

Abstract
Background:
Hemangioblastomas are associated with Von Hippel-Lindau disease (VHLD) in 10-40% of cases. Based upon a literature review we state the core features the neurosurgeon should be aware of.
Methods:
We performed a selective literature (Cochrane and Medline) search for hemangioblastoma, both sporadic and VHL associated. We reviewed general characteristics (epidemiology, symptomatology, diagnosis, and management) and focused on follow-up as well as screening modalities for sporadic and VHL associated lesions.
Results:
Based upon our literature search, we established guidelines for screening and follow-up in both sporadic and VHL associated hemangioblastoma patients.
Conclusions:
Screening for retinal angiomas, abdominal masses, and pheochromocytomas as well as genetic analysis is recommended for every patient with a newly diagnosed hemangioblastoma. Follow-up is by magnetic resonance imaging (MRI) of the clinical neuronal region at 6 and at 12-24 months postoperatively. For VHL-associated hemangioblastomas yearly investigation for craniospinal hemangioblastoma by MRI and yearly screening and follow-up for retinal angiomas is recommended. Annual abdominal ultrasound with triennial computed tomography (CT) imaging for abdominal masses is postulated. Annual audiometry is to be performed for possible endolymphatic sac tumor, detailed radiographic imaging of the skull base should be performed upon abnormality in auditory testing. Investigations for cystadenomas of the epidydimis and broad ligament only are mandatory on indication. Annual investigation for pheochromocytoma is recommended.
Keywords: Diagnosis, follow-up, hemangioblastoma, Von Hippel-Lindau disease, work-up
INTRODUCTION
Von Hippel-Lindau disease (VHLD) was first described at the beginning of the 20thcentury by the German ophthalmologist Eugene von Hippel and the Swedish pathologist Avrid Lindau.[6,10,12,14,15,20,25,27] VHLD has an autosomal dominant transmission and an incidence of 1 in 36000 to 1 in 53000 newborns.[6,10,12,14,15,20,25,27] It is caused by a mutation in the VHL gene on chromosome 3p25-6 resulting in the loss of the pVHL tumor suppressor protein function. The main function of the pVHL protein is the negative regulation of VEGF. This loss leads to a myriad of tumors: Central nervous system (CNS) hemangioblastomas, retinal angiomas, renal cysts and clear cell carcinomas, pheochromocytomas, pancreatic tumors, epidydimis cysts, cystadenomas of the broad ligament and endolymphatic sac tumors of the middle ear.[6,10,12,14,15,20,25,27]
Hemangioblastomas are highly vascular tumors of the CNS with a preponderance for the posterior fossa. Despite their frequent sporadic appearance, hemangioblastomas can be the first presentation of VHLD. Early diagnosis of VHLD is crucial for adequate screening and follow-up for the other manifestations of this condition and for genetic counseling.[6,12,13,14,15,27]
MATERIALS AND METHODS
We performed a selective literature (Cochrane and Medline) search for hemangioblastoma, both sporadic and VHL associated. We review general characteristics (epidemiology, symptomatology, diagnosis, and management) and focused on follow-up as well as screening modalities for sporadic and VHL associated lesions.
DISCUSSION
General comments
Hemangioblastomas are uncommon vascular tumors of the CNS [Figure 1]. They account for less than 3% of all CNS tumors and are generally benign, well-circumscribed but highly vascular, neoplasms. They mostly occur in infratentorial structures such as the cerebellum, the brainstem, and the spinal cord. Approximately 5% of all spinal cord tumors and 7.5% of all tumors arising in the adult posterior fossa are accounted to be a hemangioblastoma.[13,15,27]
Figure 1.
