

Abstract
Sodium valproate (VPA) is widely used throughout the world to treat epilepsy, migraine, chronic headache, bipolar disorder, and as adjuvant chemotherapy. VPA toxicity is an uncommon but potentially fatal cause of idiosyncratic liver injury. Rare mutations in POLG, which codes for the mitochondrial DNA polymerase γ (polγ), cause the Alpers-Huttenlocher syndrome (AHS). AHS is a neurometabolic disorder associated with an increased risk of developing fatal VPA-hepatotoxicity. We therefore set out to determine whether common genetic variants in POLG explain why some otherwise healthy individuals develop VPA-hepatotoxicity. We carried out a prospective study of subjects enrolled in the Drug Induced Liver Injury Network (DILIN) from 2004 to 2008 through five US centres. POLG was sequenced and the functional consequences of VPA and novel POLG variants were evaluated in primary human cell lines and the yeast model system Saccharomyces cerevisiae. Heterozygous genetic variation in POLG was strongly associated with VPA-induced liver toxicity (odds ratio = 23.6, 95% CI = 8.4 – 65.8, P = 5.1 × 10−7). This was principally due to the p.Q1236H substitution which compromised polγ function in yeast. Therapeutic doses of VPA inhibited human cellular proliferation, and high doses caused non-apoptotic cell death which was not mediated through mitochondrial DNA depletion, mutation, or a defect of fatty acid metabolism. These findings implicate impaired liver regeneration in VPA toxicity, and show that prospective genetic testing of POLG will identify individuals at high risk of this potentially fatal consequence of treatment.
Keywords: Liver failure, mitochondria, DNA polymerase, drug toxicity, pharmacogenomics
Over 1 in 37,000 subjects exposed to sodium valproate (valproic acid, VPA) develop idiosyncratic liver toxicity, with the risk reaching ~1 in 500 in young children on polytherapy.1 Increased awareness has contributed to a decline in fatal VPA-induced liver failure,2 but the worldwide use of VPA continues to increase through its use in other clinical contexts. In addition to its use as a first-line anticonvulsant, VPA is now in regular use for migraine, bipolar disorder, chronic headache, and as adjuvant chemotherapy. The prompt recognition of early symptoms and immediate discontinuation of the drug can prevent fulminant liver failure,2 but initial clinical clues are often mild and non-specific, making is difficult to identify individuals before significant liver damage occurs. Liver biopsy characteristically reveals microvesicular steatosis, and occasionally severe hepatocellular necrosis.3 Fever, rash, lymphadenopathy and/or peripheral eosinophilia are rarely present during VPA-hepatotoxicity, consistent with a direct toxic effect of the drug, rather than an immune-mediated hypersensitivity reaction typical of other anti-epileptic drugs.4
The recent description of mutations in mitochondrial DNA (mtDNA) polymerase γ (POLG) as a major cause of Alpers-Huttenlocher syndrome (AHS)5 provides a clue to the underlying mechanism of VPA-hepatotoxicity. AHS is a rare childhood encephalopathy characterised by developmental delay and intractable epilepsy and liver disease.6,7 Most cases have homozygous or compound heterozygote mutations in POLG,5 and ~1/3 of AHS patients develop liver failure within 3 months of exposure to VPA.8, 9 This raises the possibility that common genetic variation in POLG predisposes individuals to VPA-induced liver failure in the absence of a recognizable AHS-phenotype.
Materials and Methods
Participants
Patients with suspected VPA-hepatotoxicity were enrolled in the Drug Induced Liver Injury Network (DILIN) from 2004 to 2008 through five US centres involved at that time: North Carolina at Chapel Hill, the Universities of Connecticut, Michigan, Indiana, and California at San Francisco, and the co-ordinating center at Duke Clinical Research Institute.3 All had one of the following on presentation: jaundice or serum bilirubin > 2.5 mg/dL and elevation in alanine aminotransferrase (ALT), aspartate amino transferrase (AST), or alkaline phosphatase (ALP); no jaundice and serum bilirubin < 2.5 mg/dL, but elevations in ALT or AST (> 5 fold more than the upper limit of normal, ULN) or elevations in ALP (> 2xULN, Table 1). Laboratory and clinical data were captured by the site investigator who crafted a clinical narrative describing the outcome. A committee of three experienced hepatologists then reviewed the cases, blind to the results of the study, and ranked the likelihood of causality on a scale of 1 (definite) to 5 (unlikely), as described.3 The study was conducted with local ethical and institutional review board approval in accordance with the Declaration of Helsinki.
