

Abstract
Chronic kidney disease (CKD) is a prevalent life-threatening disease frequently associated with hypertension, progression to renal fibrosis and eventual renal failure. While the pathogenesis of CKD remains largely unknown, an increased inflammatory response is known to be associated with the disease and has long been speculated to contribute to disease development. However, the causative factors, the exact role of the increased inflammatory cascade in CKD and the underlying mechanisms for its progression remain unidentified. Here we report that interleukin-6 (IL-6) expression levels were significantly increased in the kidneys collected from CKD patients and further elevated in CKD patients characterized with hypertension. Functionally, we determined that angiotensin II (Ang II) is a causative factor responsible for IL-6 induction in the mouse kidney and that genetic deletion of IL-6 significantly reduced hypertension and key features of CKD including renal injury and progression to renal fibrosis in Ang II-infused mice. Mechanistically, we provide both human and mouse evidence that IL-6 is a key cytokine functioning downstream of Ang II signaling to directly induce fibrotic gene expression and preproendothelin-1 (prepro-ET-1) mRNA expression in the kidney. Overall, both the mouse and human studies reported here provide evidence that Ang II induces IL-6 production in the kidney and that, in addition to its role in hypertension, increased IL-6 may play an important pathogenic role in CKD by inducing fibrotic gene expression and ET-1 gene expression. These findings immediately suggest the IL-6 signaling is a novel therapeutic target to manage this devastating disorder affecting millions worldwide.
Keywords: Renin Angiotensin System, Cytokines, Hypertension, Chronic Kidney Disease, Endothelin-1
Introduction
Chronic kidney disease (CKD) is a life-threatening condition frequently associated with hypertension, renal dysfunction, progression to renal fibrosis and eventual chronic renal failure (CRF) 1-4. It affects twenty-six million American adults and is the 9th leading cause of mortality in the United States5. Available strategies used to manage CKD are poor and currently limited to dialysis or kidney transplantation, thus making CRF one of the most expensive diseases to treat on a per-patient basis3. Despite improvement in the knowledge of diverse aspects related to CKD, the pathogenesis and the initial molecular events leading to the chronic renal fibrosis and eventual CRF remain elusive. By understanding the molecular basis of the pathogenesis of CKD, we will identify novel therapeutic targets to treat this harmful disease and prevent its progression.
It has been long speculated that pathogenesis of chronic renal fibrosis likely results from a combination of prolonged hypoxia, ischemic-mediated inflammatory response, vascular damage and attempted tissue repair6, 7. A growing body of evidence supports a novel concept that elevated inflammation contributes to the pathogenesis of CKD.6 For example, several recent studies have shown that patients with hypertension and CKD exhibit high levels of diverse proinflammatory cytokines8-12. Some hypothesize that the activation of leukocytes and upregulation of certain cytokines propagate a state of chronic inflammation in CKD patients which likely contributes to progression of the disease9, 13. Among the cytokines identified, IL-6 is a multifunctional proinflammatory cytokine that is associated with a number of cardiovascular disorders including CKD with or without hypertension and pulmonary vascular disease.14, 15 Thus, IL-6 is now considered a major biomarker for cardiovascular risk16. A critical role of IL-6 in pathogenesis of various forms of CKD was suggested by both human and animal studies. Of note, elevated IL-6 levels in the serum of patients with CKD with or without hypertension have been reported16. Significantly, angiotension II (Ang II)-induced hypertension is attenuated in IL-6-deficient mice17-19 and knocking down IL-6 by RNA interference blocks cold-induced hypertension.20 Notably, elevated IL-6 also contributes to hypertension in pregnant rats with chronic reductions in uterine perfusion21. These findings strongly support the essential role of IL-6 in hypertension. However, specific factors to induce IL-6 elevation in CKD are unknown, the exact contribution of this cytokine to the disease features remains unclear and the molecular mechanisms underlying progression to renal fibrosis in CKD is still elusive. Here we sought to investigate causative factors responsible for the elevation of IL-6, the role of IL-6 in the pathogenesis of CKD, underlying mechanisms for the disease and potential therapeutic strategies.
