

Abstract
Damage to an epithelial surface disrupts its mechanical and immunologic barrier function and exposes underlying tissues to a potentially hostile external environment. Epithelial restitution occurs quickly to re-establish the barrier, and comprises a major part of the immediate host response to injured tissue. Pathways involving transforming growth factor beta and activation of epidermal growth factor receptor are both of critical importance, although cross-pathway interactions have been poorly characterized. Agent-based modeling has been demonstrated to be useful in integrating disparate bodies of knowledge and demonstrating the dynamic consequences of pathway structures and cellular population behavior, and is used herein to create an in-silico analog of an in-vitro scratch assay. The In-Vitro Scratch Agent-Based Model (IVSABM) consists of agents representing individual epithelial cells in a simulated extracellular matrix. Agents sense signals from the damaged environment and produce effector molecules, leading to their healing behavior. The IVSABM qualitatively matched wound healing dynamics when compared against data from traditional experiments. Putative crosstalk mechanisms were then instantiated into the IVSABM and their relative plausibility examined, suggesting interaction at the receptor tyrosine kinase level. This highlights the utility of dynamic knowledge representation in the integration of pathways previously studied in separate contexts.
Keywords: agent-based modeling, epithelial restitution, EGFR, TGF-β
INTRODUCTION
Damage to an epithelial surface disrupts its mechanical and immunologic barrier function and exposes underlying tissues to a potentially hostile external environment1. Repair of the epithelial defect involves restitution, proliferation and differentiation. The first of these processes, restitution (re-apposition of opposing sides of the damaged epithelium), occurs quickly and independently of cell proliferation to re-establish the barrier, and comprises a major part of the immediate host response to injured tissue2-5. A thorough understanding of the mechanisms governing restitution is of critical importance in the study of a myriad of disease processes involving loss of epithelial barrier function, from inflammatory bowel disease to corneal injury to chronic, non-healing wounds. The signaling pathways involved in epithelial restitution have largely been studied on an individual-pathway basis, most notably transforming growth factor beta (TGF-β) and epidermal growth factor receptor (EGFR). These two canonical pathways are each recognized as critical and necessary components of epithelial cell restitution; however, cross-pathway interactions between the two have been poorly characterized. Developing a coherent picture of epithelial restitution requires integrating the disparate knowledge bases concerning these pathways. Mathematical and computational models represent useful translational tools to help synthesize and integrate such knowledge. Dynamic knowledge representation through computational modeling and simulation can augment traditional investigational studies by generating and instantiating novel hypotheses, integrating otherwise disparate information, and bridging gaps in the current knowledge base6. Since the biological systems being examined are generally robust over a wide range of conditions, a qualitative approach to model development using relatively abstracted descriptions of the biological processes can often provide usefully sufficient representations of overall system behavior. Dynamic knowledge representation has been described and validated as a means of “conceptual model verification” in various biological contexts6.
Agent-Based Modeling (ABM) is an object-oriented, rule-based, discrete-event computational modeling technique well suited for dynamic knowledge representation and conceptual model verification. Agents representing individual system components encapsulate computational rules based on published knowledge of their intra- and inter-component interactions. A population of agents is simulated in a “virtual world” representing an in-silico experimental model7. ABMs for dynamic knowledge representation often contain a significant amount of component-level molecular and mechanistic detail to examine qualitative behaviors, while using only modular abstractions and estimations for system kinetics. Agent rules in the ABM are often written as conditional (“if-then”) statements, which is well suited for the representation of hypotheses generated by traditional basic science research. ABMs have been previously used to study a wide array of biological processes including sepsis8 and inflammatory cell trafficking9. ABM rules can include stochastic components that allow individual agents to have different behavioral trajectories yielding differential population-level dynamics within each run of the ABM. When viewed in aggregate an overall view of generalized system behavior often emerges. These characteristics of ABM make it well suited to the study of epithelial restitution, allowing the researcher to visualize behaviors at a level of resolution not currently possible with traditional techniques.
Previous computational models of epithelial wound healing have focused largely on mathematical models of growth characteristics and cell behavior10. ABMs are useful adjuncts to these types of models by yielding more general dynamic information about agent and system behavior and allowing for instantiation of specific mechanistic hypotheses. In order to investigate the dynamics of epithelial restitution in this context, we developed an ABM in NetLogo11 that is an in-silico analog of an in-vitro scratch assay, in which a confluent epithelial monolayer is scratched with a pipette tip or other instrument to create a linear defect and restitution dynamics are evaluated by time-lapse microscopy12. We term this model the In-Vitro Scratch ABM (IVSABM), and it consists of agents representing individual epithelial cells (IEC agents) moving in a simulated extracellular matrix (SECM). Note that by using a mucosal epithelial cell line we are focusing on the healing of mucosal epithelial deficits, where restitution is the primary process involved. We recognize that epidermal/keratinocyte healing processes are more complex (involving proliferation and differentiation), and plan to add additional functional modules/components as our research progresses. Rules governing IEC agent behavior in the IVSABM were extrapolated from known signaling pathways in the literature, allowing system dynamics to be examined and validated against data from traditional experiments. We used the IVSABM to propose hypothetical crosstalk mechanisms whereby TGF-β signals serve to augment the necessary EGFR signals that lead to healing. We suggest possible points of regulation, specifically at the receptor tyrosine kinase level, and propose and examine specific laminin-laminin interactions through simulation of the extracellular matrix as it forms with varying proportions of stable-type and healing-type laminins. This provided additional insight into the effects of such interactions on the dynamics of epithelial restitution.