Table 1.
Clinical and genetic data for the 17 patients with suspected valproate-induced liver injury. Causality assessment key: 2 – highly likely (75-95% likely); 3 – probable (50-75% likely); 4 – possible (25-50% likely) or 5 – unlikely (5-25% likely). ALT = serum alanine transferase, INR = international normalized coagulation ratio, n.a. = not available.
| ID No |
Age
(years) |
Race/ethnicity | Indication for valporate |
Days from
drug start to onset of hepatotoxicity |
POLG Change |
Causality
Assessment |
Peak ALT (U/L) |
Peak bilirubin
(mg/dL) |
Peak INR | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seizures | Bipolar disorder |
Epilepsy | Severe headaches |
cDNA substitution | Amino acid substitution | ||||||||
| 1 | 2 | Caucasian | + | 65 | 2 | 370 | 0.9 | n.a. | |||||
| 2 | 5 | Caucasian | + | 219 | c.3708G>T | p.Q1236H | 2 | 2471 | 2.7 | 1.9 | |||
| 3 | 9 | Caucasian | + | 74 | n.a. | 302 | n.a. | 1.2 | |||||
| 4 | 15 | Hispanic | + | 2788 | c.3708G>T | p.Q1236H | 3 | 525 | 3.9 | 1.0 | |||
| 5 | 15 | Caucasian | + | 124 | c.911T>G; c.1399G>A | p.L304R; p.A467T | 2 | 287 | 26.7 | 4.4 | |||
| 6 | 16 | Caucasian | + | 13 | 5 | 69 | 1.0 | 1.4 | |||||
| 7 | 19 | Caucasian | + | 48 | 3 | 1813 | 7.1 | 1.3 | |||||
| 8 | 22 | Caucasian | + | 4 | c.3708G>T | p.Q1236H | 3 | 76 | 0.4 | 1.6 | |||
| 9 | 23 | Caucasian | + | 720 | 3 | 835 | 0.5 | 1.5 | |||||
| 10 | 26 | Caucasian | + | + | n.a. | c.3428A>G | p.E1143G | 3 | 130 | 1.2 | 1.2 | ||
| 11 | 33 | Caucasian | + | 30 | c.3428A>G | p.E1143G | 3 | 860 | 1.0 | n.a. | |||
| 12 | 33 | Caucasian | + | 1617 | 3 | 66 | 1.7 | 1.0 | |||||
| 13 | 36 | Hispanic | + | 314 | c.3708G>T | p.Q1236H | 2 | 2947 | 7.7 | 1.7 | |||
| 14 | 36 | Caucasian | + | 51 | 4 | 106 | 32.0 | 1.5 | |||||
| 15 | 36 | Caucasian | + | 85 | 2 | 831 | 13.1 | 0.9 | |||||
| 16 | 41 | Caucasian | + | 147 | 3 | 142 | 22.4 | 2.0 | |||||
| 17 | 47 | Caucasian | + | 31 | c.3708G>T | p.Q1236H | 2 | 90 | 0.5 | 2.1 | |||
Molecular Genetic analysis
POLG exons and flanking intronic regions (BC050559) were forward and reverse sequenced (Applied Biosciences Big Dye 3.1, ABI3100). Cellular mtDNA levels were measured (MTND1) relative to the nuclear-encoded B2M (AC025270) by real-time PCR (iQ™ Sybr Green, BioRad ICycler, CA).10 MtDNA deletions were detected by long-range PCR.
Functional studies in mammalian cell models
Human hepatocyte cell lines from patients with POLG variants are not available. Given the direct toxic effect of VPA on skeletal muscle,11 we studied human primary myoblasts and myotubes from a p.Q1236H heterozygote, and a compound heterozygous for p.A467T/p.K1191N with AHS with local ethical approval (not DILIN subjects). Muscle cell culture was carried out as described.12 Myotubes were treated with 300 μM Didanosine (Sigma) or 300 μM Stavudine (Sigma) for 3-days prior to and 6 days during differentiation.12 Trypan-blue negative (viable) cells were counted using a Mod-Fuchs haemocytometer. Apoptosis was determined using the Roche Apoptosis ladder kit. Cytochrome c oxidase (COX) activity was evaluated histochemically on day 10, and intermediary metabolites of fatty acid β-oxidation were analysed by tandem mass spectrometry in culture media collected at days 0, 5 and 10.13 All cell culture studies were done in triplicate.