Materials and Methods
An expanded Methods section is available in the online Data Supplement (please see http://hyper.ahajournals.org)
Human subjects
IL-6 expression levels were measured in kidney biopsies collected from normal control individuals (n=15), CKD patients associated with hypertension (n=38) and without hypertension (n=28). CKD Patients admitted to First XiangYa Hospital were identified by the nephrologists of the Central South University at Changsha, Hunan, China. Normal individuals were selected on the basis of having normal blood pressure and kidney function with acute kidney rupture. The research protocol was approved by the Central South University Ethics Committee for the Protection of Human Subjects. The normal individuals and patients’ clinical data are listed in Table 1.
Table 1.
Information of control individuals and CKD patients
| Information | Control (N=15) | CKD Without Hypertension (N=28) | CKD With Hypertension (N=38) |
|---|---|---|---|
| Age (Years) | 41.7±14.1 | 46.4±13.5 | 49.8±12.5 |
| Gender (M/F) | 9/6 | 16/12 | 24/14 |
| SBP (mmHg) | 106.1±12.6 | 109.3±14.3 | 155.4±19.5* |
| DBP (mmHg) | 74.1±13.5 | 72.3±10.5 | 95.4±12.3* |
| BUN (mmol/L) | 4.56±1.07 | 5.82±1.13 | 8.29±2.47* |
| Bcr (μmol/L) | 70.2±23.34 | 69.36±28.84 | 117.67±29.50* |
| eGFR (ml/min) | 84.73±19.65 | 78.32±16.51 | 53.77±19.78* |
| UA (μmol/L) | 287.86±88.79 | 278.67±93.86 | 348.16±111.70 |
compared with controls, p<0.05. Kidneys of controls with normal kidney function and blood pressure were collected after 3 hours of acute kidney rupture.
SBP: systolic blood pressure; DBP: diastolic blood pressure; BUN: blood urea nitrogen; Bcr: blood creatinine; eGFR: estimated glomerular filtration rate; UA: uric acid
Animals
Wide type 8-10 week old C57BL/6 mice were purchased from Harlan Laboratories (Indianapolis, IN). IL-6-deficient mice (IL-6-/-) congenic on a C57BL/6 background were generated and genotyped as described.22 The mice were housed in the animal care facility of the University of Texas, Houston and had access to food and water ad libitum. All the protocols involving animal studies were reviewed and approved by the Institutional Animal Welfare Committee of the University of Texas Houston Health Science Center. 6-10 mice for each group were used.
Immunohistochemistry for IL-6 in kidney biopsies of human and quantification
Immunohistochemistry for IL-6 was carried out similar to ET-1 (for details see online supplement) except that the slides were incubated with rabbit anti-human IL-6 antibody (Lifespan, 1:100 dilution) in a humidified chamber at 4°C overnight. After the primary antibody incubation, anti-rabbit IgG ABC-AP staining system kit (VECTOR LAB, USA) was used. Slides were stained with vector Red alkaline phosphatase substrate (VECTOR LAB, USA) after being washed in PBS for 5 min, washed in PBS and DAB (BIOS) for 5 min and counterstained with hematoxylin. Vector Red produces a red reaction product that can be seen using either bright field or fluorescent microscopy. For negative controls, the primary antibody was replaced with the corresponding affinity-purified pre-immune IgG. Quantification of the immunohistochemical staining was performed using the Image-Pro Plus software (Media Cybernetics, Bethesda, MD). The density of the red staining (positive for IL-6) was measured. The average densities of 25 areas per kidneys were determined and the SEM is indicated. n=6 for each group.
Chronic Ang-II infusion
Mice were infused with vehicle (saline) or Ang II at a rate of 1.5 mg/kg body weight per day into the mice by osmotic minipumps (Alzet model 2001; Alza, Palo Alto, CA) subcutaneously for 2-weeks as described23.