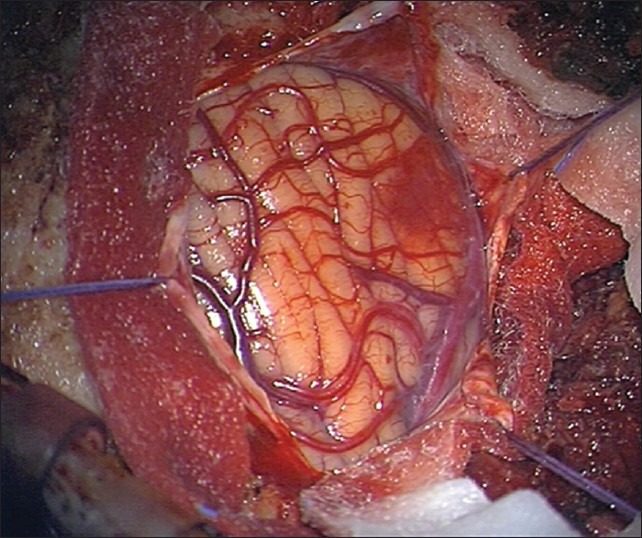
Perioperative image of a hemangioblastoma. Note the high degree of vascularity
Symptomatology occurs by local compression of neural structures and rarely because of bleeding or as a paraneoplastic complication.[6,13,15,27]
Diagnosis is suspected by gadolinium-enhanced magnetic resonance imaging (MRI). The characteristic MRI feature is a contrast enhancing nodule associated with a peritumoral cyst located in the cerebellum [Figure 2] or a homogeneously enhancing lesion on the surface of or within the spinal cord with an associated syrinx [Figure 3]. These lesions appear as a low signal on T1-weighted images and as a high signal on T2-weighted sequences.[7,13,27] These features, however, are not pathognomonic, so the definitive diagnosis is made on histopathological examination. Microscopic investigation shows an extensive vascular network with neoplastic stromal cells [Figure 4]. The neoplastic stromal cells have abundant cytoplasm with lipid vacuoles resulting in a typical clear cell morphology. Nuclear hyperchromatism and atypia, in contrast to absence of mitotic activity, are a very typical pattern of hemangioblastomas.[3,5,27]
Figure 2.
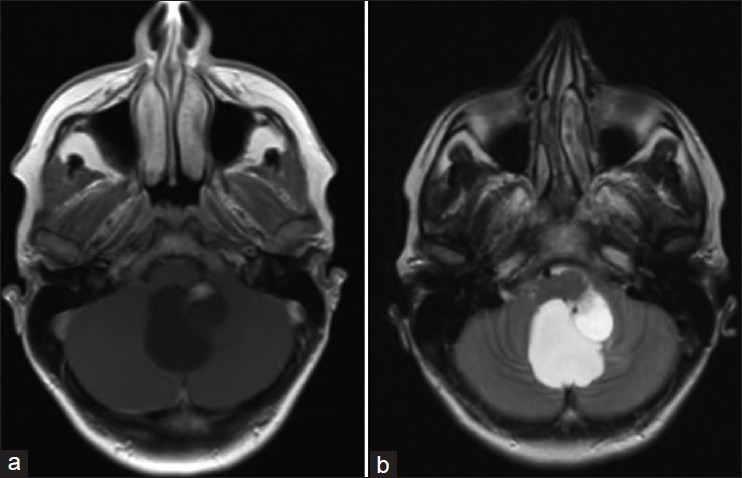
MR imaging of a cerebellar hemangioblastoma. Note the characteristic contrast enhancing nodule associated with a peritumoral cyst in the cerebellum. These lesions appear as a low signal on T1-weighted images (a) and as a high signal on T2-weighted sequences (b)
Figure 3.
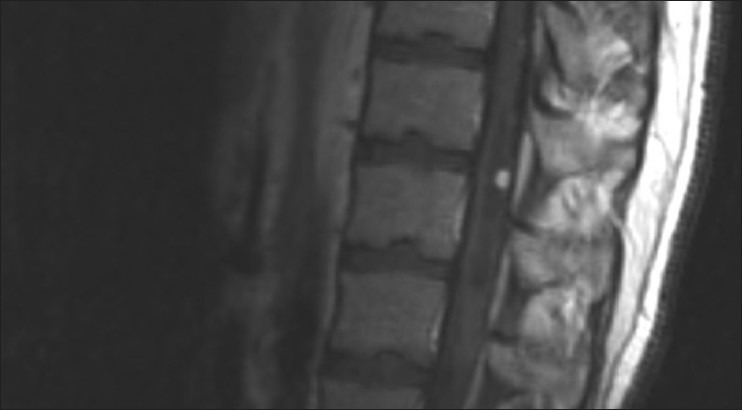
MR imaging of a spinal hemangioblastoma. Note the homogeneously contrast enhancing lesion in the spinal cord with the associated syrinx
Figure 4.