Functional studies in yeast
MIP1-human POLG chimera (MIP1C allele) was constructed through substitution of nucleotides 2911-2964 of MIP1661T wt allele with nucleotides 3658-3709 of POLG encoding sequence. p.Q1236H was introduced by site specific mutagenesis. Frequency of petite mutants and of erythromycin resistant (EryR) mutants were measured as described.14
Results
Genetic variation in POLG is common in patients with VPA-hepatotoxity
POLG substitutions were identified in 8 of the 17 patients with suspect VPA-induced hepatotoxicty (Fig.1a). One harbored compound heterozygous mutations: c.1399G>A/p.A467T, predicted to change alanine to threonine in the linker region of the protein (p.A467T); and c.911T>G predicted to alter a conserved leucine to an arginine residue also in the linker region of polγ (p.L304R, Fig.1b), previously reported in AHS. This patient was prescribed VPA for unexplained seizures and was known to have a peripheral neuropathy and clumsiness. With hindsight, these features were the first stage of the AHS, although this was not obvious on clinical presentation. This patient required a liver transplant following his initial exposure to VPA and then developed intractable seizures leading to an early death, highlighting the importance of identifying patients at risk of VPA-hepatotoxicity before commencing treatment.
Figure 1.
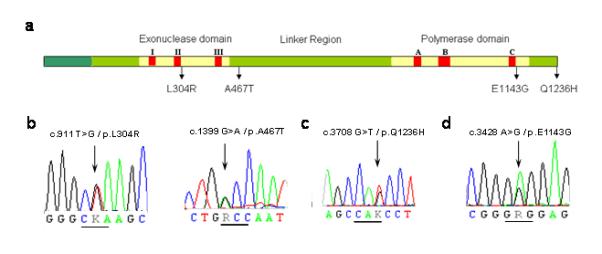
(a) Gene structure of POLG showing the location of the (b) c.911T>G/p.L304R, c.1399G>A/p.A467T, (c) c.3708G>T/p.Q1236H, and (d) c.3428A>G/p.E1143G substitutions. The exonuclease domain extends from amino acid residue 1 to 418. The polymerase domain extends from amino acid residue 756 to 1239. The linker region lies between amino acid residues 418 and 756.
The remaining seven (41%) had a single heterozygous POLG substitution. Five harbored c.3708G>T, predicted to alter a glutamine in the polymerase domain (p.Q1236H, Fig.1c), and two harbored c.3428A>G, predicted to alter a glutamic acid in the polymerase domain (p.E1143G, Fig.1d). Both the frequency of p.Q1236H (P = 1.9 × 10−4), and the combined frequency of p.Q1236H and p.E1143G (P = 5.1 × 10−7) were significantly greater than in ethnically matched population controls, giving a combined odds ratio, OR = 23.6 (95% CI = 8.4 – 65.8) (Supplementary Table S1). The strongest association was in patients where VPA-induced liver toxicity was highly likely (≥1 variants in 4/6, or 66%), and likely (4/8, or 50%) compared to unlikely (0/2 or 0%).
Functional consequences of the p.Q1236H substitution
The functional effects of p.Q1236H have not been previously studied. We therefore constructed a Polg-Mip1 chimera (Mip1C) in the model system yeast Saccharomyces cerevisiae in which 971-988 amino acids of Mip1 were substituted with the corresponding 1220-1237 amino acids of polγ (Fig.2b). This was mutagenized to introduce the substitution p.Q1236H. The mip1CQ1236H strain showed a ~1.5 fold increase in petite frequency (18.0% (±1.3) vs. 12.4% (±1.6)) (Fig.2c), indicating extended mtDNA mutability; and a 2 fold increase of EryR mutant frequency, indicating increased mtDNA point mutability, (19.7×10−8 (±2.0) vs. 10.9×10−8 (±1.2)). p.Q1236H is therefore highly likely to alter human polγ function. However, treatment with sublethal concentrations of VPA (1, 2, 5, 8 and 10mM) did not alter the yeast phenotype. The functional effects of p.E1143G have been previously described.14, 15 The phenotype of both substitutions is mild, explaining why these alleles are common throughout the world (p.Q1236H ≤8.6%, and p.E1143G ≤4%). p.Q1236H and p.E1143G may only be disadvantageous in specific contexts, such as exposure to VPA.
Figure 2.