METHODS
IVSABM Implementation
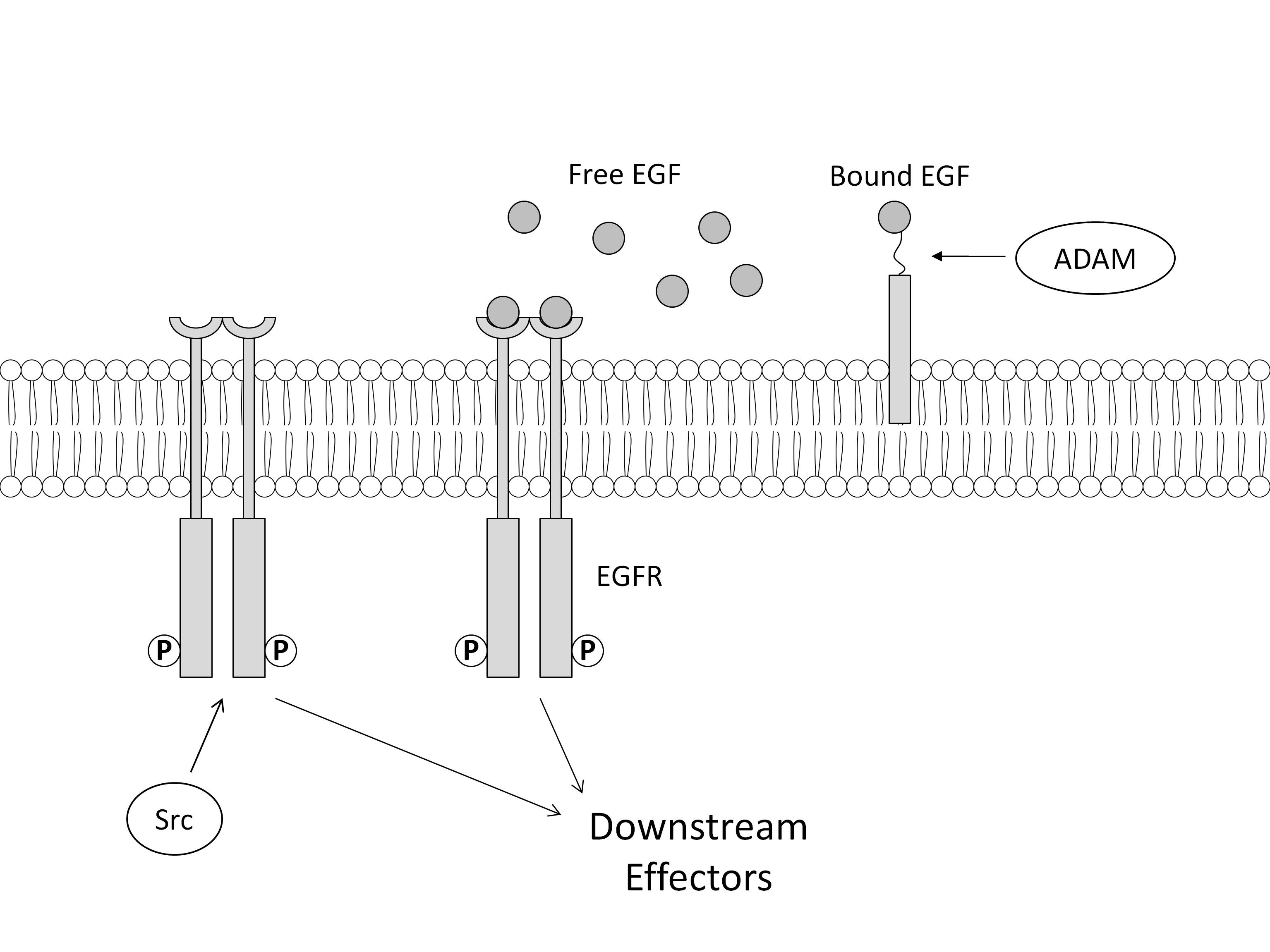
Development of the IVSABM required collecting and integrating the widespread published data describing mechanisms of epithelial signaling and processes associated with restitution, specifically those involving migration, cell spreading, and cell-matrix interactions. The following section will be divided into brief descriptions of the rules for IEC agent behavior, the properties of the SECM, overall scheduling of the IVSABM, and putative mechanisms of TGF-β-EGFR crosstalk which were examined using the model. An overall schematic of the molecular mechanisms included in the IVSABM can be seen in Figure 1. More detailed descriptions of the implemented mechanisms can be seen in Supplement 1.
Figure 1. Aggregate mechanism of IEC signaling in the IVSABM.

Pathways were extracted from published literature, and programmed as computational rules for the behavior of individual epithelial cell (IEC) agents in the model. Input signals (DAMPs, ROS, ATP, Adenosine, left) stimulate IECs to signal through intracellular pathways involving ERK 1/2 to EGFR. EGFR is also activated by free EGF in media and autocrine signaling through ligand shedding. Downstream effectors including Rac1, mTOR, FAK and paxillin lead to IEC spreading and migration. TGF-β effects govern local extracellular matrix laminin concentrations.
Individual Epithelial Cell (IEC) Agent Rules
IEC agent rules include representations of the activation and transduction of EGFR and TGF-β, and external behaviors manifest as movement and spreading rules. The primary events have been divided into the following steps:
Rules for EGFR activation and signaling. EGFR is activated by intracellular signaling cascades resulting from IEC stimulation by damage signals in the simulated media. EGFR activation occurs by both ligand-dependent and -independent mechanisms. Downstream signaling from EGFR leads to production of effector molecules for healing behaviors.
Rules for ERK 1/2 signaling. ERK 1/2 acts a key signaling molecule, acting as a nexus between the various stimulatory damage signals and EGFR, as well as between EGFR and downstream effectors. ERK is critical in maintaining the forward feedback loop in signaling propagation.