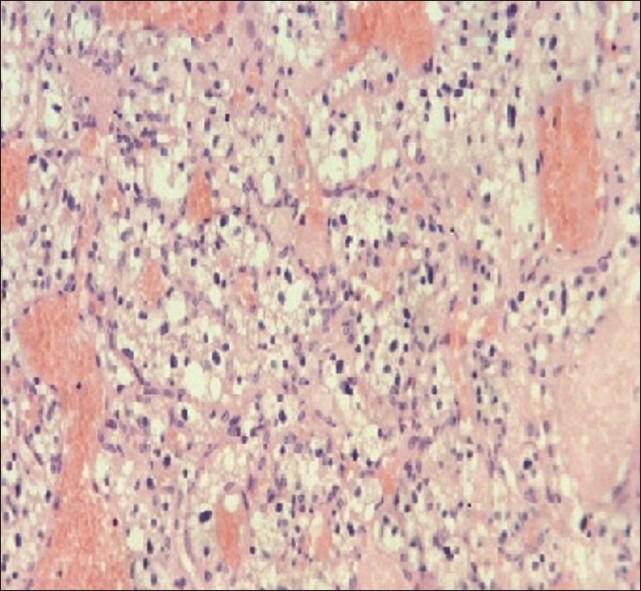
Histopathological examination of a hemangioblastoma. Note the extensive vascular network with neoplastic stromal cells. The neoplastic stromal cells have abundant cytoplasm with lipid vacuoles resulting in a typical clear cell morphology”
Hemangioblastomas do not have a gender predisposition.[13] They can occur either sporadically or as a component in VHLD.[13]
Approximately 75% of all hemangioblastomas are sporadic. The average age at presentation of a sporadic hemangioblastoma is in the fourth and fifth decade of life. Sporadic hemangioblastomas are, in general, solitary.[9,13,14,27,29] Surgical resection can offer definitive therapy in these hemangioblastomas. A complete surgical resection is usually feasible and the lesion normally does not reoccur. When tumor localization is invading vital structures and a tumor progression or neurological deficit is observed, operative intervention should be considered. As such, in 82-98% of cases, symptoms will improve or stabilize. Stereotactic radiosurgery or conventional radiation therapy can be considered in case of an inoperable lesion.[9,16,18,23,24]
On the contrary, hemangioblastomas associated with VHLD are generally diagnosed at a younger age, in the second and third decade of life. They usually are multiple and are the most common lesions associated with VHLD affecting 60-84% of these patients. While a complete surgical resection usually provides a definitive cure in sporadic hemangioblastomas, VHL associated hemangioblastomas tend to recur. Therefore therapeutic measure should focus on careful timing of surgical intervention(s). Surgical intervention should be reserved for symptomatic lesions, lesions with a demonstrated accelerated growth pattern or lesions that would compromise important neurological structures in the near future. Stereotactic gamma knife radiosurgery and radiation therapy may play a role in avoiding multiple neurosurgical interventions or in lesions that are not accessible by surgery.[6,14,20,27,29]
In VHLD, due to the underlying genetic mechanism, a variety of other tumors occur: Retinal angiomas, renal cysts and clear cell carcinomas, pheochromocytomas, pancreatic tumors, epidydimis cysts, cystadenomas of the broad ligament, and endolymphatic sac tumors of the middle ear. Neurosurgeons must be aware of these and be able to help counseling their patients.
