

SUMMARY
Macrophages possess numerous mechanisms to combat microbial invasion, including sequestration of essential nutrients, like Zn. The pleiotropic cytokine granulocyte macrophage-colony stimulating factor (GM-CSF) enhances antimicrobial defenses against intracellular pathogens such as Histoplasma capsulatum, but its mode of action remains elusive. We have found that GM-CSF activated infected macrophages sequestered labile Zn by inducing binding to metallothioneins (MTs) in a STAT3 and STAT5 transcription factor-dependent manner. GM-CSF upregulated expression of Zn exporters, Slc30a4 and Slc30a7 and the metal was shuttled away from phagosomes and into the Golgi apparatus. This distinctive Zn sequestration strategy elevated phagosomal H+ channel function and triggered reactive oxygen species (ROS) generation by NADPH oxidase. Consequently, H. capsulatum was selectively deprived of Zn, thereby halting replication and fostering fungal clearance. GM-CSF mediated Zn sequestration via MTs in vitro and in vivo in mice and in human macrophages. These findings illuminate a GM-CSF-induced Zn-sequestration network that drives phagocyte antimicrobial effector function.
INTRODUCTION
Zn is an abundant transition metal in living organisms. It is essential in immune homeostasis and function and Zn deficiency has been associated with thymic atrophy, impaired B, T and NK cell responses and T helper-1 (Th1) cytokine production (Fraker et al., 1986). Zn also functions in intracellular signaling (Hirano et al., 2008). Such a wide association with cellular functions requires strict regulation of Zn.
Availability of Zn to biomolecules is tightly regulated by binding proteins, metallothioneins (MTs) and Zn transporters. MTs are cysteine-rich proteins that bind up to seven Zn ions with picomolar affinity and carry the labile Zn fraction to cellular compartments. MTs are induced during oxidative stress, scavenge reactive oxygen species (ROS) and reduce heavy metal intoxication (Coyle et al., 2002).
Macrophages are crucial role in immune defense; they phagocytose and kill intracellular pathogens by oxidative burst, nitric oxide production (Fang, 1997) and T cell activation (Mosser, 2003). ‘Nutritional immunity’ signifies deprivation of essential nutrients to pathogens, thereby exerting an antimicrobial effect (Appelberg, 2006). In this regard, interferon gamma (IFN-γ) deprives Fe in macrophages by regulating transferrin receptors and the transporter Slc11a1 (Nairz et al., 2008; Zwilling et al., 1999). In contrast, macrophages may intoxicate Mycobacterium tuberculosis with Zn in phagosomes (Botella et al., 2011). Zn competitively inhibits Mn binding to an essential virulence determinant in Streptococcus pneumoniae (McDevitt et al., 2011), rendering it inactive. Thus, both metal starvation and intoxication are part of the arsenal employed by macrophages in host defenses.
Apart from directly exploiting metals for combating pathogens, immune cells also need them for activation of defense mechanisms. In this regard, modulation of Zn activates microglia (Kauppinen et al., 2008) and dendritic cells (Kitamura et al., 2006) by altering surface markers and cytokine expression. Thus, Zn regulation is crucial in determining the outcome of an infectious process.
The fungal pathogen, H. capsulatum, is distributed worldwide and causes pulmonary and disseminated histoplasmosis, particularly in immunocompromised patients. For clearance of infection, coordinated action of innate and adaptive immunity is essential. The fungus gains entry into the host primarily via pulmonary route, is phagocytosed and replicates within resting macrophages (Howard, 1964). Activation of macrophages with IFN-γ or GM-CSF promotes fungal growth inhibition. While IFN-γ acts only on murine macrophages in vitro, GM-CSF inhibits growth of yeasts in both murine and human macrophages (Newman and Gootee, 1992). A lack of GM-CSF is lethal during infection and treatment with recombinant GM-CSF accelerates fungal clearance (Deepe et al., 1999).
We have reported that Zn is essential for survival of H. capsulatum within macrophages and chelation causes drastic retardation of fungal growth. (Winters et al., 2010). Here, we elucidate a mechanism by which GM-CSF activates macrophages to preferentially sequester Zn while enhancing ROS production and denying the intracellular pathogen, access to this element. Using a murine pulmonary model of H. capsulatum infection in vivo, we reveal the significance of GM-CSF in Zn sequestration by MTs in infected macrophages. Finally, we have shown that this effect of GM-CSF is conserved in human macrophages.
RESULTS
GM-CSF modulates total Zn and Zn proteome distribution
An unbiased inductively coupled plasma-mass spectrometry (ICP-MS) total elemental analysis was performed to evaluate changes in the concentration of Zn, Fe, Cu and Mn in macrophage lysates and H. capsulatum itself. The total Zn concentration increased in GM-CSF activated peritoneal macrophages compared to the resting control, and increased further in activated peritoneal and bone marrow macrophages during infection with 5 yeasts per macrophage. In contrast, Zn concentration was significantly reduced (p<0.001) in H. capsulatum recovered from activated macrophages in comparison to yeasts from resting peritoneal macrophages, and media-cultured H. capsulatum, suggesting that GM-CSF activation induced deprivation of yeast associated Zn. GM-CSF also altered Fe within macrophages, but not in intracellular yeasts. Meanwhile, activation did not alter the concentration of Cu and Mn in macrophages and yeasts (Figures 1A and 1B). Thus, GM-CSF specifically altered Zn flux in H. capsulatum infected peritoneal and bone marrow macrophages.
Figure 1. GM-CSF distinctly modulates Zn-distribution in infected macrophages.

Total concentration of Zn by ICP-MS in (A, B) lysates from peritoneal and bone marrow macrophages; RP, resting peritoneal, Act, GM-CSF activated, and, total Zn analysis by ICP-MS in H. capsulatum (Hc) recovered from macrophages and compared with yeasts grown in Ham’s F12 media, data are mean ± SD, 4 independent experiments. The cell lysate concentrations are based on 2.5 × 106 cells in ~200 μl; H. capsulatum concentration was calculated based on mass of yeasts recovered; (C, D) typical SEC-ICP-MS chromatograms of activated peritoneal and bone marrow macrophages, compared to resting conditions; (E, F) Left panel, Graphical representation of color coded peaks depicting Zn distribution in various MW fractions in the chromatograms of infected activated peritoneal and bone marrow macrophages; middle panel, Zn distribution calculated as area under the curve of individual peaks against the total area; red and green boxes with **, ## symbol indicate significant differences in the corresponding color coded fractions between different groups; data are mean ± SD, 4 independent experiments; (G, H) Typical SEC-ICP-MS chromatograms of TNF-α treated macrophages, 3 independent experiments, Y axis in all chromatograms is off-set to allow easy comparison under the same scale. Related to Figure S1 and Table S1.
To examine the Zn-proteome distribution in macrophages, we used size exclusion chromatography based on molecular size via hydrodynamic radii coupled to ICP-MS (SEC-ICP-MS). The Zn signal increased upon exposure to GM-CSF compared to resting peritoneal macrophages and a greater increase was seen upon activation and infection (Figure 1C). A large increment in the Zn signal was observed in infected activated bone marrow macrophages compared to uninfected control (Figure 1D). The relative Zn distribution between fractions was altered; the principal change occurred at ~20 min (≈20–7 kDa), representing a major portion of total Zn. This correlated with a reduction in the labile, exchangeable Zn pool appearing after 22 min in infected activated peritoneal and bone marrow macrophages (Figures 1E and 1F). Zn distribution was altered dynamically as early as 2 h and held a similar trend for the 24 h time period in infected activated peritoneal macrophages (Figures S1A and S1B).
To determine the specificity of GM-CSF in Zn modulation, macrophages were treated with the pro-inflammatory cytokine, tumor necrosis factor-α (TNF-α). TNF-α did not alter the Zn signal in macrophages (Figures 1G and 1H).