Rules for secondary signaling for propagation of restitution: The roles of extracellular ATP and adenosine. ATP and adenosine released by damaged tissue at the wound edge can acvtivate the ERK 1/2 pathway via binding to their respective receptors. ATP is converted to adenosine by CD73, which is upregulated in simulated hypoxia.
Rules for simulated extracellular matrix effects: The role of laminins. The SECM is composed of two laminin sub-types, LM-511 and LM-332. LM-511 acts as the major structural component of the SECM, delivering pro-stability signals to IECs. Conversely, LM-332 is secreted at the wound edge, renders the SECM more plastic, and conveys pro-motility signals to IECs.
Rules for epithelial cell migration and spreading: The link to cytoskeletal components. Signaling via EGFR and integrin-laminin interactions at the cell surface lead to pro-motility effectors that confer the healing phenotype on IECs. Specifically, expression of Rac1, FAK and paxillin lead to migration behavior while mTOR leads to cell spreading.
Each of these processes involves an extensive series of molecular components (as depicted in Figure 1); for a detailed description of these components and their interactions see Supplement 1. When compiled together, these multiple signaling pathways yield an extraordinarily complex overall picture of restitution mechanics.
Overall Architecture of the ABM
Computational agents representing individual epithelial cells (IECs) were used to populate an in-silico analog of an in-vitro epithelial cell culture (Figure 2A). Agents were created on a simulated background of culture media, containing soluble factors with which the agents interact. The resultant arrangement represents a confluent epithelial monolayer, with each agent mapped to a distinct space (patch) on the grid. Intracellular proteins and cell surface receptors were assigned to agents (Table 1), and diffusible factors were similarly assigned to patches (Table 2). IECs interact with their environment via surface receptors, and secrete factors that then become patch variables. IECs have intact tight junctions with all of their immediate neighboring cells. Overall model logic is outlined in Figure 3.
Figure 2. Epithelial restitution in the IVSABM.
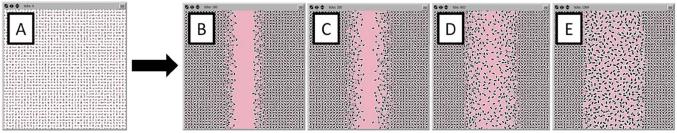
The intact simulated monolayer is represented in panel A. At the beginning of each run, there is a linear defect across the center of the IEC monolayer representing a scratch wound (B). As the model progresses, IECs migrate inwards to close the wound space until the two sides are re-approximated (C-E).
Table 1.
Agent variables in the IVSABM. A summary of the variables encoded in each IEC in the IVSABM. This includes membrane bound receptors which can interact with diffusible variables in the environment, as well as intracellular molecules which act as signaling intermediates and effectors.
| IEC Agent Variable | Description | Effect in IVSABM |
|---|---|---|
| EGFR | Membrane-bound receptor | Binds EGF, leads to effectors of motility, cell spreading |
| TLR-4 | Membrane-bound receptor | Binds DAMPs/ROS, activates MEK/ERK pathway |
| P2Y | Membrane-bound receptor | Binds ATP, activates PLC/PKC pathway |
| A2B | Membrane-bound receptor | Binds adenosine, activates PI3K pathway |
| TGF-β-R | Membrane-bound receptor; available only in absence of tight junctions |
Binds TGF-β, leads to LM-332 deposition in SECM |
| Integrin-α3β1 | Membrane-bound receptor; induced by wounding |
Binds LM-332, activates Rac1 |
| Integrin-α6β4 | Membrane-bound receptor | Binds LM−511, activates RhoA |
| CD73 | Membrane-bound receptor; upregulated 10-fold by HIF-1α |
Converts ATP to adenosine |
| Intracellular kinases (MEK, ERK, PLC, PKC, PI3K, Src, Akt, SMAD) |
Signaling intermediates | Lead to various pathways and effector molecules |
| HIF−1α | Transcription factor; induced by hypoxia |
Upregulation of CD73 |
| NFκB | Transcription factor | Leads to EGFR expression |
| ADAM-17 | Metalloprotease | Releases membrane-bound EGF into media |
| FAK/Paxillin | Effectors | Form a complex to induce simulated lamellopodial protrusion |
| mTOR | Effector | Cell spreading |
| Rac1 | Effector; induced by integrin- α3β1 |
Pro-motility |
| RhoA | Effector; induced by integrin- α6β4 |
Pro-stability |
Table 2.
Extracellular (patch) variables in the IVSABM. A summary of the variables encoded to the extracellular environment in the IVSABM. These variables include diffusible mediators and components of the SECM.
| Patch Variable | Description | Effect in IVSABM |
|---|---|---|
| DAMPs/ROS | Induced by wounding | Binds to TLR-4 |
| ATP | Induced by wounding | Binds to P2Y |
| Adenosine | Converted from ATP by CD73 in hypoxia | Binds to A2B |
| TGF-β | Induced by ROS | Activates LM-332 secretion |
| LM-511 | Matrix component; constitutive | Leads to Rho-A |
| LM-332 | Matrix component; induced by TGF-β | Leads to Rac1 |
| EGF | Growth factor; present in serum-based media and released from membrane by ADAM-17 |
Binds to EGFR |
Figure 3. Logic of the IVSABM.

IEC agents receive inputs from their environment in the form of diffusible damage-associated variables, SECM components and neighboring cells. On sensing these inputs IECs carry out signaling, activation and secretion processes in order to effect behaviors including migration and spreading.
