosci 27: 6781–6787, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alford S, Frenguelli BG, Schofield JG, Collingridge GL. Characterization of Ca2+ signals induced in hippocampal CA1 neurones by the synaptic activation of NMDA receptors. J Physiol 469: 693–716, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato L, Pignataro G, Di Renzo GF. Pharmacology of brain Na+/Ca2+ exchanger: from molecular biology to therapeutic perspectives. Pharmacol Rev 56: 633–654, 2004 [DOI] [PubMed] [Google Scholar]
- Armisén R, Sierralta J, Vélez P, Naranjo D, Suárez-Isla BA. Modal gating in neuronal and skeletal muscle ryanodine-sensitive Ca2+ release channels. Am J Physiol Cell Physiol 271: C144–C153, 1996 [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron 21: 13–26, 1998 [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Bernardi G. Activation of M1-like muscarinic receptors is required for the induction of corticostriatal LTP. Neuropharmacology 38: 323–326, 1999 [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Pisani A, Bernardi G. Endogenous ACh enhances striatal NMDA-responses via M1-like muscarinic receptors and PKC activation. Eur J Neurosci 10: 2887–2895, 1998 [DOI] [PubMed] [Google Scholar]
- Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci 12: 4224–4233, 1992a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Pisani A, Mercuri NB, Bernardi G. Long-term potentiation in the striatum is unmasked by removing the voltage-dependent magnesium block of NMDA receptor channels. Eur J Neurosci 4: 929–935, 1992b [DOI] [PubMed] [Google Scholar]
- Calabresi P, Pisani A, Mercuri NB, Bernardi G. Post-receptor mechanisms underlying striatal long-term depression. J Neurosci 14: 4871–4881, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canitano A, Papa M, Boscia F, Castaldo P, Sellitti S, Taglialatela M, Annunziato L. Brain distribution of the Na+/Ca2+ exchanger-encoding genes NCX1, NCX2, and NCX3 and their related proteins in the central nervous system. Ann NY Acad Sci 976: 394–404, 2002 [DOI] [PubMed] [Google Scholar]
- Carter AG, Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron 44: 483–493, 2004 [DOI] [PubMed] [Google Scholar]
- Carter AG, Vogt KE, Foster KA, Regehr WG. Assessing the role of calcium-induced calcium release in short-term presynaptic plasticity at excitatory central synapses. J Neurosci 22: 21–28, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Peterson JD, Gertler TS, Glajch KE, Quintana RE, Cui Q, Sebel LE, Plotkin JL, Shen W, Heiman M, Heintz N, Greengard P, Surmeier DJ. Strain-specific regulation of striatal phenotype in Drd2-eGFP BAC transgenic mice. J Neurosci 32: 9124–9132, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavis P, Fagni L, Lansman JB, Bockaert J. Functional coupling between ryanodine receptors and L-type calcium channels in neurons. Nature 382: 719–722, 1996 [DOI] [PubMed] [Google Scholar]
- Christie BR, Magee JC, Johnston D. Dendritic calcium channels and hippocampal long-term depression. Hippocampus 6: 17–23, 1996 [DOI] [PubMed] [Google Scholar]
- Cohen AS, Moore KA, Bangalore R, Jafri MS, Weinreich D, Kao JP. Ca2+-induced Ca2+ release mediates Ca2+ transients evoked by single action potentials in rabbit vagal afferent neurones. J Physiol 499: 315–328, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M, Wokosin D, Plotkin JL, Tian X, Surmeier DJ. Differential excitability and modulation of striatal medium spiny neuron dendrites. J Neurosci 28: 11603–11614, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulhunty AF, Haarmann CS, Green D, Laver DR, Board PG, Casarotto MG. Interactions between dihydropyridine receptors and ryanodine receptors in striated muscle. Prog Biophys Mol Biol 79: 45–75, 2002 [DOI] [PubMed] [Google Scholar]
- Emptage N, Bliss TV, Fine A. Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron 22: 115–124, 1999 [DOI] [PubMed] [Google Scholar]
- Fagni L, Chavis P, Ango F, Bockaert J. Complex interactions between mGluRs, intracellular Ca2+ stores and ion channels in neurons. Trends Neurosci 23: 80–88, 2000 [DOI] [PubMed] [Google Scholar]