GM-CSF alters Zn transporter expression in H. capsulatum infected macrophages
We hypothesized that GM-CSF alters macrophage Zn distribution by regulating transporters. In an unbiased analysis of expression of Zn importers, ZIPs (Slc39a1-14) and exporters, ZNTs (Slc30a1-10), ~60 fold and 40 fold upregulation in Slc39a2, was observed in infected activated peritoneal and bone marrow macrophages respectively, compared to resting uninfected control (Figures 2A and 2C). A significant 2–4 fold increase in Slc304 and Slc30a7 (p<0.001 for peritoneal and p<0.05 for bone marrow macrophages) was also observed upon activation and infection (Figures 2B and 2D). The expression of another importer, Slc39a8 was unchanged (Figure S2A). TNF-α treatment of did not alter Slc39a2, Slc30a4 and Slc30a7 expression (Figure 2E). These data suggest that GM-CSF activated-infected macrophages may increase Zn import and mobilize it into intracellular organelles via exporters.
Figure 2. GM-CSF alters Zn transporter and MT expression.

qRT-PCR of Slc39a and Slc30a in (A, B) peritoneal; (C, D) bone marrow; (E) TNF-α treated macrophages, data are mean ± SEM, 3 independent experiments; (F, G) Mt1, Mt2 and Mt3 expression in macrophages; RP, resting peritoneal; Act, GM-CSF activated; Hc, H. capsulatum,, data are mean ± SEM, 8 independent experiments; ND, not detected. Related to Figure S2.
Proteomics analysis of SEC fractions
GM-CSF altered the Zn-binding proteome in macrophages. For proteomics analysis, five fractions identified by SEC-ICP-MS were collected with the plasma turned-off and analyzed using a bottoms-up proteomics scheme. The list of identification parameters and proteins in all fractions and Zn-binding proteins highlighted in the fractions at ~10 and ~20 min are in Table S1. The CCCH zinc finger protein, ZC3H12A, calreticulin and MT2 were selected as being potentially associated with immune response and Zn binding. MT2 was detected in the peak that scavenged a majority of Zn at 20 min. MT1, MT3 and ZIP2 were not identified in database search. ZIP2 is located on the cell membrane. The association of membrane proteins with hydrophobic lipids and their insolubility poses a challenge in proteomic analysis (Santoni et al., 2000). Peptide matching is compromised by complex covalent hetero-oligomerization of MT isoforms, likely explaining the absence of MT1/MT3 as hits in database search. We observed oligomerization at ~20 min based on a shift in retention time towards the high molecular weight region, with the peak showing maximal absorbance at 260 nm, typical of -S-S- bonds formed by MTs during oligomerization, and not 280 nm, representative of Trp and Tyr containing proteins (Figure S1C).
GM-CSF, but not TNF-α induces MT expression in macrophages
GM-CSF enhanced Zn binding to the ~20 min fraction in infected macrophages (Figures 1C and 1D). Proteomic analysis and UV-Vis spectral characteristics suggested a major contribution of MTs to this Zn signal (Figure S1C, S1D, and S1E). Therefore, we examined the effect of GM-CSF on Mt expression. GM-CSF, but not TNF-α increased Mt1, Mt2 and Mt3 expression in macrophages during infection (Figures 2F and 2G), Mt3 is predominantly expressed in the brain (Falnoga et al., 2012). To ensure that our findings are valid, Mt3 was examined in hepatocytes and thymocytes and was not upregulated (Figure S2B). Expression of the Zn finger ribonuclease, Zc3h12a was enhanced in infected macrophages but was not specific to GM-CSF (Figures S2D and S2E). GM-CSF did not alter the expression of calreticulin and of the metal response element binding transcription factor-1 (Mtf1) which regulates Mt expression (Figure S2C and S2F). Thus, GM-CSF specifically induced Mt expression.
GM-CSF mobilizes labile Zn away from H. capsulatum
Confocal microscopy was used to visualize labile Zn with the fluorescent probe, Zinpyr-1 (Cao et al., 2001). Uninfected resting peritoneal macrophages showed labile Zn distribution in the cytoplasm and epinuclear space (Figure 3A); however, Zn staining was diffuse in infected resting peritoneal macrophages (Figure 3C). GM-CSF activation caused focal Zn accumulation (Figures 3B and 3F) and mobilized Zn away from H. capsulatum (Figures 3D and 3G) into the Golgi apparatus (Figure 3I). The reduced Zn fluorescence of yeasts observed in GM-CSF activated macrophages was not due to an impermeability of the dye (Figure S3A). Previous reports suggest that Zinpyr-1 may localize in the Golgi (Walkup et al., 2000). To ensure that labile Zn localization was a result of cytokine stimulation, we evaluated it in TNF-α treated macrophages. Zn distribution was non-focal, no or very little Zn staining was observed in the Golgi and labile Zn was observed in yeast (Figures 3E and 3H). This is consistent with lack of changes in Zn regulation in TNF-α treated macrophages (Figures 1G, 1H, 2E and 2G). Peritoneal macrophages treated with Zn and the ionophore, pyrithione, yielded bright fluorescence and very dim staining was observed in cells treated with the Zn chelator N,N,N′,N′-tetrakis-(2-pyridylmethyl)-ethylenediamine (TPEN) (Figures S3B and S3C). Labile Zn was examined in alveolar macrophages, the initial phagocyte that encounters H. capsulatum in vivo. While staining occurred in the cytosol and Golgi in resting alveolar macrophages, GM-CSF activation caused Zn localization in the Golgi (Figures S3D-S3G). Collectively, these data suggest that GM-CSF induced Zn redistribution in macrophages, which may limit its accessibility to H. capsulatum.
Figure 3. GM-CSF, but not TNF-α triggers Zn mobilization.

Confocal microscopic analysis of Zn (green), nucleus (blue) and H. capsulatum (Hc) (red) in (A–E) peritoneal; (F–H) bone marrow macrophages; merge, overlay of Zn, nucleus and H. capsulatum; (I) Staining for Zn (green), nucleus (blue) and Golgi (red); merge, overlay of Zn, nucleus and Golgi; yellow, co-localization of Golgi and Zn dyes; white arrows point at the Golgi; scale bars, 20 μm; RP, resting peritoneal; Act, activated; 3 independent experiments. Related to Figure S3.
Metallothioneins sequester and deprive H. capsulatum of Zn
Since Zn regulation was similar in both types of macrophages, we utilized bone marrow macrophages for further studies. To determine if Zn sequestration in macrophages and deprivation in H. capsulatum was caused by MTs, we took two approaches. First, we silenced the expression of all four Mt genes (Figure 4A). Mt-silenced macrophages manifested reduced Zn binding to the chromatographic fraction at 20 min accompanied by an increase in the labile Zn fraction at ~25 min. (Figure 4B and zoom in). H. capsulatum recovered from Mt silenced macrophages had higher Zn content (p<0.01) (Figure 4C). Next, we examined Zn sequestration in Mt1−/−Mt2−/− macrophages. These cells exhibited a lack of Zn-binding to the fraction at 20 min, accompanied by an increase in labile Zn in GM-CSF treated and infected macrophages (Figure 4D and zoom in). H. capsulatum recovered from Mt1−/−Mt2−/− macrophages had higher Zn concentration (p<0.01) compared to wild type (WT) cells (Figure 4E). Thus, Zn deprivation within H. capsulatum resulted preponderantly from sequestration by MTs.
Figure 4. GM-CSF engages the STAT5 and STAT3 transcriptional program to regulate MT-Zn sequestration.