At baseline conditions representing the intact monolayer, IECs were assigned the following receptors in an unbound form: EGFR, P2Y, TLR-4, CD73, A2B, and integrins α3β1 and α6β4 (see Supplement 1 and Table 1). With regard to the SECM, LM-511 was assigned to each patch with an IEC. EGF was added to the media in the case of experiments where “serum-based” media or “EGF-enriched” media is used. LM-332 was not assigned to any patches at baseline. LM-511 was secreted by IECs and degraded by patches to simulate a low baseline turnover level. A “scratch” was then introduced, coded as a linear defect of cells and matrix across the center of the grid (Figure 2B). IECs at the wound edge were considered to be “damaged” and able to elaborate ROS, DAMPs, and ATP into the media as stimulatory variables for IECs. These elements can diffuse freely throughout the media during simulated experiments. Matrix was removed from the wound space along with the IEC agents upon scratching.
During each run, IECs signal through their surface receptors based on the presence of available stimulatory ligand on the patch they occupy. Bound receptors are no longer able to interact with ligands, but after signaling to intracellular components they are re-constituted at the cell surface in an unbound form to restart the pathway if further ligand is available. Ligand is removed from the patch on binding to the appropriate receptor. Intracellular signaling cascades were modeled as 1:1 interactions, where downstream molecules are assigned a value equal to their corresponding upstream molecule with each tick. Intracellular molecules were degraded at a set rate to simulate the action of phosphorylases and other degradative pathways. Pathway redundancy was integrated by allowing similar molecules to interact with those molecules directly downstream in multiple pathways. Cell spreading was implemented as an increase in the 2-dimensional “size” of the epithelial cell, coded as an increase in size with each tick, limited by and never reaching a maximum diameter of pi, the largest a spherical cell (radius = 1 unit) could theoretically achieve with flattening; in practice the greatest spreading seen in the simulation was ~ 70% maximal flattening. “Migrating” IECs move onto adjacent patches not currently occupied by an IEC (randomly determined if there is more than one empty adjacent patch), leading to re-population of the damaged area of the grid. As IECs move away from their neighbors tight junctions are broken, and then re-formed when they come back in contact with other IECs. This allows for further TGF-β signaling by those IECs without a full complement of tight junctions, and consequently increased LM-332 production and secretion. Migrating IECs become “stopped” when they reach a distance of greater than 1 patch from any neighbor and are able to restart once the trailing cells had closed the gap. This simulates the “sheet-like” movement behavior of cell monolayers observed in culture (Figures 2C-E). The IVSABM is considered to be “healed” when IECs from one side of the scratched monolayer become apposed with cells from the opposite side and signaling mechanisms cease (Figure 2E).
The time scale of the IVSABM is such that ‘virtual time’ represents a distinct amount of real time, in that 1 “tick” of the system corresponds to approximately 1 minute. The model was successfully calibrated such that the condition using serum-based media (described below), which takes approximately 24 hrs (1440 min) to heal in-vitro, heals in approximately 1300-1400 ticks.
In IVSABM laminins were coded as patch variables, but in order to further study laminin interactions a second ABM was constructed in NetLogo at a finer, molecular level of resolution. In this ABM, agent variables representing the full length LM-511 and the truncated LM-332 were introduced into a grid. LM-511 agents were coded to aggregate and form a matrix-like lattice based on the presence of other LM-511 agents in their immediate neighborhood. LM-332 agents were coded to move about randomly, but to interfere with LM-511-511 interactions when in close proximity. Simulations were performed with LM-511 alone, as well as with LM-332 in order to study the ability of the system to form a stable, regular matrix and the dynamics of LM-332 inhibition of this process.
Process of model development and calibration
Once the basic rule structures are determined (as noted above) further model development involves an iterative process by which the model parameters undergo refinement. Such refinement falls into two categories: refinement of model structure (i.e. what are the forms of the rules being included), and refinement of model parameters (i.e. adjusting the constants involved in implementing the rules). Generally, the simplest, most limited and most widely accepted rules are used as the starting point. Then the parameters of the rules are adjusted during the refinement process to see if the model can be made to produce recognizable behaviors: this process is termed calibration. If this can be done with parameters that are plausible (for example, a non-plausible parameter would be one that causes a migrating epithelial cell to move at a non-plausible rate) then we consider this to be a plausible, minimally sufficient model, and then subsequent in-silico experiments can be performed using this ABM. If, however, the model cannot be made to generate a set of recognizable behaviors (as is often the case particularly in the earlier stages of development), the “model structure” is insufficient/incomplete, and more mechanisms/rules need to be invoked. To a great degree, this is the art of making a computational model, one that very closely mimics the overall process of biomedical research. Each iteration of the ABM represents a “hypothesis” that when deemed to be insufficient, requires tailoring to attempt to explain the data. However, the key here is that all of these adjustments and refinements occur during the construction/validation/calibration stage of ABM building; once the ABM has been “accepted” as trustworthy and in-silico experiments intended to test a new hypothesis are performed, the core internal structure of the ABM cannot be changed (as described below).
Putative mechanisms of TGF-β-EGFR pathway cross-talk
To study interactions between EGFR and TGF-β based signaling pathways using the IVSABM, Rac1 was identified as a common downstream effector of the two pathways, and its activity was used to evaluate restitution. We proposed and implemented 3 hypothetical mechanisms for interaction and control between the two pathways at the level of integrin-EGFR cross-phosphorylation and activation (Figure 4) to examine their respective plausibility. Mechanism 1: LM-332 binding to integrin α3β1 leading to intracellular signaling through Src; this rule was used for the baseline model. Mechanism 2: Direct signaling from LM-332 binding to an as-yet unidentified receptor to Rac1 independent of EGFR activity. Mechanism 3: Extracellular binding of an EGF-like domain of LM-332 directly to EGFR. The rationale for this potential mechanism is due to the recognition that post-translational processing of LM-332 uncovers an EGF-like sequence which has been shown to be capable of independently binding EGFR13. Each of these three mechanisms was implemented separately, and the pattern of restitution resulting from each was evaluated against behavior of the reference in-vitro scratch assay.