Blood Pressure
We used two methods to measure systolic blood pressure. First, systolic blood pressure was measured by a carotid catheter-calibrated tail-cuff system (CODA, Kent Scientific, Torrington, CT) before and after minipump implantation as described.23-25 In addition to the tail cuff system to measure blood pressure in live animals, we also measured blood pressure in anesthetized animals21. Specifically, on the final day of Ang II infusion the intracarotid mean arterial blood pressure was measured in the mice after anesthesia with isoflurane (2%). The carotid artery was isolated and cannulated with a PE-50 microtip catheter. The intracarotid mean artery blood pressure was measured with a pressure transducer connected to a Grass Model 7B chart recorder (AD Instrument Co, USA). Blood pressure was recorded and averaged over a 10-minute period.
Urine analysis
Twenty four hour urine was collected on different days from mice with or without Ang II infusion using a metabolic cage (Nalgene) as previously described.23-25
Enzyme Immunometric Assay (EIA) for mouse kidney ET-1
The kidneys were homogenized with an Ultrasonic homogenizer (Model W-220F, Heat systems Ultrasonics) for 60 s in 10 volumes of NP-40 lysis buffer containing protease inhibitor cocktail (Roche Diagnostics, Germany). The homogenate was centrifuged at 12,000 g for 15 min at 4°C and the supernatant was stored at -80°C until it was used. The protein concentration was measured by BCA assay. About 400μl kidney protein was placed into a micro-centrifuge tube containing same volume of 20% acetic acid, the mixture was centrifuged at 3000 g for 10 min at 4°C, and the supernatant was kept. Subsequently, ET-1 was extracted with a Sep-Pak C-18 cartridge (Honeywell, USA). Finally, the elutants were reconstituted with 0.25 mL of assay buffer and subjected to sensitive Enzyme Immunometric Assay (EIA) for ET-1 (Life Science, USA). The ET-1 level was normalized to protein concentration.
Statistical analyses
All data were expressed as the mean ± SEM. Data were analyzed for statistical significance using GraphPad Prism 4 software (GraphPad Software, San Diego, CA). Student’s t tests (paired or unpaired as appropriate) were applied in two-group analysis. Differences between the means of multiple groups were compared by the one-way analysis of variance (ANOVA), followed by a Tukey’s multiple comparisons test. A value of P < 0.05 was considered significant and was the threshold to reject the null hypothesis.
Results
IL-6 expression levels are increased in the kidneys of CKD patients and further elevated in CKD patients with hypertension
To determine whether IL-6 is increased in the kidneys of CKD patients, we examined IL-6 expression profiles in kidney biopsies collected from normal controls (n=15) and CKD patients with (n=38) and without hypertension (n=28) (see Table 1 for detailed information of human subjects). We found that the IL-6 expression level was low in the kidneys of normal control individuals. However, IL-6 levels were elevated in both glomeruli and tubules of kidney biopsies isolated from CKD patients with or without hypertension (Figure 1A). Quantitative image analysis demonstrate that increased IL-6 staining in the kidneys of CKD patients was significantly higher than controls and that IL-6 levels were further elevated in CKD patients with hypertension (Figure 1B). These studies show that elevated IL-6 is associated with CKD.
Figure 1. IL-6 expression level is elevated in the kidneys of CKD patients and is further increased in CKD patients with hypertension.