(A) qRT-PCR of Mt genes in activated (Act) macrophages treated with scramble or Mt siRNA, % decrease in expression compared to scramble siRNA control, data are mean ± SEM, 2 independent experiments; (B, D) SEC-ICP-MS chromatograms of Mt silenced activated infected (Hc) macrophages and from WT and Mt1−/Mt2−/− mice and zoom in of labile Zn fraction, 2 independent experiments; (C, E) Total metal analysis of Zn in H. capsulatum (Hc) from Mt silenced and WT, Mt1−/−Mt2−/− macrophages, data are mean ± SD, 2 independent experiments; (F) Colony forming units (CFU) of H. capsulatum 24 h post infection from WT and Mt1−/−Mt2−/− activated macrophages compared to untreated control; data are mean ± SEM, 4 independent experiments; (G–J) SEC-ICP-MS chromatograms of chemically inhibited and Stat3 and Stat5 silenced macrophages, % decrease in peak area is compared to scramble siRNA treated infected macrophages; Y axis, Offset Zn-signal, 3 independent experiments; (K) p-STAT3, p-STAT5 and β-actin from silenced macrophages 24 h post activation, n=2; (L) Western blot of p-STAT3, STAT3 and β-actin from lysates 10 min post activation and infection, n=2.
We queried if MT1 and MT2 participated in antimicrobial defense. We postulated that the lack of Zn sequestration in the absence of MT1 and MT2 would diminish the ability of GM-CSF to inhibit fungal growth. While H. capsulatum growth was inhibited in WT activated macrophages, Mt1−/−Mt2−/− activated cells showed decreased ability to limit growth of yeasts (Figure 4F).
GM-CSF engages the STAT5 and STAT3 transcriptional program to mediate Zn regulation
We sought to determine the signaling mechanism that resulted in Zn modulation. GM-CSF signals through STAT5 (Bhattacharya et al., 2001) and STAT binding sites are present on the Mt promoter (Lee et al., 1999). Inhibition of STAT5 and STAT3 reduced MT-Zn binding (Figure 4G and 4H). To confirm our observations with chemical inhibitors, we silenced Stat5 and Stat3, which decreased Zn binding to MTs in the chromatograms of activated infected macrophages (Figures 4I, 4J and 4K).
These findings led us to postulate that GM-CSF may directly activate STAT5 and STAT3 signaling. GM-CSF increased STAT3 phosphorylation 10 min post-activation and infection, proving that the cytokine directly activated STAT3 (Figure 4L). There was >2.5 and >5 fold increase in Mt1 and Mt2 expression, as early as 10 min post infection and remained high for 24 h; Mt3 was modestly altered (Figure 5A). These data indicate that Zn regulation by GM-CSF occurs via STAT5 and STAT3 signaling.
Figure 5. Early regulation of Mt genes and role of Slc39a2 in MT-Zn sequestration.
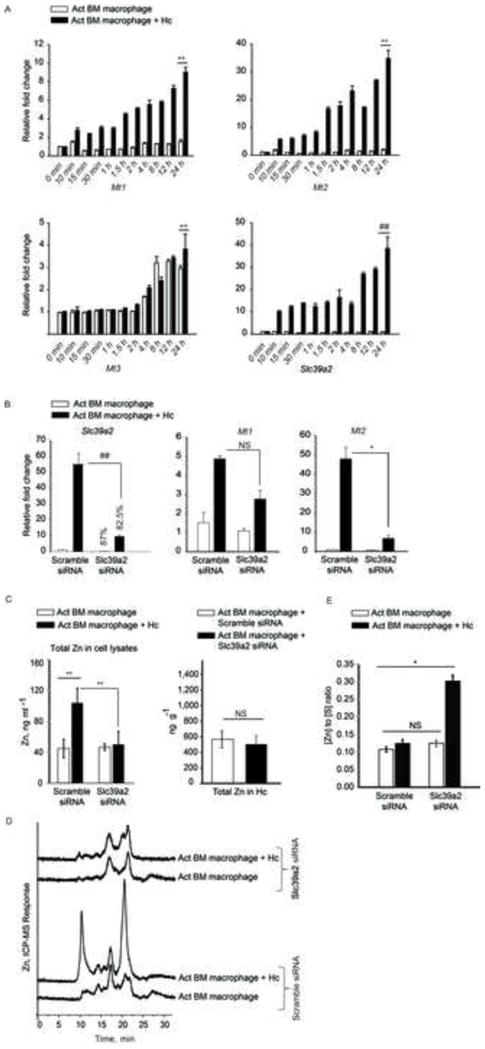
(A) qRT-PCR of Mt1, Mt2, Mt3 and Slc39a2, time points are min and h post infection, data are mean ± SEM, n=3 at 0–12 h and n=10 independent experiments at 24 h; (B) qRT-PCR of Slc39a2, Mt1 and Mt2 in Slc39a2 silenced macrophages normalized to scramble siRNA control, % decrease in expression compared to respective scramble controls, data are mean ± SEM, 3 independent experiments; (C) Total metal analysis of Zn in lysates (left) and H. capsulatum (right) from scramble siRNA and Slc39a2 silenced macrophages, data are mean ± SD, 3 independent experiments; (D) SEC-ICP-MS chromatogram of scramble siRNA and Slc39a2 silenced activated macrophages, Y axis, Offset Zn- signal; right panel, zoom in of labile Zn fraction, 3 independent experiments; (E) Ratio of Zn to sulfur in the MT fraction collected from 19 to 21 min based on the areas under the chromatogram corrected by differences in sensitivity through calibration with S and Zn standards, data are mean ± SD, 3 independent experiments. Related to Figure S4.
We investigated the importance of Slc39a2 in regulating GM-CSF function. Activated infected macrophages manifested a ~10 fold increase in Slc39a2 expression 10 min post infection (Figure 5A). We questioned whether the regulation of Slc39a2 was a prerequisite in macrophages defense. Upregulation of Mt genes was partially dependent on Slc39a2, but silencing Slc39a2 did not alter Zn deprivation in H. capsulatum, ROS production or growth inhibition (Figures 5B, 5C, S4A and S4B). Slc39a2 silencing reduced Zn influx and MT-Zn signal in macrophages, but did not affect labile Zn sequestration (Figures 5C and 5D). To probe how MTs in these macrophages established a defensive state against the pathogen, we compared the amount of Zn bound per MT by determining the [Zn] to [S] ratio (Zn2+ to MTs) between control and Slc39a2 silenced macrophages. As expected, scramble siRNA treated macrophages had increased MT and sequestered labile Zn (Figure 5D). But, the amount of Zn bound to MTs in Slc39a2 silenced infected macrophages was ~2.4 fold greater than scramble siRNA treated control, thereby enabling efficient Zn sequestration (Figure 5E), even with lower production of MTs. Silencing Slc39a14 did not affect GM-CSF driven macrophage defense (Figure S4C–F).
Zn sequestration enhances oxidative burst
GM-CSF enhances ROS production (Weisbart et al., 1987). Therefore, we asked if Zn sequestration affected oxidative burst during infection. GM-CSF increased ROS in macrophages. Zn chelation by TPEN in serum-free medium augmented ROS production by ~40%, which was significantly higher (p<0.05) compared to activated infected macrophages (Figure 6A). This effect was reversed upon addition of ZnSO4 (p<0.001). To ensure that this was not due to chelation of other metals by TPEN, we specifically depleted Zn from serum-free and serum-containing media. Low-Zn serum containing media did not show a significant increase in ROS over activated infected macrophages (Figure 6A), possibly due to the presence of residual Zn contributed by serum or serum-LPS that may have an inhibitory effect on NADPH oxidase (Nox) activity. However, macrophages exposed to Zn-depleted serum-free media manifested ~36% increase in ROS, significantly higher (p<0.01) than activated infected macrophages (Figure 6A). These data emphasize that a labile Zn-deprived state induced by GM-CSF facilitated increased oxidative burst, which was entirely reversible upon addition of Zn.
Figure 6. Zn sequestration enhances ROS production.