Figure 4. Putative mechanisms of TGF-β and EGFR crosstalk in epithelial cell signaling.

TGF-β signaling proceeds through SMADs, leading to deposition of laminins in the extracellular matrix. Thereafter, signaling can lead to Rac1 though 3 proposed mechanisms: (1) interaction of laminins with cell surface integrins leading to intracellular (ligand-independent) phosphorylation of EGFR through Src kinase, (2) direct Rac1 activation independent of EGFR, or (3) direct binding of proteolytically cleaved EGF-like fragments of LM-332 to EGFR and subsequent Rac1 activation.
RESULTS
Observed System Dynamics
The IVSABM qualitatively reproduced epithelial wound healing dynamics when compared to in-vitro studies. Activation of epithelial cell via EGFR expression and signaling as described led to cell migration and spreading in order to heal the damaged area of the model (Figures 2B-E). Temporally, the DAMP/ROS signal reaches its peak intensity first, at approximately 20 minutes (Figure 5). This corresponds to an initial burst of EGFR production and autocrine signaling via released EGF. This is closely followed by peak of ATP signal, which maintains its intensity until approximately 100 minutes. Once the ligand source has been exhausted, these signals fade out and return to zero (Figure 5). Thereafter, the EGFR signal is maintained by integrin-laminin interactions through Src. When the amount of EGFR available for downstream signaling is examined there is an early peak corresponding to the DAMP and ROS signals, a second peak corresponding to the initial burst of LM-332 secretion, and then a subsequent decline to a new steady level until the cells reach “confluence” and discontinue their production of LM-332. At this point the remaining stimulus for EGFR production is effectively extinguished and degradation of EGFR brings its value to zero (Figure 6, Letter A). Alternatively, when the laminin-integrin effect is removed from the code there is an overall reduction in the peak of EGFR signal, no second steady level of EGFR, and the system fails to heal (Figure 6, Letter B).
Figure 5. Damage signals in the IVSABM.

DAMPs/ROS in the simulated media reach peak levels approximately 20 minutes following wounding, and then are quickly exhausted as they bind to their receptors and return to zero. ATP reaches its peak slightly later at 100 minutes, and returns to zero thereafter. Representative curves are displayed.
Figure 6. EGFR activation in the IVSABM.

Under baseline conditions EGFR levels quickly increase and reach three distinct peaks, corresponding to damage signals, initial laminin-332 burst, and steady-state, respectively (Letter A). When the laminin-integrin effect is removed from the code the overall EGFR level is decreased and the steady-state is not reached (Letter B). Representative curves are displayed.
Calibration and initial validation of the ABM
The IVSABM was calibrated to known rates of restitution from in-vitro studies, in which the rates of epithelial migration were examined in the presence and absence of serum-based and serum-free media and the model parameters tuned to produce qualitatively similar behavior. Wound closure was measured as percent wound healed using 25 iterations of the model in each condition, and compared to reported data from Reference 14 (Figure 7). In serum-free media, closure percentages were 31 ± 1.1% at 12h, and 37 ± 1.2% at 18h in the IVSABM. This is comparable to closure percentages of 28% at 12h and 38% at 18h observed in reference serum-free in-vitro experiments14. In serum media, percent wound healed was measured at 56 ± 0.9% at 12h and 68 ±1.3% at 18h in the IVSABM, compared to 57% and 82% for the reference serum in-vitro data14. Each value for the IVSABM was statistically significant from initial wound size and from all other values (p < 0.0001). Rule parameter values remained unchanged for all subsequent in-silico experiments, barring differences associated with implementing the set of proposed hypothetical mechanisms and specific experimental conditions (as explained below). All in-silico experiments were conducted using the serum-based media condition.
Figure 7. Calibration of IVSABM restitution rates to known in-vitro data.

Wound healing was measured as percent of wound healed at 12 and 18 hours in both serum-based and serum-free media (n = 25), and compared to reference data from Kheradmand et al (1994), specifically wound closure percentages from the “Image Analysis System Wound Closure Assay” experiments 14 (*Note: there are no error bars from the reference data set as they are reported only as percentages). In serum media, IVSABM wounds were 56 ± 0.9% healed at 12h and 68% ± 1.32% healed at 18h, compared to 57% and 82% respectively in the reference data. In serum-free media, IVSABM wounds were 31 ± 1.1% healed at 12h and 37 ± 1.2% healed at 18h, compared to 28% and 38% respectively in the reference data. Each data pair showed a significant difference between the 2 timepoints (p < 0.0001).
Initial validation examined the necessity of EGFR for healing in the IVSABM. This behavior was selected given that EGFR activation is absolutely required for epithelial healing in-vitro, and the same condition must therefore be met in the IVSABM15,16. When an “EGFR knockout” was implemented by removing activation of the EGFR variable the healing capacity of the IVSABM was indeed completely abolished.
TGF-β and EGFR Signaling Crosstalk
Rac1 was identified as the common downstream molecule for potential interactions between EGFR and TGF-β based signaling mechanisms (Figure 4). Implementation of Mechanism 1, whereby interaction of LM-332 with integrin α3β1 leads to EGFR through Src kinase and EGFR, led to mean Rac1 levels of 7726.34 ± 153.72 units. Deletion of TGF-β led to a significant reduction to 4100 ± 0 units (p < 0.0001), and deletion of EGFR led to complete abolition of Rac1 expression (p < 0.0001) (Figure 8). Observation of IVSABM simulation runs revealed that in the TGF-β (−) condition fewer IECs became activated and produced Rac1 (i.e. gained the capacity to migrate) compared to baseline, while in the EGFR (−) condition no cells were able to do so.