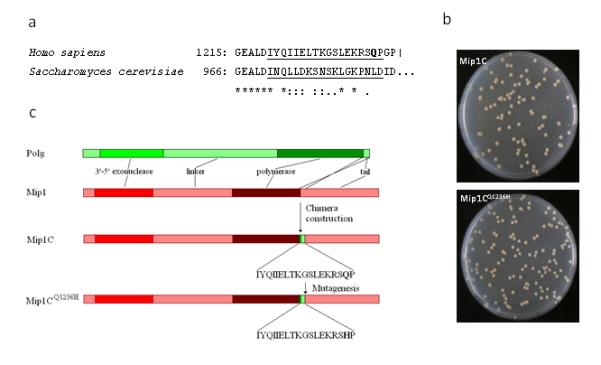
(a) Alignment of C-terminal stretch of human polγ and the corresponding stretch of yeast Mip1. Q1236 amino acid is in bold; the region which is changed in Mip1C is underscored. (b) Linear representation of polγ, Mip1, Mip1C and Mip1CQ1236H organization. Q1236 amino acid is in bold. (c) Petri dish images showing the normal and petite colonies from the parental (Mip1C) and the Mip1CQ1236H strain (bottom).
No evidence of a secondary mtDNA defect in whole blood
Given the role of POLG in mtDNA replication we looked for evidence of a qualitative or quantitative defect of mtDNA in whole-blood cellular mtDNA because liver tissue was not available from the affected individuals. No mtDNA deletions were detected by long-range PCR and the mtDNA content was no different to age-matched controls (83.9 copies/cell, SD 58.8; vs 85.8, SD 28.3, Supplementary Fig.1a).
Cellular effects of sodium valproate
Treatment of control and patient myoblasts with 50mM and 100mM VPA compromised cell proliferation, with extensive cellular ballooning, vacuolization and detachment within 3 days of treatment. Despite the observed cell death, there was no significant decrease in mtDNA content (Fig.3a), nor detectable mtDNA deletions (Supplementary Fig.1b) following treatment for ten days with 2mM and 10mM VPA. By contrast, ethidium bromide treated cells grown in parallel showed the expected decrease in mtDNA content after 10 days but no defect of cellular proliferation and no evidence of cell death (Fig.3b). There was no evidence of apoptosis in any of the cell lines after 10 days of treatment. Multiple mtDNA deletions were not detected in any of the cell pellets, there were no differences in COX activity observed, and β-oxidation metabolites remained within normal limits (Supplementary Table.2). We therefore extended our studies to post-mitotic myotubes, which more closely model mtDNA depletion in vivo.12 MtDNA levels were significantly lower in AHS and Q1236H myotubes than in controls (Fig.3c). To determine whether mtDNA depletion itself predisposes to further mtDNA loss after VPA-exposure, we depleted the myotubes with didanosine and stavudine, which induce less severe myotube mtDNA depletion than ethidium bromide.12 MtDNA depletion levels in Q1236H myotubes were less than in controls, and similar to the AHS cell-lines, but there was no further decrease in mtDNA content with the addition of 10mM VPA (Fig.3c).
Figure 3.
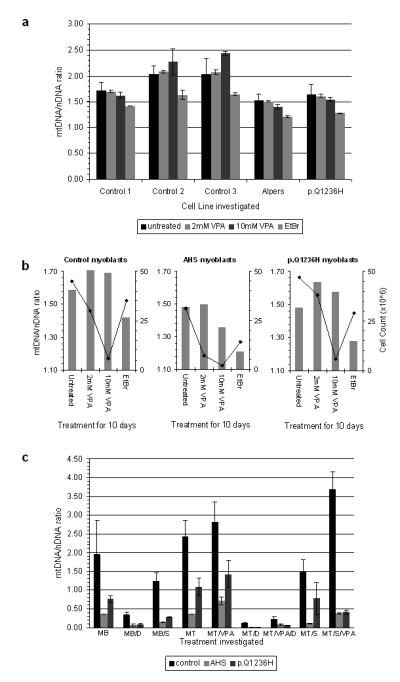
(a) Ratio of mtDNA to nuclear DNA for each cell line following ten days of treatment with either 2mM VPA, 10mM VPA, 50ng/ml Ethidium bromide (EtBr) or untreated. AHS = p.A467T/p.K1191N compound heterozygote. (b) Relationship between mtDNA level and cell viability after ten days of treatment. For comparison, mtDNA ratios (mtDNA/nDNA) are represented as columns on the left hand y-axis and cell count (×106) as a line plot on the right hand y-axis. VPA = sodium valproate. EtBr = ethidium bromide. AHS = p.A467T/p.K1191N compound heterozygote. (c) Ratio of mtDNA to nuclear DNA for each myotube cell line following 9 days of treatment with/without 300 μM didanosine (D) or stavudine (S) and additionally with/without 10mM VPA. MB= myoblasts, MT=myotubes, AHS = p.A467T/p.K1191N compound heterozygote. VPA = sodium valproate. Error bars = SD in all panels.