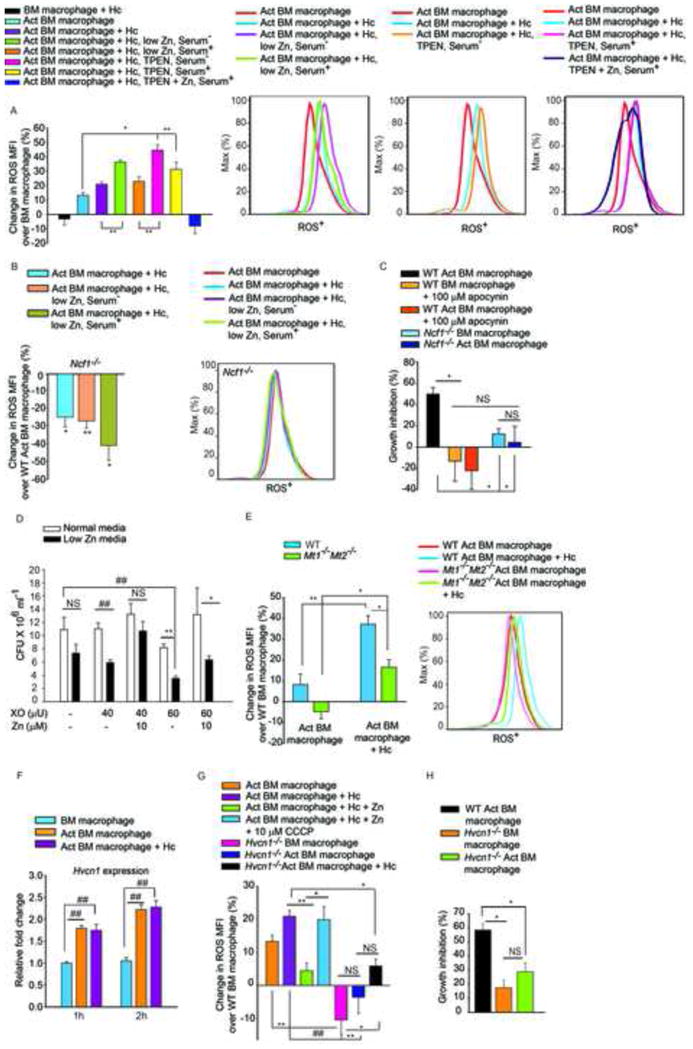
(A) % Change in ROS MFI in cells cultured in Zn replete, low Zn serum−, low Zn serum+, 10 μM TPEN serum−, 20 μM TPEN serum+, and 20 μM TPEN with 100 μM ZnSO4 treated serum+ media, serum− media was included as serum containing media has considerably higher Zn. Where serum+ media was used, a higher concentration of TPEN was added (20μM), last group is significantly different (p<0.01) from all groups; ROS histograms demonstrating the effect of Zn sequestration; Act, activated, Hc, H. capsulatum, 2–3 independent experiments; (B) % change in ROS MFI in Ncf1−/− activated macrophages, compared to WT activated uninfected cells, 3 independent experiments, histogram from Ncf1−/− macrophages; (C) % inhibition of yeast growth in apocynin treated or Ncf1−/− activated over WT macrophages, 3 independent experiments; (D) CFU of H. capsulatum in normal or low Zn media with increasing concentration of XO, with or without addition of Zn, 3 independent experiments; (E) % change in ROS MFI in WT and Mt1−/−Mt2−/− macrophages compared to WT uninfected cells, ROS histogram of WT and Mt1−/−Mt2−/− macrophages, 3 independent experiments; (F) Hvcn1 expression normalized to untreated macrophages, 3 independent experiments; (G) % change in ROS MFI in WT activated macrophages in response to infection, Zn and CCCP, 2 independent experiments, and Hvcn1−/− activated cells compared to WT, 3 independent experiments; (H) % inhibition of H. capsulatum growth in infected and activated WT vs Hvcn1−/− macrophages over WT control, 3 independent experiments; all data are mean ± SEM. Related to Figure S5.
The sources of cellular ROS include mitochondrial respiration (Drose and Brandt, 2012) and ROS produced by Nox (Minakami and Sumimotoa, 2006). Nox contributes to phagosomal ROS production that targets pathogens contained within these vacuoles. Activated macrophages from Ncf1−/− mice failed to generate an increased ROS response upon H. capsulatum challenge or Zn depletion (Figure 6B), indicating that reduction in labile Zn specifically enhanced ROS generation via Nox. We postulated that ROS production would be compromised in Mt1−/−Mt2−/− macrophages as a consequence of reduced Zn sequestration. While WT activated infected macrophages effectively induced an oxidative burst, this was dampened in Mt1−/−Mt2−/− macrophages (Figure 6E).
ROS was essential for H. capsulatum growth inhibition; macrophages in which Nox was inhibited by apocynin and Ncf1−/− macrophages had decreased ability to inhibit H. capsulatum growth (Figure 6C). To discern whether the defensive action of GM-CSF was a direct result of Zn limitation or enhanced ROS caused by Zn deprivation, we subjected H. capsulatum to ROS in vitro under Zn limitation. Though xanthine oxidase (XO) at a higher concentration reduced growth in normal media, Zn limitation greatly enhanced the inhibitory effect of ROS on H. capsulatum growth and this was reversible upon addition of Zn (Figure 6D). To determine the mechanism by which Zn sequestration facilitated increased Nox activity, we examined the regulation of hydrogen voltage gated proton channel (Hvcn1). Macrophages enhanced Hvcn1 expression in response to GM-CSF in a STAT3 and STAT5 dependent manner (Figures 6F and S5A). Proton flux by Hvcn1 accelerates ROS generation via Nox and Zn inhibits Hvcn1 function (DeCoursey et al., 2003). Addition of the protonophore, carbonyl cyanide m-chlorophenyl hydrazone (CCCP) reversed the inhibitory effect of Zn on ROS generation (Figure 6G). ROS production was attenuated in Hvcn1 silenced and Hvcn1−/− macrophages upon GM-CSF activation and infection. Hvcn1−/− activated macrophages showed compromised ability to control H. capsulatum growth compared to WT control (Figures 6G, 6H, S5B, S5C). These data signify the impact of Zn sequestration in enhancing phagocyte defense via ROS production.
GM-CSF drives Zn binding to MT in vivo
GM-CSF has a profound influence on the fate of infection in vivo (Deepe et al., 1999). We hypothesized that GM-CSF signaling was required for Zn sequestration in macrophages in vivo. In a pulmonary model of H. capsulatum infection, F4/80+ CD11b+ GFP+ and F4/80+ CD11b+ GFP− macrophages were sorted from lung leukocytes of green fluorescent protein (GFP) H. capsulatum infected mice (Schematic, Figure 7A). F4/80+ CD11b+ GFP+ macrophages showed enhanced Zn binding to MTs compared to F4/80+ CD11b+ GFP− cells (Figure 7B). To evaluate the importance of GM-CSF in Zn binding to MTs in vivo, it was neutralized and Zn profiles of macrophages were evaluated 7 days post infection (p.i). There was a sharp reduction in Zn bound to MTs in F4/80+ CD11b+ GFP+ macrophages in GM-CSF neutralized mice making it comparable to F4/80+ CD11b+ GFP− macrophages (Figure 7D). No alteration was seen in Rat IgG controls (Figure 7C). We asked whether GM-CSF neutralization enhanced fungal burden in macrophages. The percent and MFI of GFP+ cells in F4/80+ CD11b+ macrophages were higher in anti-GMCSF treated mice (Figure 7F). To further examine the importance of GM-CSF signaling in Zn regulation, WT macrophages were adoptively transferred into Csf2ra−/− mice lacking the GM-CSF receptor, infected with GFP H. capsulatum and WT vs. Csf2ra −/− macrophages were sorted 7 days post infection. Mt1, Mt2 and Slc39a2 expression was enhanced in infected WT macrophages, while Csf2ra−/− macrophages showed over 60, 93 and 97% decrease in Mt1, Mt2 and Slc39a2 expression respectively compared to infected WT macrophages (Figures 7G and 7H). Moreover, the percent infectivity of F4/80+ CD11b+ GFP+ cells in Csf2ra−/− mice was higher (16.7%) as compared to infected WT CD45.1 macrophages (1.26%). These data highlight the importance of GM-CSF in regulating Zn during infection in vivo.
Figure 7. GM-CSF triggers Zn binding to MTs in vivo and in human macrophages.