Figure 8. Rac1 levels with knockout of TGF-β and EGFR in the IVSABM.

TGF-β and EGFR were each knocked out with alterations in the code, and maximum Rac1 levels examined. When compared to baseline conditions, deletion of TGF-β led to significantly decreased max Rac1 levels from 7726.34 ± 153.72 to 4100 ± 0 units (* p < 0.0001), and deletion of EGFR led to complete abolition of Rac1 expression (** p < 0.0001) (n=100 iterations).
Implementation of Mechanism 2 (direct signaling from integrin-α3β1 to Rac1) resulted in significant elevation of Rac1 levels to 47210.34 ± 829.11 units. When Mechanisms 1 and 2 were implemented together, there was a synergistic increase in Rac1 levels seen to 57246.52 ± 879.78 units. When Mechanism 3 was implemented, the wound was unable to close, with Rac1 levels at 1322 ticks peaking at 4179.59 ± 22.02 units (Figure 9). Each of these differences was statistically significant (p < 0.0001). With regard to time to wound closure, when compared to Mechanism 1, neither Mechanism 2 (1321.11 ± 16.87 minutes, p = 0.1136) nor Mechanisms 1 and 2 together (1324.4 ± 1607 minutes, p = 0.8050) were significantly different (data not shown). This suggests that although Rac1 levels were increased with the inclusion of Mechanism 2 (both alone and in conjunction with Mechanism 1), overall epithelial healing dynamics were not changed.
Figure 9. Rac1 levels on instantiation of putative mechanisms of TGF-β-EGFR pathway crosstalk in the IVSABM.

Mechanisms 1-3 were instantiated into the IVSABM (refer to Figure 4). When compared to mechanism 1, mean peak Rac1 levels were elevated in mechanism 2 from 7726.34 ± 153.72 units to 47210.34 ± 829.11 units (p < 0.001). Mechanisms 1 and 2 together yielded a synergistic increase in Rac1 levels seen to 57246.52 ± 879.78 units vs. mechanism 1 and vs. mechanism 2 individually. Mechanism 3 yielded peak Rac1 levels of 4179.59 ± 22.02 units. Each of these differences was statistically significant (p < 0.0001) when compared to all other groups (n = 100 iterations).
Matrix Dynamics: Laminins 332 and 511
LM-511 is present at the beginning of each run and is both degraded and secreted at a low constant rate. LM-332 is secreted by IECs in response to TGF-β, leading to an exponential rise in LM-332 to a peak at 400-500 ticks (400-500 minutes). Patches close to the wound edge tend to have higher relative LM-332 concentrations than those within the monolayer. A second peak of LM-332 occurs at approximately 1000 ticks. Once IECs make sufficient contacts with the opposing edge IECs in the wound, TGF-β signaling is shut off and LM-332 concentrations fall to zero (Figure 10A).
Figure 10. SECM laminin subtype levels in the IVSABM.

(A) LM-511 maintains a fairly steady level throughout each run. LM-332 levels are exponentially increased to a peak at approximately 400-500 minutes, followed by a second peak at 1000 minutes. LM-332 levels fall below LM-511 and eventually to zero at the end of the run. Representative curves are displayed. (B) Simulation of spontaneous matrix assembly in the presence of only LM-511 molecules (white crosses); LM-511 is able to form a regular lattice. (B) When LM-332 is added (blue crosses), the matrix assembly is disrupted at the edge but maintains its lattice structure throughout its bulk.
The second, molecular level ABM was run with varying amounts of LM-511 and LM-332. In the absence of LM-332, LM-511 molecules are able to form a stable matrix-like lattice (Figure 10B). In order to recreate the situation seen in the scratch wound, LM-511 was allowed to form a lattice on one side of the grid, and LM-332 was introduced at the free edge. As the simulation proceeds, LM-332 predominates at the edge of the matrix, which becomes disorganized, while LM-511 predominates in the bulk of the matrix with maintenance of a stable lattice structure. With increasing amounts of LM-332, the matrix becomes increasingly disrupted and a gradient develops with increasing levels of LM-332 and resultant matrix disorganization nearer to the wound edge (Figure 10C). In the IVSABM the same gradient developed. This distribution led to propagation of the migratory phenotype of epithelial cells from the wound edge inward, and subsequently to healing of the system.
DISCUSSION
Restitution is the process through which epithelial cells re-establish barrier function following wounding, and involves cell spreading and migration but not differentiation or proliferation4,5,14. This work focuses specifically on mechanisms of mucosal epithelial restitution, and although the complete wound healing picture is more complex and involves other cell types and processes such as proliferation, understanding epithelial restitution is of critical importance to the study of many disease states, as well as healthy wound healing dynamics as a whole. Knowledge concerning the multiple cell signaling pathways and cell-matrix interactions involved is scattered throughout the literature and presents a challenge to attempts to produce a cohesive picture of the restitution process. ABM is a useful means of integrating disparate mechanistic knowledge, and here we describe an ABM that integrates known mechanistic components of epithelial restitution and computationally recreates a healing epithelial monolayer in an in-vitro system.
Using mechanistic molecular details as rules for behavior, IECs were able to signal using cues from the external environment and produce effector molecules leading to a migratory phenotype. Cells are initially stimulated by the presence of DAMPs, ROS, and ATP. This signal acts to “kick start” the system by leading to an initial surge of EGFR signaling. This signal fades out quickly though, by about 200 minutes. Subsequently EGFR is maintained at a relatively constant level by a signaling loop from laminin-integrin interactions. This seems reasonable, since it is then easily shut off when the number of neighboring cells reaches a sufficient such that TGF-β signaling turns off and the cell is able to revert to a non-migratory phenotype (i.e. contact inhibition). With regard to TGF-β signaling, computational knockout leads to reduction in the total amount of Rac1 in the system and decreased restitution. Although in this minimally sufficient signaling model the cells are unable to migrate under knockout conditions (since laminin secretion is based solely on TGF-β), it seems reasonable to assume that there are other factors which contribute to laminin deposition in-vitro which would allow for migration, and TGF-β serves to augment the EGFR signal necessary for healing.