Discussion
VPA is a branched medium chain fatty acid known to inhibit mitochondrial β-oxidation,16 possibly through the microsomal production of toxic metabolites including 4-ene-VPA,17 or cytosolic and mitochondrial CoA sequestration effects.18 However, we saw no evidence of a β-oxidation defect, making this mechanism unlikely in this context. We also saw no evidence of a secondary mtDNA defect, despite the VPA dose-related growth inhibition and cell death. By contrast, treating identical cell lines with ethidium bromide, didanosine or stavudine caused profound but recoverable mtDNA depletion without cell death. VPA-toxicity is therefore unlikely to be mediated through a direct effect on mtDNA, explaining why we did not observe a COX defect in VPA-treated cells. Although it is possible that these aspects are specifically deranged in the liver, we observed the morphological characteristics of VPA hapatotoxicity in human myoblasts,3 implicating the same mechanism in our in vitro model. Moreover, an elevated serum creatine kinase in patients with VPA-toxicity points to a similar direct toxic effect on skeletal muscle.11
Unlike mature skeletal muscle and brain, the liver can proliferate in response to damage, and there is clear evidence of hepatocyte proliferation in patients with AHS. We have shown that VPA impairs cellular proliferation in vitro, and that p.Q1236H increases mtDNA mutability in yeast and may decrease mtDNA copy number in myotubes. This raises the possibility that both mechanisms independently compromise the regenerative capacity of the liver, thus inhibiting the endogenous capacity for liver repair in response to an external insult. For VPA, this could be through the inhibition of histone deacetylases which regulate gene expression by relaxing chromatin structure and facilitating access to DNA by the transcriptional machinery.19
Supplementary Material
Acknowledgements
PFC is a Wellcome Trust Senior Fellow in Clinical Science who also receives funding from the Medical Research Council (UK), the UK Parkinson’s Disease Society, and the UK NIHR Biomedical Research Centre for Ageing and Age-related disease award to the Newcastle upon Tyne Foundation Hospitals NHS Trust. Telethon-Italy Foundation (Grant No. GGP07019) to IF. RH and SB are supported by the Deutsche Forschungsgemeinschaft HO 2505/2-1. The Muscle Tissue Culture Collection is part of the German network on muscular dystrophies (MD-NET, service structure S1, 01GM0601) funded by the German ministry of education and research (BMBF, Bonn, Germany). The Muscle Tissue Culture Collection is a partner of EuroBioBank (www.eurobiobank.org) and TREAT-NMD (EC, 6th FP, proposal # 036825). RH and is also supported by the Newcastle upon Tyne Hospitals NHS Charity (RES0211/7262). Global and Hispanic control data for p.Q1236H was kindly supplied by Dr Andy Singleton, NIH Bethesda.
References
- 1.Dreifuss FE, Langer DH, Moline KA, Maxwell JE. Valproic acid hepatic fatalities. II. US experience since 1984. Neurology. 1989 Feb;39(2 Pt 1):201–7. doi: 10.1212/wnl.39.2.201. [DOI] [PubMed] [Google Scholar]
- 2.Bryant AE, 3rd, Dreifuss FE. Valproic acid hepatic fatalities. III. U.S. experience since 1986. Neurology. 1996 Feb;46(2):465–9. doi: 10.1212/wnl.46.2.465. [DOI] [PubMed] [Google Scholar]
- 3.Fontana RJ, Watkins PB, Bonkovsky HL, Chalasani N, Davern T, Serrano J, et al. Drug-Induced Liver Injury Network (DILIN) prospective study: rationale, design and conduct. Drug Saf. 2009;32(1):55–68. doi: 10.2165/00002018-200932010-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gopaul S, Farrell K, Abbott F. Effects of age and polytherapy, risk factors of valproic acid (VPA) hepatotoxicity, on the excretion of thiol conjugates of (E)-2,4-diene VPA in people with epilepsy taking VPA. Epilepsia. 2003 Mar;44(3):322–8. doi: 10.1046/j.1528-1157.2003.07202.x. [DOI] [PubMed] [Google Scholar]