Retinal angiomas are hemangioblastomas which develop in the retina and the optic nerve. They are encountered in up to 60% of VHLD patients who survive to the age of 60 years and are often multifocal or bilateral. If left untreated they can lead to loss of vision by hemorrhage, subsequent retinal detachement and glaucoma. In order to prevent these complications, retinal angiomas should be systematically screened and treated upon detection. Treatment is by laser photocoagulation and cryotherapy, effective in 70% of cases. If this modality fails, radiation therapy may be useful.[6,10,14,20,21,26,28]
Renal lesions occur in approximately two-thirds of VHLD patients. Mean age at onset is 44 years and an estimating 69% of patients surviving to the age of 60 years will develop renal cysts or renal clear cell carcinomas.[21] If multiple lesions are present, a more frequent examination is required. The therapeutic approach to renal involvement in VHLD varies from careful subsequent surveillance in renal cysts to kidney-sparing surgical interventions such as partial nephrectomy or ablation techniques for renal cell carcinoma. Resection is usually reserved for tumors >3 cm in diameters. Tumors <3 cm in diameter can be safely monitored because of their low metastatic potential.[4,6,14,30]
Pheochromocytomas are catecholamine producing tumors that arise from chromaffin cells of the adrenal medulla and sympathetic ganglia. Diagnosis is suspected when a patient presents with a classical pattern of paroxysmal hypertension, headache, sweating, and tachycardia. In literature it is estimated that, in sporadic cases, the annual incidence of pheochromocytomas is approximately 0.8 per 100000 person years. In a sporadic population, the average age at diagnosis is 47 years. In VHLD patients, however, pheochromocytomas tend to be seen in younger patients and they are often multiple or extraadrenal and less likely to be associated with symptoms or biochemical evidence of catecholamine production. Incidence in literature varies around 10% for the VHLD patient population.[1,6,14] The importance of the possibility of a pheochromocytoma lies in the preoperative work-up of a VHLD patient as there is a consequent risk of sympathetic overdrive and increased perioperative morbidity.
Pancreatic lesions including cysts, serous cystadenomas and neuroendocrine tumors are common in patients with VHLD. Up to 70% of patients with VHLD have associated pancreatic abnormalities according to the literature. Simple cysts and cystadenomas are, in general, asymptomatic. Neuroendocrine tumors can metastasize to the liver and subsequently may produce symptoms due to peptide secretion in approximately 8% of cases.[6,14] Because of the general lack of symptomatology many of these lesions are diagnosed incidentally during renal surveillance. Therapeutic intervention for these pancreatic lesions lies between watchful surveillance for small (<3 cm in diameter) asymptomatic lesions and surgical resection for symptomatic and/or lesions >3 cm in diameter in the pancreatic tail and body and/or lesions >2 cm in the head of the pancreas.[2,19]
Cystadenomas of the epidydimis and broad ligament are generally asymptomatic and the real incidence is unknown. Symptomatic treatment only is required when symptoms (swelling, pain, dyspareunia or menorrhagia) occur.
Papillary cystadenomas of the endolymphatic sac of the middle ear are highly vascular lesions arising within the posterior temporal bone. Symptoms include hearing loss, tinnitus, vertigo, aural pain and less often facial paresis. These lesions are seen with variable penetrance in VHLD. Up to 15% of patients with VHLD have been reported with this type of tumor.[6,14] Management of these lesions needs to be based on individual risk stratification between severity of symptoms and the potential complications associated with surgery. Treatment as such is primarily surgical: On complete excision, surgery is curative. For recurrent disease, stereotactic radiosurgery offers new perspectives. In case of hearing loss due to bilateral tumoral location, cochlear implants may be an option.[6,17]
Screening for VHLD in hemangioblastoma patients
Diagnosis of VHLD is made based upon clinical criteria of Melmon and Rosen and more recently on genetic detection of the germline mutation in peripheral blood leukocytes. Based upon clinical criteria, patients with a family history of VHLD and a VHL associated tumor meet the clinical criteria for diagnosis of VHLD. Patients with a negative family history fulfill the clinical diagnostic criteria if they have two or more CNS hemangioblastomas or one hemangioblastoma and a VHL associated tumor. The sensitivity and specifity of the genetic testing is nearly 100%. In patients presenting with VHL associated tumors and a negative genetic test based upon peripheral blood, a somatic mosaicism should be considered.[6,12,20,25]
The main difficulty in the work-up for hemangioblastoma is to differentiate between a sporadic and a VHL associated lesion. The true proportion of hemangioblastoma associated with VHLD is diversely estimated. Figures from 10% to 40% have been reported in the literature.[13,15,27] One should take care not to underdiagnose VHLD in the presence of an apparently isolated lesion without clear evidence of family history. A systematic clinical and genetic screening therefore are strongly recommended for every patient with a newly diagnosed CNS hemangioblastoma.[13,15,27,31]
Upon presentation of an apparently sporadic hemangioblastoma, the literature suggests to perform a fixed work-up.[8,20]
The literature suggests to consistently perform craniospinal imaging, ophtalmoscopy, abdominal ultrasound, pheochromocytoma screening by metanephrine, and VMA detection in urine and auditory testing (possible endolymphatic sac tumor of the middle ear) for VHL associated hemangioblastoma.[8,20] Screening for papillary cystadenomas of the epidydimis and broad ligament are only to be performed upon indication in patients with VHLD.[6,7,11,14,17,22]
Follow-up for sporadic and VHL associated hemangioblastoma
Evidence-based suggestions for follow-up after treatment for sporadic CNS hemangioblastoma were not found in the literature. Based upon our experience we suggest to perform a MRI scan of the involved neuronal axis at 6 and at 12-24 months in the postoperative period. These follow-up modalities are to be advanced or extended based upon clinical indication.