With all of the IECs accurately signaling and interacting with one another and the environment, the IVSABM is qualitatively able to accurately represent a healing in-vitro epithelial culture. Simulated cells from opposing sides of the monolayer migrate in a sheet-like pattern inwards through the described intracellular, cell-cell and cell-matrix interactions, and once the injury is healed cells lose their migratory capacity and the model stops.
Also consistent with in-vitro models, loss of EGFR in the ABM leads to complete abolition of wound healing. In the computational code, signals must go through EGFR in order to reach the effector molecules for migration (Rac1) and spreading (mTOR). This must in fact be the case in-vitro as well; otherwise there would be some minimal level of healing seen through pathways which circumvent EGFR. As such, the IVSABM presents at least a minimally sufficient and plausible overall mechanism for epithelial restitution.
An examination of the interactions between the extracellular matrix and cell surface integrins was of particular interest in constructing the IVSABM. It is well described that TGF-β is intimately involved in deposition of matrix at the leading edge of a healing wound17, and this may be a key link to EGFR based mechanisms of signaling. We hypothesize several mechanisms of TGF-β-EGFR crosstalk, each of which produces unique outcomes in the model. Mechanism 1, in which LM-332 interactions with integrin α3β1 lead to EGFR through Src, produces accurate wound healing dynamics, suggesting that produced levels of Rac1 are sufficient for healing. Furthermore, this satisfies the condition that EGFR must be absolutely required since there is no way for these cells to signal directly from integrins to Rac1. Mechanism 2, where integrin signals lead directly to Rac1, also produces accurate healing dynamics but with substantially higher levels of Rac1. However, this does not satisfy the EGFR requirement, which suggests that while integrin signaling leads to pro-motility effectors, there must be an intermediate step involving EGFR. Therefore Mechanism 2 is deemed implausible by not fitting with known observations and can be discarded. Mechanism 3, where a fragment of LM-332 binds directly to EGFR, does not lead to complete wound healing in the ABM. This mechanism satisfies the EGFR requirement, but at levels of LM-332 which are normally sufficient to allow healing there is insufficient activation of EGFR downstream pathways. Since the overall behavior resulting from Mechanism 3 does not match the reference experimental behavior, it too can be deemed implausible and also be discarded. Given these findings, Mechanism 1 seems to be the most consistent with current knowledge.
Interactions among the ECM laminins LM-332 and LM-511 are relatively poorly characterized to this point. It is known is that LM-511 is able to self-polymerize into a stable matrix18,19. When the molecular-level ABM is run using only LM-511, this is in fact the case. The proteolytically processed LM-332 is thought to interfere with LM-511 self-assembly, and this phenomenon was accurately represented in this model. As the system runs, LM-511 is still able to form a stable matrix at in the area not at the wound edge. However, LM-332 at the edge intercalates itself between the LM-511 molecules, and a “transitional” area emerges where the matrix is more loosely organized and in continuous flux. This seems a reasonable mechanism for laminin organization in a healing wound: LM-332 is more predominant at the wound edge, and a more plastic matrix would be more conducive to cell migration. When LM-332 production is shut off, LM-511 again predominates in relative abundance and the cells are stabilized in their new positions on the matrix. Although relatively coarse in detail, again this could represent a minimally sufficient mechanistic explanation of extracellular matrix dynamics.
The IVSABM represents the first computational, integrative representation of the two canonical signaling pathways, TGF-β and EGFR, involved in epithelial restitution. The IVSABM sufficiently reproduced the dynamics of a healing in-vitro epithelial monolayer, and allowed the instantiation of putative mechanisms for TGF-β-EGFR pathway crosstalk. In-silico experiments in the IVSABM allowed examination of the plausibility of those hypotheses, with results that suggest that signaling through Src is the point of crosstalk between TGF-β and EGFR. While this hypothesis requires further experimental testing for confirmation, we see the role of such in-silico experiments as increasing the efficiency of laboratory investigations by eliminating implausible hypotheses and helping to focus the direction of subsequent laboratory experiments. Going forward, given its modular design, we anticipate that the IVSABM will serve as a framework to which much detail can be added, specifically related to proliferation and differentiation and the role of progenitor cell lines20, and serve a useful adjunct for future inquiry into possible mechanisms of wound healing.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 11.

ACKNOWLEDGEMENTS
Funding support for this work was provided by: NIH P50 2P50GM053789-13, NIDRR H133E070024 and NSF 0830-370-V601. The authors would like to acknowledge the members of the Alverdy Lab for intellectual contributions in the construction of the model. We would also like to thank the University of Chicago, Department of Surgery, for funding this resident research. Disclosure: GA serves as a consultant for Immunetrics, Inc.