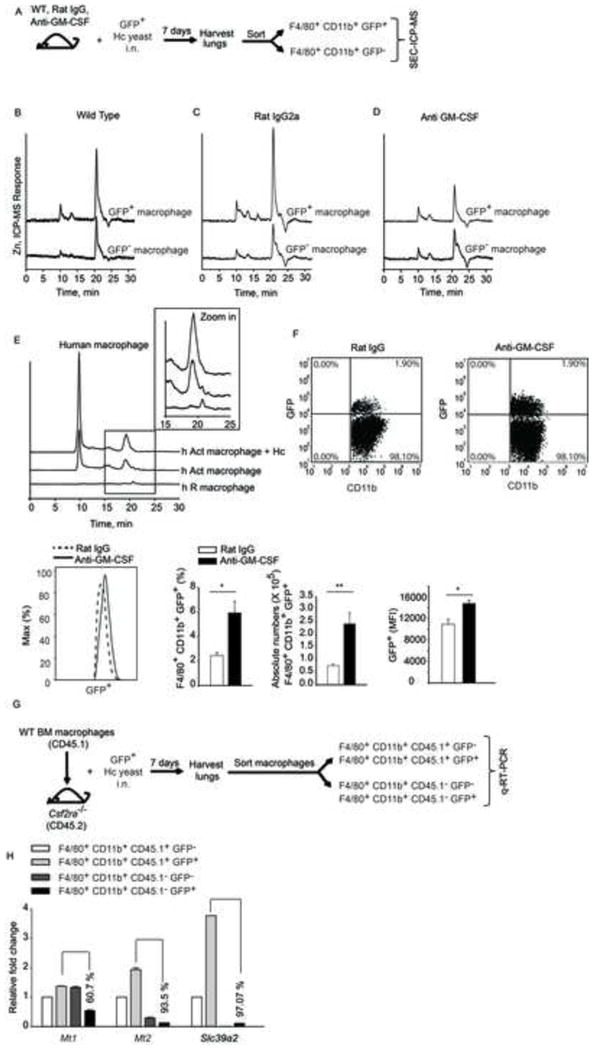
(A) Schematic, mice were left untreated or treated with rat IgG or anti-GM-CSF and infected i.n. with 2 × 106 GFP+ yeasts (Hc); lungs were harvested 7 days p.i. and F4/80+CD11b+GFP+ and F4/80+CD11b+GFP− cells were sorted. SEC-ICP-MS profiles of (B) F4/80+CD11b+GFP+ and F4/80+ CD11b+GFP− cells isolated from lungs; (C) rat IgG treated and (D) anti-GM-CSF treated mice, 2 independent experiments; (E) SEC-ICP-MS profiles of lysates from human resting (h R) and GM-CSF activated (h Act) macrophages; Y axis, offset Zn signal; inset, zoom in of 15–25 min fraction, 2 independent experiments; (F) Dot plot of lung leukocytes gated on F4/80+CD11b+ macrophages; histogram of F4/80+CD11b+GFP+ macrophages; bar graphs are quantification of %, absolute numbers and MFI of F4/80+CD11b+GFP+ macrophages in rat IgG vs anti-GM-CSF treated mice, data are mean ± SEM, n=4; (G) Schematic representing transfer of CD45.1 WT macrophages into CD45.2 Csf2ra−/− mice, infection and sorting of WT vs Csf2ra−/− macrophages from lungs for gene expression analysis; (H) qRT-PCR of Mt1, Mt2 and Slc39a2 from sorted CD45.1+ (WT) and CD45.1− (Csf2ra−/−) GFP− and GFP+ macrophages normalized to CD45.1+GFP− macrophages, % values are decrease in gene expression in Csf2ra−/− CD45.1− GFP+ macrophages compared to WT CD45.1+ GFP+ macrophages, data are mean ± SEM, data are from 1 experiment. Related to Figure S6.
GM-CSF mediates Zn sequestration in macrophages from Slc11a1+/+ mice and from humans
C57BL/6 mice used in this study lack a functional Slc11a1 protein (White et al., 2004). This prompted us to determine if Zn regulation was altered in the presence of this Fe transporter. GM-CSF induced a similar Zn profile in macrophages from CBA/J mice that express functional Scl11a1 (Figure S6A–S6C). We asked if this phenomenon occurred in human macrophages. Zn distribution was analyzed in GM-CSF activated infected human macrophages compared with resting control. There was a similarity between Zn profiles in human and murine macrophages. Activation of human macrophages with GM-CSF elevated MT-Zn binding during infection (Figures 7E and S6G and S6H). These data indicate that GM-CSF broadly induces modulation of Zn in macrophages and that this is a conserved mechanism between mice and humans. We also determined if GM-CSF promoted Zn sequestration to a distinct clade of H. capsulatum, G186R. Activated macrophages strongly induced Zn modulation via MTs and Zn transporters upon infection with G186R H. capsulatum (Figure S6D-S6F). Thus, Zn sequestration by GM-CSF arrests pathogen survival by dually targeting fungal Zn acquisition and augmenting oxidative damage to the fungus.
DISCUSSION
This study revealed an underlying mechanism for the action of GM-CSF in combating infection with an intracellular pathogen. To date, the mechanism of GM-CSF function has not been fully elucidated. We have demonstrated that GM-CSF signaling empowers macrophage defense by facilitating a dual outcome of limitation of bioavailable Zn and simultaneous enhancement of superoxide burst which renders the pathogen susceptible to the host.
During infection, GM-CSF reduced total Zn in yeasts, but enhanced Zn influx in macrophages. Although this finding appears counterintuitive, GM-CSF induced an increase in the Zn binding proteome suggesting an enhancement of Zn dependent cellular functions such as transcription, enzyme function and metabolism (Beyersmann, 2002). To cope with pathogen invasion, activated macrophages sequestered the exchangeable Zn fraction away from intracellular fungus, while concurrently increasing ROS production.
We have reported that GM-CSF decreased total Zn in macrophages (Winters et al., 2010). The discrepancy between the current and past study is a result of an improvement in the analytical method, specifically in the blank correction of lysate analysis. We went from a flow injection analysis in a metal containing system, to a continuous flow analysis in a metal free system. The SEC-ICP-MS chromatograms manifest the same results in both studies with more total Zn in GM-CSF activated and infected peritoneal and bone marrow macrophages than the resting states by calculation of area under the curve. Also, total Zn in H. capsulatum was consistent, since the metal free system was used in both studies. Thus, the observation that GM-CSF activation induces a state of Zn sequestration and reduces total Zn in H. capsulatum has been reproducibly demonstrated in this study.
Confocal microscopy revealed that GM-CSF, but not TNF-α treated macrophages route labile Zn into the Golgi, in association with increased expression of Slc30a4 and Slc30a7 transporters that channel Zn into this organelle (Palmiter and Huang, 2004). Zn deprivation in H. capsulatum was unique to GM-CSF activated macrophages demonstrating the specificity of this cytokine in Zn mobilization. The Golgi acts as a site of labile Zn storage (McCormick et al., 2010), and be ‘Zn-storage depot’ in restricting Zn access to intracellular pathogens, while reserving it for phagocyte function. Fluorescent Zn probes are an invaluable tool in imaging Zn, but their application is affected by signal variation due to pH, ionic strength and propensity to localize in acidic compartments (Tomat and Lippard, 2010). While total cellular Zn is in micromolar range, the labile Zn pool identified using Zinpyr-1 is in the picomolar range (Eide, 2006). Hence, the observed Zn signal may represent a fraction of total Zn. Nevertheless, labile Zn is a crucial component of the total Zn pool. Immune cells deliver Zn signals upon pathogen contact and changes in labile Zn pool may reflect signal transducing properties of this metal ion (Hirano et al., 2008).