After intervention for a VHL associated lesion, there are detailed follow-up schedules available. The literature suggests to perform a yearly MRI driven craniospinal control as well as an annual ophtalmoscopy, a yearly abdominal ultrasound with triennial computed tomography (CT) imaging, a yearly audiometry and pheochromocytoma investigation by urine analysis (metanephrine – VMA).[7,8,11,17,20,22] Based upon clinical indication these follow-up modalities should be advanced or extended.
CONCLUSION
Hemangioblastomas are seen as an early presentation of VHLD in 10-40% of cases.
Screening for retinal angiomas, abdominal masses and pheochromocytomas as well as genetic analysis is recommended for every patient with a newly diagnosed hemangioblastoma. Follow-up is by MRI of the clinical neuronal region at 6 and at 12-24 months postoperatively.
For VHL associated hemangioblastomas, yearly investigation for craniospinal hemangioblastoma by MRI and yearly screening and follow-up for retinal angiomas is recommended. Annual abdominal ultrasound with triennial CT imaging for abdominal masses is postulated. Annual audiometry is to be performed for possible endolymphatic sac tumor, detailed radiographic imaging of the skull base should be performed upon abnormality in auditory testing. Investigations for cystadenomas of the epidydimis and broad ligament only are mandatory on indication. Annual investigation for pheochromocytoma is recommended.
Footnotes
Available FREE in open access from: http://www.surgicalneurologyint.com/text.asp?2013/4/1/145/121110
Contributor Information
Sven Bamps, Email: sven.bamps@uzleuven.be.
Frank Van Calenbergh, Email: frank.vancalenbergh@uzleuven.be.
Steven De Vleeschouwer, Email: steven.devleeschouwer@uzleuven.be.
Johannes Van Loon, Email: Johannes.vanloon@uzleuven.be.
Raf Sciot, Email: raf.sciot@uzleuven.be.
Eric Legius, Email: eric.legius@uzleuven.be.
Jan Goffin, Email: jan.goffin@uzleuven.be.