- Fino E, Glowinski J, Venance L. Bidirectional activity-dependent plasticity at corticostriatal synapses. J Neurosci 25: 11279–11287, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Paillé V, Cui Y, Morera-Herreras T, Deniau JM, Venance L. Distinct coincidence detectors govern the corticostriatal spike timing-dependent plasticity. J Physiol 588: 3045–3062, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci 34: 441–466, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425: 917–925, 2003 [DOI] [PubMed] [Google Scholar]
- Goussakov I, Miller MB, Stutzmann GE. NMDA-mediated Ca2+ influx drives aberrant ryanodine receptor activation in dendrites of young Alzheimer's disease mice. J Neurosci 30: 12128–12137, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Sabatini BL. Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci 13: 958–966, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JM, Meyer T. Control of action potential-induced Ca2+ signaling in the soma of hippocampal neurons by Ca2+ release from intracellular stores. J Neurosci 17: 4129–4135, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Cooper G, Dunne SF, Dusel B, Luan CH, Surmeier DJ, Silverman RB. CaV1.3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson's disease. Nat Commun 3: 1146, 2012 [DOI] [PubMed] [Google Scholar]
- Katanosaka Y, Iwata Y, Kobayashi Y, Shibasaki F, Wakabayashi S, Shigekawa M. Calcineurin inhibits Na+/Ca2+ exchange in phenylephrine-treated hypertrophic cardiomyocytes. J Biol Chem 280: 5764–5772, 2005 [DOI] [PubMed] [Google Scholar]
- Kim YT, Namkung YL, Kwak J, Suh CK. Involvement of Na+-Ca2+ exchanger on metabotropic glutamate receptor 1-mediated [Ca2+]i transients in rat cerebellar Purkinje neurons. Neuroscience 146: 170–177, 2007 [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci 25: 10537–10545, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature 445: 643–647, 2007 [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron 60: 543–554, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner TN, Kreitzer AC. RGS4 is required for dopaminergic control of striatal LTD and susceptibility to parkinsonian motor deficits. Neuron 73: 347–359, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscombe D, Madison DV, Poenie M, Reuter H, Tsien RW, Tsien RY. Imaging of cytosolic Ca2+ transients arising from Ca2+ stores and Ca2+ channels in sympathetic neurons. Neuron 1: 355–365, 1988 [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology 58: 951–961, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, Partridge JG, Tang KC. Plastic control of striatal glutamatergic transmission by ensemble actions of several neurotransmitters and targets for drugs of abuse. Ann NY Acad Sci 1003: 226–240, 2003 [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science 275: 209–213, 1997 [DOI] [PubMed] [Google Scholar]
- Martone ME, Alba SA, Edelman VM, Airey JA, Ellisman MH. Distribution of inositol-1,4,5-trisphosphate and ryanodine receptors in rat neostriatum. Brain Res 756: 9–21, 1997 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Barbara JG, Nakamura K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron 24: 727–737, 1999 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Nakamura K, Lasser-Ross N, Barbara JG, Sandler VM, Ross WN. Inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ release evoked by metabotropic agonists and backpropagating action potentials in hippocampal CA1 pyramidal neurons. J Neurosci 20: 8365–8376, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci 26: 11001–11013, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature 408: 584–588, 2000 [DOI] [PubMed] [Google Scholar]
- Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I, Surmeier DJ. G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J Neurosci 25: 1050–1062, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillé V, Picconi B, Bagetta V, Ghiglieri V, Sgobio C, Di Filippo M, Viscomi MT, Giampà C, Fusco FR, Gardoni F, Bernardi G, Greengard P, Di Luca M, Calabresi P. Distinct levels of dopamine denervation differentially alter striatal synaptic plasticity and NMDA receptor subunit composition. J Neurosci 30: 14182–14193, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak V, Kerr JN. Dopamine receptor activation is required for corticostriatal spike-timing-dependent plasticity. J Neurosci 28: 2435–2446, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 102: 89–97, 2000 [DOI] [PubMed] [Google Scholar]