Our observations with Mt silenced and Mt1−/−Mt2−/− macrophages reinforce the critical function of MTs in antimicrobial defenses. In activated macrophages, MTs powered the loss of pathogen-associated Zn. We demonstrate that MTs are essential mediators of GM-CSF function and their absence presents a survival advantage to the pathogen. Of note, the WT control used in Mt1−/−Mt2−/− studies (SvJ strain) depicted a Zn regulatory response similar to C57BL/6 macrophages. Thus, interference with MT regulation impairs the antimicrobial effect induced by GM-CSF.
We showed that both STAT5 and STAT3 orchestrate Zn modulation guided by GM-CSF. Disruption of STAT5 and STAT3 signaling abrogated MT-Zn sequestration. STAT5 and STAT3 heterodimerize in the presence of granulocyte-colony stimulating factor (Novak et al., 1996); in this context, GM-CSF may also induce heterodimerization to regulate MT expression. We sought to explain Zn alteration in infected macrophages by examining transporters. ZIP2 imports Zn into the cytosol from the extracellular milieu (Eide, 2006). Slc39a2 silencing reduced MT expression, but did not affect the ability of MTs to sequester labile Zn. Upregulation of Slc39a2 may indicate its involvement in cellular processes that do not directly affect macrophage defense functions within the analyzed 24h period of infection. It is also plausible that the pathogen triggers Zn influx via ZIP2, an effect, counteracted by Zn sequestration in a GM-CSF primed macrophage.
Our findings reveal that Zn sequestration exerts a broader impact on macrophage antifungal defense. Conflicting evidence exists regarding the ability of Zn to trigger or dampen ROS production (Bishop et al., 2007; DeCoursey et al., 2003). In activated macrophages, a reduction in the labile Zn pool was clearly associated with an increase in ROS that was dependent on Nox and reversed by exposure to Zn. GM-CSF achieved this feat by a two-fold regulatory mechanism: first, the cytokine upregulated expression of Hvcn1 that elevates Nox activity; second, it induced a Zn limiting environment, that promotes proton pumping by Hvcn1 and Nox function. Ncf1−/− and Hvcn1−/− activated macrophages exhibited reduced growth inhibitory effect on H. capsulatum. Apocynin inhibits Nox, but may also interfere with NO production, and that may explain the increased reversal of inhibition (Muijsers et al., 2000). Charge compensation by K+ channels may account for the partial growth inhibitory capacity seen in Hvcn1−/− activated macrophages (Nauseef, 2007). An increased ROS triggers a variety of second messenger signals, such as activation of STAT3 (Simon et al., 1998) and nuclear factor kappa B (Gloire et al., 2006) that possibly converge into potent and sustained macrophage defense. From the pathogen perspective, Zn paucity may compromise microbial protein functions such as superoxide dismutase activity that mediates resistance to the host (Kehl-Fie et al., 2011). Metal binding sites on proteins may exhibit degeneracy (Heizmann and Cox, 1998). Zn limitation to the pathogen can therefore, potentially enforce substitution of Zn dependent metal binding sites on microbial proteins with more reactive metals, such as Cu and Fe. Indeed, the ROS generated in vitro by XO exerted a profound effect on H. capsulatum survival under Zn limitation.
Emphasizing the impact of Zn sequestration on ROS generation, we demonstrated that MTs are essential for an optimal ROS response induced by GM-CSF. In the absence of MT1 and MT2, GM-CSF failed to effectively elevate ROS. These studies were performed at an early stage, because GM-CSF triggers MTs immediately following infection. This observation is surprising, since MTs bind metals but also scavenge ROS (Maret, 2000). However, GM-CSF activated macrophages remarkably elevated Zn sequestration by MTs. The oligomeric shift in MTs may result from oxidation of Cys residues only during later stages of infection.
We have reported that lack of GM-CSF leads to detrimental fungal burden (Deepe et al., 1999). In this study, we found that blockade of GM-CSF in vivo was sufficient to diminish Zn sequestration and elevate macrophage fungal burden, signifying that among several cytokines, GM-CSF uniquely modulates MT-Zn homeostasis. This phenomenon may play a central role in inhibition of fungal growth in vivo. These mechanisms are conserved among clades of H. capsulatum, strains of mice and humans.
In view of diversity in defense mechanisms against distinct pathogens, human macrophages employ a unique Zn-intoxication strategy to limit growth of M. tuberculosis. Labile Zn deprivation by GM-CSF reveals a distinct mechanism by which the host may combat intracellular pathogens. With these contrasting mechanisms of Zn intoxication versus deprivation, it can be postulated that cytokine-activated macrophages evolved to use specialized and distinct defense arsenals against different pathogens. On the other hand, microbes have evolved means to cope with nutrient paucity. Salmonella typhimurium resists calprotectin mediated Zn deprivation by upregulating a high affinity Zn transporter (Liu et al., 2012). In contrast, H. capsulatum fails to cope with Zn sequestration and enhanced free radical challenge. Little is known about Zn transporters in H. capsulatum, but one may derive that, while the organism strives to acquire Zn by increasing import, the amount is insufficient to drive intracellular growth. Of note, H. capsulatum does not have to confront an elevated calprotectin in macrophages (Winters et al., 2010). On the other hand, it has to compete for Zn acquisition with MTs, which may be more potent than calprotectin in that each molecule of the former scavenges 7 ions with picomolar affinity (Coyle et al., 2002), while the latter binds 2 Zn ions per molecule with nanomolar affinity. (Kehl-Fie et al., 2011; Korndorfer et al., 2007).
In summary, using a combination of gene expression analysis, microscopy, ICP-MS, SEC coupled to ICP-MS and proteomics, we have dissected a mechanism by which GM-CSF activated macrophages pose a dual defense system simultaneously challenging microbial tolerance to restricted Zn availability coupled to increased oxidative damage. Lastly, we propose that labile Zn deprivation may be a global defense strategy that can be extended to growth restriction of other intracellular fungi. Whether macrophages preferentially employ labile Zn deprivation over Zn intoxication for defense against distinct microbial classes remains to be determined.
EXPERIMENTAL PROCEDURES
See also supplementary procedures.
Mice
C57BL/6, CBA/J, Mt1tm1BriMt2tm1Bri (Mt1−/−Mt2−/−), 129S1/SvImJ WT (control for Mt1−/−Mt2−/−) and B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) mice were obtained from Jackson Laboratory. We thank Dr. Bruce Trapnell, Cincinnati Children’s Hospital Medical Center for the Csf2ra−/− mice; Dr. John Engelhardt, Dr. Yulong Zhang (University of Iowa), Dr. Brahm Segal, Dr. Nazmul Khan (Roswell Park Cancer Institute) and Dr. Long-Jun Wu and Dr. Jiyun Peng (Rutgers University) for providing bone marrow from Ncf1−/− and Hvcn1−/− mice. Animal experiments were in accordance with Animal Welfare Act guidelines of National Institutes of Health.
Activation, STAT inhibition and infection
Bone marrow macrophages were activated with cytokines for 24 h before infection. Peritoneal macrophages were rested for 24 h, followed by activation for 24 h before infection and were activated again during infection. Where indicated, macrophages were cultured throughout with 100μM STAT3 or STAT5 inhibitors, S3I-201 and N′-((4-Oxo-4H-chromen-3-yl)methylene) nicotinohydrazide (Calbiochem) respectively. For infection, G217B or G186R H. capsulatum were cultured in Ham’s F12 (Zn concentration, 250 ± 18 ng/ml or 4 μM) and washed before use.
Microscopy
Macrophages were infected with PKH-26 stained H. capsulatum for 24 h followed by staining with nuclear, Golgi and Zn dyes where indicated for 30 min. Images were acquired on a Zeiss LSM710 confocal and analyzed using ZEN 2011 software.
Silencing
Genes were silenced using TransIT-TKO transfection reagent (Mirus Bio LLC) and 100nM Mt (Santa Cruz), Slc39a2, Slc39a14, Stat3 or Stat5 (50nM each of Stat5a and Stat5b), Hvcn1 or scrambled siRNA (Dharmacon) as per manufacturer’s instructions.