REFERENCES
- 1.Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc. 1983;58:802–4. [PubMed] [Google Scholar]
- 2.Blansfield JA, Choyke L, Morita SY, Choyke PL, Pingpank JF, Alexander HR, et al. Clinical, genetic and radiographic analysis of 108 patient with von Hippel-Lindau disease manifested by pancreatic neuroendocrine neoplasms. Surgery. 2007;142:814–8. doi: 10.1016/j.surg.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bohling T, Hatva E, Plate K, Haltia M, Alitalo K. Tumor of the Nervous system: Pathology and genetics. Lyon, France: International Agency for Research on Cancer; 1997. Von Hippel-Lindau disease and capillary haemangioblastoma; p. 179. [Google Scholar]
- 4.Bratslavsky G, Liu JJ, Johnson AD, Sudarshan S, Choyke PL, Linehan WM, et al. Salvage partial nephrectomy for hereditary renal cancer: Feasibility and outcomes. J Urol. 2008;179:67–70. doi: 10.1016/j.juro.2007.08.150. [DOI] [PubMed] [Google Scholar]
- 5.Burger P, Scheithauer B. Washington: Armed Forces Institute of Pathology; 1994. Tumors of the Central nervous System; p. 239. [Google Scholar]
- 6.Butman JA, Linehan WM, Lonser RR. Neurologic manifestations of von Hippel-Lindau disease. JAMA. 2008;300:1334–42. doi: 10.1001/jama.300.11.1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choyke PL, Glenn GM, Wagner JP, Lubensky IA, Thakore K, Zbar B, et al. Epididymal cystadenomas in von Hippel-Lindau disease. Urology. 1997;49:926–31. doi: 10.1016/s0090-4295(97)00074-5. [DOI] [PubMed] [Google Scholar]
- 8.Choyke PL, Glenn GM, Walther MM, Patronas NJ, Linehan WM, Zbar B. Hippel-Lindau disease: Genetics, clinical and imaging features. Radiology. 1995;194:629–42. doi: 10.1148/radiology.194.3.7862955. [DOI] [PubMed] [Google Scholar]
- 9.Conway JE, Chou D, Clatterbuck RE, Brem H, Long DM, Rigamonti D. Hemangioblastomas of the central nervous system in von Hipel-Lindau syndrome and sporadic disease. Neurosurgery. 2001;48:55–62. doi: 10.1097/00006123-200101000-00009. [DOI] [PubMed] [Google Scholar]
- 10.Dollfus H, Maasin P, Taupin P, Nemeth C, Amara S, Giraud S, et al. Retinal hemangioblastoma in von Hippel-Lindau Disease: A clinical and molecular study. Invest Opthalmol Vis Sci. 2002;43:3067–74. [PubMed] [Google Scholar]
- 11.Gersell DJ, King TC. Papillary cystadenoma of the mesosalpinx in von Hippel-Lindau disease. Am J Surg Pathol. 1988;12:145–9. doi: 10.1097/00000478-198802000-00008. [DOI] [PubMed] [Google Scholar]
- 12.Gijtenbeek JM, Jacobs B, Sprenger SH, Eleveld MJ, van Kessel AG, Kros JM, et al. Analysis of von hippel-lindau mutations with comparative genomic hybridization in sporadic and hereditary hemangioblastomas: Possible genetic heterogeneity. J Neurosurg. 2002;97:977–82. doi: 10.3171/jns.2002.97.4.0977. [DOI] [PubMed] [Google Scholar]
- 13.Gläsker S. Central nervous system manifestations in VHL: Genetics, pathology and clinical phenotypic features. Fam Cancer. 2005;4:37–42. doi: 10.1007/s10689-004-5347-6. [DOI] [PubMed] [Google Scholar]
- 14.Hill P, Maxwell P. Von Hippel-Lindau disease: Insights and advances. Adv Clin Neurosci Rehabil. 2003;3:15. [Google Scholar]
- 15.Innus C, Patterson J. Hemangioblastoma without von Hippel-Lindau disease. JAAPA. 2007;20:28–31. doi: 10.1097/01720610-200711000-00017. 2. [DOI] [PubMed] [Google Scholar]
- 16.Jagannathan J, Lonser RR, Smith R, DeVroom HL, Oldfield EH. Surgical management of cerebellar hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2008;108:210–22. doi: 10.3171/JNS/2008/108/2/0210. [DOI] [PubMed] [Google Scholar]
- 17.Kim HJ, Butman JA, Brewer C, Zalewski C, Vortmeyer AO, Glenn G, et al. Tumors of the endolymphatic sac in patients with von Hippel-Lindau disease: Implications for their natural history, diagnosis and treatment. J Neurosurg. 2005;102:503–12. doi: 10.3171/jns.2005.102.3.0503. [DOI] [PubMed] [Google Scholar]