- Plotkin JL, Day M, Surmeier DJ. Synaptically driven state transitions in distal dendrites of striatal spiny neurons. Nat Neurosci 14: 881–888, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JL, Wu N, Chesselet MF, Levine MS. Functional and molecular development of striatal fast-spiking GABAergic interneurons and their cortical inputs. Eur J Neurosci 22: 1097–1108, 2005 [DOI] [PubMed] [Google Scholar]
- Richter TA, Kolaj M, Renaud LP. Low voltage-activated Ca2+ channels are coupled to Ca2+-induced Ca2+ release in rat thalamic midline neurons. J Neurosci 25: 8267–8271, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Konnerth A. Stores not just for storage. Intracellular calcium release and synaptic plasticity. Neuron 31: 519–522, 2001 [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca2+ ions in dendritic spines. Neuron 33: 439–452, 2002 [DOI] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science 321: 848–851, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindou T, Ochi-Shindou M, Wickens JR. A Ca2+ threshold for induction of spike-timing-dependent depression in the mouse striatum. J Neurosci 31: 13015–13022, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmigol A, Verkhratsky A, Isenberg G. Calcium-induced calcium release in rat sensory neurons. J Physiol 489: 627–636, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung KW, Choi S, Lovinger DM. Activation of group I mGluRs is necessary for induction of long-term depression at striatal synapses. J Neurophysiol 86: 2405–2412, 2001 [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Plotkin J, Shen W. Dopamine and synaptic plasticity in dorsal striatal circuits controlling action selection. Curr Opin Neurobiol 19: 621–628, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper JM, Sharpe NA, Koós TZ, Trent F. Postnatal development of the rat neostriatum: electrophysiological, light- and electron-microscopic studies. Dev Neurosci 20: 125–145, 1998 [DOI] [PubMed] [Google Scholar]
- Testa CM, Standaert DG, Young AB, Penney JB. Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J Neurosci 14: 3005–3018, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topolnik L, Chamberland S, Pelletier JG, Ran I, Lacaille JC. Activity-dependent compartmentalized regulation of dendritic Ca2+ signaling in hippocampal interneurons. J Neurosci 29: 4658–4663, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully K, Treistman SN. Distinct intracellular calcium profiles following influx through N- versus L-type calcium channels: role of Ca2+-induced Ca2+ release. J Neurophysiol 92: 135–143, 2004 [DOI] [PubMed] [Google Scholar]
- Uryu K, Butler AK, Chesselet MF. Synaptogenesis and ultrastructural localization of the polysialylated neural cell adhesion molecule in the developing striatum. J Comp Neurol 405: 216–232, 1999 [DOI] [PubMed] [Google Scholar]
- Usachev YM, Thayer SA. All-or-none Ca2+ release from intracellular stores triggered by Ca2+ influx through voltage-gated Ca2+ channels in rat sensory neurons. J Neurosci 17: 7404–7414, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A. The endoplasmic reticulum and neuronal calcium signalling. Cell Calcium 32: 393–404, 2002 [DOI] [PubMed] [Google Scholar]
- Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, Tkatch T, Lovinger DM, Surmeier DJ. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron 50: 443–452, 2006 [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Kawaguchi Y. The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J Neurosci 16: 2397–2410, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ. The role of the basal ganglia in habit formation. Nat Rev Neurosci 7: 464–476, 2006 [DOI] [PubMed] [Google Scholar]
- Zhang H, Maximov A, Fu Y, Xu F, Tang TS, Tkatch T, Surmeier DJ, Bezprozvanny I. Association of CaV1.3 L-type calcium channels with Shank. J Neurosci 25: 1037–1049, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Köhler M, Yang SN, Zhang F, Larsson O, Berggren PO. Growth hormone promotes Ca2+-induced Ca2+ release in insulin-secreting cells by ryanodine receptor tyrosine phosphorylation. Mol Endocrinol 18: 1658–1669, 2004 [DOI] [PubMed] [Google Scholar]