- 5.Naviaux RK, Nguyen KV. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann Neurol. 2004 May;55(5):706–12. doi: 10.1002/ana.20079. [DOI] [PubMed] [Google Scholar]
- 6.Alpers BJ. Diffuse progressive degeneration of the gray matter of the cerebrum. Arch Neurol Psychiatry. 1931;25:469–505. [Google Scholar]
- 7.Huttenlocher PR, Solitare GB, Adams G. Infantile diffuse cerebral degeneration with hepatic cirrhosis. Arch Neurol. 1976 Mar;33(3):186–92. doi: 10.1001/archneur.1976.00500030042009. [DOI] [PubMed] [Google Scholar]
- 8.Nguyen KV, Sharief FS, Chan SS, Copeland WC, Naviaux RK. Molecular diagnosis of Alpers syndrome. J Hepatol. 2006 Feb 20;45:108–16. doi: 10.1016/j.jhep.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 9.Chinnery PF, Zeviani M. 155th ENMC workshop: Polymerase gamma and disorders of mitochondrial DNA synthesis, 21-23 September 2007, Naarden, The Netherlands. Neuromuscul Disord. 2007 Dec 19;18(3):259–67. doi: 10.1016/j.nmd.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Durham SE, Bonilla E, Samuels DC, DiMauro S, Chinnery PF. Mitochondrial DNA copy number threshold in mtDNA depletion myopathy. Neurology. 2005;65:453–5. doi: 10.1212/01.wnl.0000171861.30277.88. [DOI] [PubMed] [Google Scholar]
- 11.Koenig SA, Buesing D, Longin E, Oehring R, Haussermann P, Kluger G, et al. Valproic acid-induced hepatopathy: nine new fatalities in Germany from 1994 to 2003. Epilepsia. 2006 Dec;47(12):2027–31. doi: 10.1111/j.1528-1167.2006.00846.x. [DOI] [PubMed] [Google Scholar]
- 12.Bulst S, Abicht A, Holinski-Feder E, Muller-Ziermann S, Koehler U, Thirion C, et al. In vitro supplementation with dAMP/dGMP leads to partial restoration of mtDNA levels in mitochondrial depletion syndromes. Hum Mol Genet. 2009 Feb 16;18:1590–9. doi: 10.1093/hmg/ddp074. [DOI] [PubMed] [Google Scholar]
- 13.Gempel K, Kiechl S, Hofmann S, Lochmuller H, Kiechl-Kohlendorfer U, Willeit J, et al. Screening for carnitine palmitoyltransferase II deficiency by tandem mass spectrometry. J Inher Metab Dis. 2002 Feb;25(1):17–27. doi: 10.1023/a:1015109127986. [DOI] [PubMed] [Google Scholar]
- 14.Baruffini E, Ferrero I, Foury F. Mitochondrial DNA defects in Saccharomyces cerevisiae caused by functional interactions between DNA polymerase gamma mutations associated with disease in human. Biochim Biophys Acta. 2007 Dec;1772(11-12):1225–35. doi: 10.1016/j.bbadis.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 15.Chan SS, Longley MJ, Copeland WC. Modulation of the W748S mutation in DNA polymerase {gamma} by the E1143G polymorphism in mitochondrial disorders. Hum Mol Genet. 2006 Nov 6;15:3473–83. doi: 10.1093/hmg/ddl424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turnbull DM, Bone AJ, Bartlett K, Koundakjian PP, Sherratt HS. The effects of valproate on intermediary metabolism in isolated rat hepatocytes and intact rats. Biochem Pharmacol. 1983 Jun 15;32(12):1887–92. doi: 10.1016/0006-2952(83)90054-0. [DOI] [PubMed] [Google Scholar]
- 17.Ishikura H, Matsuo N, Matsubara M, Ishihara T, Takeyama N, Tanaka T. Valproic acid overdose and L-carnitine therapy. Journal of analytical toxicology. 1996 Jan-Feb;20(1):55–8. doi: 10.1093/jat/20.1.55. [DOI] [PubMed] [Google Scholar]
- 18.Aires CC, Ruiter JP, Luis PB, ten Brink HJ, Ijlst L, de Almeida IT, et al. Studies on the extra-mitochondrial CoA -ester formation of valproic and Delta4 -valproic acids. Biochim Biophys Acta. 2007 Apr;1771(4):533–43. doi: 10.1016/j.bbalip.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 19.Kernochan LE, Russo ML, Woodling NS, Huynh TN, Avila AM, Fischbeck KH, et al. The role of histone acetylation in SMN gene expression. Hum Mol Genet. 2005 May 1;14(9):1171–82. doi: 10.1093/hmg/ddi130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.