ROS
Macrophages were activated for 24h and then incubated with 5μM Dihydroethidium (DHE) (Invitrogen) in fresh media for 15 min in dark, followed by GM-CSF and infected for 2h. Where indicated, 100μM ZnSO4 and TPEN or Zn depleted media was added 20 min prior to DHE. CCCP was added 30 min before analysis by flow cytometry (Accuri C6). Data were analyzed with FCS Express 3 De Novo Software (CA) or FlowJo.
In vivo
Eight week old C57BL/6 mice were infected with 2 × 106 GFP+ H. capsulatum yeasts i.n. Mice were injected ip. with 0.5mg control mAb Rat IgG2a (BioXcell) or MP1–22E9 mAb Rat IgG2a anti-GM-CSF a day before and on the day of infection. After 7 days, lungs were homogenized with gentleMACS Dissociator (Miltenyi Biotec, CA), collagenase treated, filtered through 60 μM nylon mesh and washed. Leukocytes were isolated using Lympholyte M (Cedarlane Laboratories, Canada) and stained with APC F4/80 (AbD Serotec) and PerCP CD11b mAbs (BD Biosciences) and CD16/CD32 blocking mAbs for 30 min at 4°C and washed; gated on F4/80+CD11b+ and sorted into GFP+ (containing phagocytosed H. capsulatum) and GFP− (not containing H. capsulatum) using 5-laser FACS Aria II (BD Biosciences) in a BSLII facility at Cincinnati Children’s Hospital; collected at 4°C in culture media and washed prior to lysis. For adoptive transfer, 2×106 bone marrow macrophages from WT (CD45.1) were transferred intratracheally into 8 week old Csfr2a−/− (CD45.2) mice 1 day prior to H. capsulatum infection. On day 7 post infection, lung leukocytes were sorted into 4 populations: F4/80+CD11b+CD45.1+GFP− or GFP+ and F4/80+CD11b+CD45.1−GFP− or GFP+ macrophages for analysis of gene expression.
Human macrophages
PBMCs were isolated from human blood (Hoxworth Blood Center, University of Cincinnati) using Ficoll-Paque. Monocytes were purified with Monocyte Isolation Kit II on a VarioMACS separator (Miltenyi Biotec) and cultured in RPMI with 12.5% human male AB serum (Sigma-Aldrich) for 7 days, and macrophages were exposed to 10 ng/ml GM-CSF (Peprotech) or vehicle for 24 h before infection, re-exposed to GM-CSF at the time of infection and lysates were prepared 24 h later.
Cell lysates for ICP-MS
Macrophages were washed and lysed with 0.1% sodium dodecyl sulfate (SDS) in HPLC water for 20 min on ice. Lysates were passed through a 0.22 μm filter or centrifuged at 13,000rpm for 5 min for analysis. For total metal analysis of H. capsulatum, lysates were centrifuged at 13,000rpm for 5 min to pellet the yeasts and the pellet was washed twice with HBSS.
Total elemental analysis and SEC-ICP-MS screening
ICP-MS analysis was performed on Agilent 7700x ICP-MS instrument (Agilent Technologies). A conventional Meinhard nebulizer, Peltier-cooled spray chamber, and shield torch constituted the sample introduction system under standard conditions. Sc was used as internal standard and SRM (DORM-3 or NIST1745) were analyzed for every digestion.
For SEC-ICP-MS, Agilent 1100 series HPLC system equipped with a binary pump, vacuum membrane degasser, thermostated auto sampler, column oven, and diode array detector (DAD), with a semi-micro flow UV-Vis cell was coupled to the ICP-MS through a 0.17mm internal diameter short PEEK tube. The system was controlled with Chemstation software. A TSK Gel 3000SW 7.5 × 300 mm column was used. For proteomics, samples were concentrated before injection using MWCO filter 3 kDa (Millipore). For analysis of MT saturation, the MTs-SEC signal (20 min) was collected and analyzed for S and Zn to determine the MT:Zn ratio in a Superdex peptide SEC-ICP-MS using O2 reaction mode.
Proteomics
The SEC-ICP-MS fractions were analyzed using an Agilent 1200 nanoHPLC, nanoChip ESI and 6300 series MSD Ion Trap XCT Ultra system. Peptide and corresponding protein identification were conducted using MASCOT server (Matrix Science).
Statistics
p-values were calculated using one way ANOVA for multiple comparisons and adjusted with Bonferroni’s or Holm Sidak correction; and non-paired Student t test where two groups were compared; *p<0.05; **p<0.01; ##p<0.001; NS, not significant.
Supplementary Material
Acknowledgments
We thank Agilent Technologies for instrumentation, ICP-MS and nanoHPLC-ESI-IT-MS and Drs. B. Klein, W. Nauseef, and D. Hildeman for their valuable feedback. This work was supported by grants from the NIH, AI-094971, AI-106269 and a Merit Review from Veterans Affairs.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
K.S.V and J.L.F. contributed equally, designed and performed experiments, analyzed data and wrote the manuscript, K.S.V performed in vitro, microscopy, in vivo and human experiments and interpreted data, J.L.F. performed chromatographic separations with UV-Vis, ICP-MS and ESI-IT-MS-MS analysis and interpreted data, A.P. performed bioinformatics analysis, J.C. and G.S.D. designed and supervised the work. All authors participated in final manuscript preparation.
References
- Appelberg R. Macrophage nutriprive antimicrobial mechanisms. J Leukoc Biol. 2006;79:1117–1128. doi: 10.1189/jlb.0206079. [DOI] [PubMed] [Google Scholar]
- Beyersmann D. Homeostasis and cellular functions of zinc. Materialwissenschaft und Werkstofftechnik. 2002;33:764–769. [Google Scholar]
- Bhattacharya S, Stout BA, Bates ME, Bertics PJ, Malter JS. Granulocyte macrophage colony-stimulating factor and interleukin-5 activate STAT5 and induce CIS1 mRNA in human peripheral blood eosinophils. American Journal of Respiratory Cell and Molecular Biology. 2001;24:312–316. doi: 10.1165/ajrcmb.24.3.4238. [DOI] [PubMed] [Google Scholar]
- Bishop GM, Dringen R, Robinson SR. Zinc stimulates the production of toxic reactive oxygen species (ROS) and inhibits glutathione reductase in astrocytes. Free Radic Biol Med. 2007;42:1222–1230. doi: 10.1016/j.freeradbiomed.2007.01.022. [DOI] [PubMed] [Google Scholar]
- Botella H, Peyron P, Levillain F, Poincloux R, Poquet Y, Brandli I, Wang C, Tailleux L, Tilleul S, Charriere GM, et al. Mycobacterial p(1)-type ATPases mediate resistance to zinc poisoning in human macrophages. Cell Host Microbe. 2011;10:248–259. doi: 10.1016/j.chom.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Bobo JA, Liuzzi JP, Cousins RJ. Effects of intracellular zinc depletion on metallothionein and ZIP2 transporter expression and apoptosis. J Leukoc Biol. 2001;70:559–566. [PubMed] [Google Scholar]
- Coyle P, Philcox JC, Carey LC, Rofe AM. Metallothionein: the multipurpose protein. Cell Mol Life Sci. 2002;59:627–647. doi: 10.1007/s00018-002-8454-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE, Morgan D, Cherny VV. The voltage dependence of NADPH oxidase reveals why phagocytes need proton channels. Nature. 2003;422:531–534. doi: 10.1038/nature01523. [DOI] [PubMed] [Google Scholar]
- Deepe GS, Jr, Gibbons R, Woodward E. Neutralization of endogenous granulocyte-macrophage colony-stimulating factor subverts the protective immune response to Histoplasma capsulatum. J Immunol. 1999;163:4985–4993. [PubMed] [Google Scholar]
- Drose S, Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol. 2012;748:145–169. doi: 10.1007/978-1-4614-3573-0_6. [DOI] [PubMed] [Google Scholar]