Abbreviations
- ABM
(Agent-Based Model)
- ATP
(Adenosine Triphosphate)
- DAMP
(Damage Associated Molecular Pattern)
- EGF
(Epidermal Growth Factor)
- EGFR
(Epidermal Growth Factor Receptor)
- ERK
(Extracellular signal Regulated Kinase)
- FAK
(Focal Adhesion Kinase)
- IEC
(Individual Epithelial Cell)
- IVSABM
(In-Vitro Scratch Agent-Based Model)
- LM
(Laminin)
- mTOR
(Mammalian Target of Rapamycin)
- Rac1
(Ras-related C3 botulinum toxin substrate 1)
- ROS
(Reactive Oxygen Species)
- SECM
(Simulated Extracellular Matrix)
- TGF-β
(Transforming Growth Factor-Beta)
REFERENCES
- 1.Podolsky D. Mucosal immunity and inflammation. V. Innate mechanisms of mucosal defense and repair: the best offense is a good defense. Am J Physiol Gastrointest Liver Physiol. 1999;277:G495–G499. doi: 10.1152/ajpgi.1999.277.3.G495. [DOI] [PubMed] [Google Scholar]
- 2.Mammen J, Matthews JB. Mucosal repair in the gastrointestinal tract. Crit Care Med. 2003;31(8 Suppl):S532–537. doi: 10.1097/01.CCM.0000081429.89277.AF. [DOI] [PubMed] [Google Scholar]
- 3.Iizuka M, Konno S. Wound healing of intestinal epithelial cells. World J Gastroenterol. 2011;17(17):2161–2171. doi: 10.3748/wjg.v17.i17.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sturm A, Dignass AU. Epithelial restitution and wound healing in inflammatory bowel disease. World J Gastroenterol. 2008;14(3):348–353. doi: 10.3748/wjg.14.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dignass A, Podolsky DK. Cytokine modulation of intestinal epithelial restitution: central role of transforming growth factor beta. Gastroenterology. 1993;105(5):1323–1332. doi: 10.1016/0016-5085(93)90136-z. [DOI] [PubMed] [Google Scholar]
- 6.An G. Dynamic knowledge representation using agent-based modeling: ontology instantiation and verification of conceptual models. Methods Mol Biol. 2009;500:445–468. doi: 10.1007/978-1-59745-525-1_15. [DOI] [PubMed] [Google Scholar]
- 7.Bonabeau E. Agent-based modeling: methods and techniques for simulating human systems. Proc Natl Acad Sci USA. 2002;99(Suppl 3):7280–7287. doi: 10.1073/pnas.082080899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.An G. Agent-based computer simulation and sirs: building a bridge between basic science and clinical trials. Shock. 2001;16(4):266–273. doi: 10.1097/00024382-200116040-00006. [DOI] [PubMed] [Google Scholar]
- 9.Bailey A, Thorne BC, Peirce SM. Multi-cell agent-based simulation of the microvasculature to study the dynamics of circulating inflammatory cell trafficking. Ann Biomed Eng. 2007;35(6):916–936. doi: 10.1007/s10439-007-9266-1. [DOI] [PubMed] [Google Scholar]
- 10.Walker D, Wood S, Southgate J, Holcombe M, Smallwood R. An integrated agent-mathematical model of the effect of intercellular signalling via the epidermal growth factor receptor on cell proliferation. J Theor Biol. 2006;242(3):774–789. doi: 10.1016/j.jtbi.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 11.Wilensky U. 1999 NetLogo. http://ccl.northwestern.edu/netlogo/. [Google Scholar]
- 12.Liang C, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2(2):329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 13.Schenk S, Hintermann E, Bilban M, Koshikawa N, Hojilla C, Khokha R, Quaranta V. Binding to EGF receptor of a laminin-5 EGF-like fragment liberated during MMP-dependent mammary gland involution. J Cell Biol. 2003;161(1):197–209. doi: 10.1083/jcb.200208145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kheradmand F, Folkesson HG, Shum L, Derynk R, Pytela R, Matthay MA. Transforming growth factor-α enhances alveolar epithelial cell repair in a new in vitro model. Am J Physiol. 1994;267:L728–L738. doi: 10.1152/ajplung.1994.267.6.L728. (Lung Cell Mol Physiol 11) [DOI] [PubMed] [Google Scholar]
- 15.Block E, Matela AR, SundarRaj N, Iszkula ER, Klarlund JK. Wounding induces motility in sheets of corneal epithelial cells through loss of spatial constraints: role of heparin-binding epidermal growth factor-like growth factor signaling. J Biol Chem. 2004;279:24307–24312. doi: 10.1074/jbc.M401058200. [DOI] [PubMed] [Google Scholar]
- 16.Tokumaru S, Higashiyama S, Endo T, Nakagawa T, Miyagawa JI, Yamamori K, Hanakawa Y, Ohmoto H, Yoshino K, Shirakata Y, Matsuzawa Y, Hashimoto K, Taniguchi N. Ectodomain shedding of epidermal growth factor receptor ligands is required for keratinocyte migration in cutaneous wound healing. J Cell Biol. 2000 doi: 10.1083/jcb.151.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moyano J, Greciano PG, Buschmann MM, Koch M, Matlin KS. Autocrine TGF-{beta}1 Activation Mediated by Integrin {alpha}V{beta}3 Regulates Transcriptional Expression of LM-332 in Madin-Darby Canine Kidney (MDCK) Epithelial Cells. Mol Biol Cell. 2010 doi: 10.1091/mbc.E10-06-0523. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng Y, Champliaud MF, Burgeson RE, Marinkovich MP, Yurchenco PD. Self-assembly of laminin isoforms. J Biol Chem. 1997;272:31525–31532. doi: 10.1074/jbc.272.50.31525. [DOI] [PubMed] [Google Scholar]
- 19.Hamill K, Kligys K, Hopkinson SB, Jones JC. Laminin deposition in the extracellular matrix: a complex picture emerges. J Cell Sci. 2009;122(Pt 24):4409–4417. doi: 10.1242/jcs.041095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walter M, Wright KT, Fuller HR, MacNeil S, Johnson WE. Mesenchymal stem cell-conditioned medium accelerates skin wound healing: an in vitro study of fibroblast and keratinocyte scratch assays. Exp Cell Res. 2010;316(7):1271–1281. doi: 10.1016/j.yexcr.2010.02.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.