- 18.Koh ES, Nichol A, Millar BA, Ménard C, Pond G, Laperriere NJ. Role of fractionated external beam radiotherapy in hemangioblastoma of the central nervous system. Int J Radiat Oncol Biol Phys. 2007;69:1521–6. doi: 10.1016/j.ijrobp.2007.05.025. [DOI] [PubMed] [Google Scholar]
- 19.Libutti SK, Choyke PL, Alexander HR, Glenn G, Bartlett DL, Zbar B, et al. Clinical and genetic analysis of patients with pancreatic neuroendocrine tumors associated with von Hippel-Lindau disease. Surgery. 2000;128:1022–7. doi: 10.1067/msy.2000.110239. [DOI] [PubMed] [Google Scholar]
- 20.Maher ER, Webster AR, Moore AT. Clinical features and molecular genetics of von Hippel-Lindau disease. Ophtalmic Genet. 1995;16:79–84. doi: 10.3109/13816819509059966. [DOI] [PubMed] [Google Scholar]
- 21.Maher ER, Yates JR, Harries R, Benjamin C, Harris R, Moore AT, et al. Clinical features and natural history of von Hippel-Lindau Disease. Quart J of Medicine. 1990;77:1151–63. doi: 10.1093/qjmed/77.2.1151. [DOI] [PubMed] [Google Scholar]
- 22.Manski TJ, Heffner DK, Glenn GM, Patronas NJ, Pikus AT, Katz D, et al. Endolymphatic sac tumors – a source of morbid hearing loss in von Hippel-Lindau disease. JAMA. 1997;277:1461–6. doi: 10.1001/jama.277.18.1461. [DOI] [PubMed] [Google Scholar]
- 23.Moss JM, Choi CY, Adler JR, Jr, Soltys SG, Gibbs IC, Chang SD. Stereotactic radiosurgical treatment of cranial and spinal hemangioblastomas. Neurosurgery. 2009;65:79–85. doi: 10.1227/01.NEU.0000348015.51685.D2. [DOI] [PubMed] [Google Scholar]
- 24.Parker F, Aghakhani N, Ducati LG, Yacubian-Fernandes A, Silva MV, David P, et al. Results of microsurgical treatment of medulla oblongata and spinal cord hemangioblastomas: A comparison of two distinct clinical patient groups. J Neurooncol. 2009;93:133–7. doi: 10.1007/s11060-009-9861-0. [DOI] [PubMed] [Google Scholar]
- 25.Priesemann M, Davies KM, Perry LA, Drake WM, Chew SL, Monson JP, et al. Benefits of screening in von Hippel-Lindau disease – Comparison of morbidity associated with initial tumours in affected parents and children. Horm Res. 2006;66:1–5. doi: 10.1159/000093008. [DOI] [PubMed] [Google Scholar]
- 26.Raja D, Benz MS, Murray TG, Escalona-Benz EM, Markoe A. Salvage external beam radiotherapy of retinal capillary hemangiomas secondary to von Hippel-Lindau disease: Visual and anatomic outcomes. Ophtalmology. 2004;111:150–3. doi: 10.1016/j.ophtha.2003.04.003. [DOI] [PubMed] [Google Scholar]
- 27.Richard S, Campello C, Taillandier L, Parker F, Resche F. Haemangioblastoma of the central nervous system in von Hippel-Lindau disease. J Intern Med. 1998;243:547–53. doi: 10.1046/j.1365-2796.1998.00337.x. [DOI] [PubMed] [Google Scholar]
- 28.Singh AD, Nouri M, Shields CL, Shields JA, Smith AF. Retinal capillary hemangioma: A comparison of sporadic cases and cases associated with von Hippel-Lindau disease. Ophtalmology. 2001;108:1907–11. doi: 10.1016/s0161-6420(01)00758-8. [DOI] [PubMed] [Google Scholar]
- 29.Slater A, Moore NR, Huson SM. The natural history of cerebellar hemangioblastomas in von Hippel-Lindau disease. AJNR Am J Neuroradiol. 2003;24:1570–4. [PMC free article] [PubMed] [Google Scholar]
- 30.Walther MM, Choyke PL, Glenn G, Lyne JC, Rayford W, Venzon D, et al. Renal cancer in families with hereditary renal cancer: Prospective analysis of a tumor size treshold for renal parenchymal sparing surgery. J Urol. 1999;161:1475–9. doi: 10.1016/s0022-5347(05)68930-6. [DOI] [PubMed] [Google Scholar]
- 31.Woodward ER, Wall K, Forsyth J, Macdonald F, Maher ER. VHL mutation analysis in patients with isolated central nervous system haemangioblastoma. Brain. 2007;130:836–42. doi: 10.1093/brain/awl362. [DOI] [PubMed] [Google Scholar]