- Eide DJ. Zinc transporters and the cellular trafficking of zinc. Biochim Biophys Acta. 2006;1763:711–722. doi: 10.1016/j.bbamcr.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Falnoga I, Zelenik Pevec A, Slejkovec Z, Znidaric MT, Zajc I, Mlakar SJ, Marc J. Arsenic Trioxide (ATO) Influences the gene expression of metallothioneins in human glioblastoma cells. Biol Trace Elem Res. 2012 doi: 10.1007/s12011-012-9431-8. [DOI] [PubMed] [Google Scholar]
- Fang FC. Perspectives series: host/pathogen interactions. mechanisms of nitric oxide-related antimicrobial activity. J Clin Invest. 1997;99:2818–2825. doi: 10.1172/JCI119473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraker PJ, Gershwin ME, Good RA, Prasad A. Interrelationships between zinc and immune function. Fed Proc. 1986;45:1474–1479. [PubMed] [Google Scholar]
- Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72:1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Heizmann CW, Cox JA. New perspectives on S100 proteins: a multi-functional Ca 2+ -, Zn 2+ - and Cu 2+-binding protein family. BioMetals. 1998;11:383–397. doi: 10.1023/a:1009212521172. [DOI] [PubMed] [Google Scholar]
- Hirano T, Murakami M, Fukada T, Nishida K, Yamasaki S, Suzuki T. Roles of zinc and zinc signaling in immunity: zinc as an intracellular signaling molecule. Adv Immunol. 2008;97:149–176. doi: 10.1016/S0065-2776(08)00003-5. [DOI] [PubMed] [Google Scholar]
- Howard DH. Intracellular behavior of Histoplasma capsulatum. J Bacteriol. 1964;87:33–38. doi: 10.1128/jb.87.1.33-38.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen TM, Higashi Y, Suh SW, Escartin C, Nagasawa K, Swanson RA. Zinc triggers microglial activation. J Neurosci. 2008;28:5827–5835. doi: 10.1523/JNEUROSCI.1236-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehl-Fie Thomas E, Chitayat S, Hood MI, Damo S, Restrepo N, Garcia C, Munro Kim A, Chazin Walter J, Skaar Eric P. Nutrient Metal Sequestration by Calprotectin Inhibits Bacterial Superoxide Defense, Enhancing Neutrophil Killing of Staphylococcus aureus. Cell Host & Microbe. 2011;10:158–164. doi: 10.1016/j.chom.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura H, Morikawa H, Kamon H, Iguchi M, Hojyo S, Fukada T, Yamashita S, Kaisho T, Akira S, Murakami M, Hirano T. Toll-like receptor-mediated regulation of zinc homeostasis influences dendritic cell function. Nat Immunol. 2006;7:971–977. doi: 10.1038/ni1373. [DOI] [PubMed] [Google Scholar]
- Korndorfer IP, Brueckner F, Skerra A. The crystal structure of the human (S100A8/S100A9)2 heterotetramer, calprotectin, illustrates how conformational changes of interacting alpha-helices can determine specific association of two EF-hand proteins. J Mol Biol. 2007;370:887–898. doi: 10.1016/j.jmb.2007.04.065. [DOI] [PubMed] [Google Scholar]
- Lee DK, Carrasco J, Hidalgo J, Andrews GK. Identification of a signal transducer and activator of transcription (STAT) binding site in the mouse metallothionein-I promoter involved in interleukin-6-induced gene expression. Biochem J. 1999;337(Pt 1):59–65. [PMC free article] [PubMed] [Google Scholar]
- Liu JZ, Jellbauer S, Poe AJ, Ton V, Pesciaroli M, Kehl-Fie TE, Restrepo NA, Hosking MP, Edwards RA, Battistoni A, et al. Zinc sequestration by the neutrophil protein calprotectin enhances Salmonella growth in the inflamed gut. Cell Host Microbe. 2012;11:227–239. doi: 10.1016/j.chom.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maret W. The Function of Zinc Metallothionein: A Link between Cellular Zinc and Redox State. The Journal of Nutrition. 2000;130:1455S–1458S. doi: 10.1093/jn/130.5.1455S. [DOI] [PubMed] [Google Scholar]
- McCormick N, Velasquez V, Finney L, Vogt S, Kelleher SL. X-ray fluorescence microscopy reveals accumulation and secretion of discrete intracellular zinc pools in the lactating mouse mammary gland. PLoS One. 2010;5:e11078. doi: 10.1371/journal.pone.0011078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDevitt CA, Ogunniyi AD, Valkov E, Lawrence MC, Kobe B, McEwan AG, Paton JC. A molecular mechanism for bacterial susceptibility to zinc. PLoS Pathog. 2011;7:e1002357. doi: 10.1371/journal.ppat.1002357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakami R, Sumimotoa H. Phagocytosis-coupled activation of the superoxide-producing phagocyte oxidase, a member of the NADPH oxidase (nox) family. Int J Hematol. 2006;84:193–198. doi: 10.1532/IJH97.06133. [DOI] [PubMed] [Google Scholar]
- Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- Muijsers RB, van Den Worm E, Folkerts G, Beukelman CJ, Koster AS, Postma DS, Nijkamp FP. Apocynin inhibits peroxynitrite formation by murine macrophages. Br J Pharmacol. 2000;130:932–936. doi: 10.1038/sj.bjp.0703401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nairz M, Fritsche G, Brunner P, Talasz H, Hantke K, Weiss G. Interferon-gamma limits the availability of iron for intramacrophage Salmonella typhimurium. Eur J Immunol. 2008;38:1923–1936. doi: 10.1002/eji.200738056. [DOI] [PubMed] [Google Scholar]
- Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- Newman SL, Gootee L. Colony-stimulating factors activate human macrophages to inhibit intracellular growth of Histoplasma capsulatum yeasts. Infect Immun. 1992;60:4593–4597. doi: 10.1128/iai.60.11.4593-4597.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak U, Mui A, Miyajima A, Paradiso L. Formation of STAT5-containing DNA binding complexes in response to colony-stimulating factor-1 and platelet-derived growth factor. J Biol Chem. 1996;271:18350–18354. doi: 10.1074/jbc.271.31.18350. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Huang L. Efflux and compartmentalization of zinc by members of the SLC30 family of solute carriers. Pflugers Arch. 2004;447:744–751. doi: 10.1007/s00424-003-1070-7. [DOI] [PubMed] [Google Scholar]
- Santoni V, Molloy M, Rabilloud T. Membrane proteins and proteomics: un amour impossible? Electrophoresis. 2000;21:1054–1070. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1054::AID-ELPS1054>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol. 1998;275:C1640–1652. doi: 10.1152/ajpcell.1998.275.6.C1640. [DOI] [PubMed] [Google Scholar]
- Tomat E, Lippard SJ. Imaging mobile zinc in biology. Curr Opin Chem Biol. 2010;14:225–230. doi: 10.1016/j.cbpa.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkup GK, Burdette SC, Lippard SJ, Tsien RY. A new cell-permeable fluorescent probe for Zn2+ J Am Chem Soc. 2000:122. [Google Scholar]
- Weisbart RH, Kwan L, Golde DW, Gasson JC. Human GM-CSF primes neutrophils for enhanced oxidative metabolism in response to the major physiological chemoattractants. Blood. 1987;69:18–21. [PubMed] [Google Scholar]
- White JK, Stewart A, Popoff JF, Wilson S, Blackwell JM. Incomplete glycosylation and defective intracellular targeting of mutant solute carrier family 11 member 1 (Slc11a1) Biochem J. 2004;382:811–819. doi: 10.1042/BJ20040808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters MS, Chan Q, Caruso JA, Deepe GS., Jr Metallomic analysis of macrophages infected with Histoplasma capsulatum reveals a fundamental role for zinc in host defenses. J Infect Dis. 2010;202:1136–1145. doi: 10.1086/656191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwilling BS, Kuhn DE, Wikoff L, Brown D, Lafuse W. Role of iron in Nramp1-mediated inhibition of mycobacterial growth. Infect Immun. 1999;67:1386–1392. doi: 10.1128/iai.67.3.1386-1392.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.