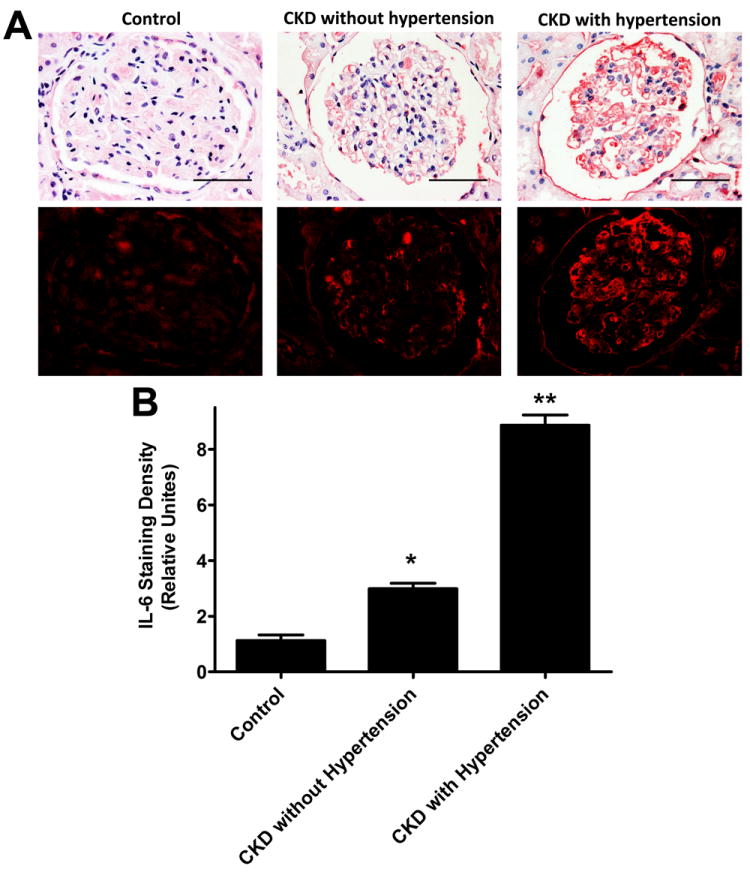
(A) Immunohistochemistry study of IL-6 expression in control individuals with acute kidney rupture, CKD patients without and with hypertension. Microscopic examination revealed that IL-6 expression was significantly elevated in glomeruli and tubules of kidneys of CKD patients without hypertension and additionally elevated in CKD patients with hypertension. Scale bar=400 μM. (B) Semiquantitative analysis of IL-6 expression levels in the kidney biopsies from controls and CKD patients with or without hypertension. The average densities ± s.e.m. of 25 areas per kidney were determined. n = 15-48 kidneys for each category. * p<0.05 compared to control with acute kidney rupture, ** p<0.05 compared to CKD without hypertension.
IL-6 deficiency attenuates Ang II-induced proteinuria in mice
Because it is difficult to determine the role of increased IL-6 in CKD patients, we took advantage of IL-6 deficient mice. To assess the significance of increased IL-6 in the pathophysiology of CKD associated with hypertension, we chose to infuse Ang II, a potent vasoconstrictor known to induce hypertension and renal damage and dysfunction,26, 27 key features seen in humans with CKD, into both wild type and IL-6-deficient mice. First, we found that IL-6 mRNA expression levels were significantly elevated in the kidneys of mice following a two week-Ang II infusion compared to controls with saline infusion (Figure 2A). Next, we assessed renal injury in each group during the two week Ang II-infusion by measuring albumin content in 24-hour collected urine. We found that the ratio of urinary albumin to creatinine in 24-hour collected urine was elevated in the Ang II-infused mice compared to that of the controls by day 7 and reached a significantly higher level by day 14 of continuous Ang II infusion (Figure 2B). In contrast, the increased proteinuria was significantly attenuated in IL-6 deficient mice on day 14 of Ang II-infusion (Figure 2C). Thus, these findings provide the in vivo evidence that IL-6 is a mediator of Ang II induced kidney damage featured with proteinuria in an intact animal.
Figure 2. Elevated IL-6 mRNA in the kidneys contributes to proteinuria in Ang II-infused mice.

Wild type (WT) and IL-6-deficient mice were infused with Ang II for 2 weeks. (A) IL-6 mRNA was elevated in the kidneys of Ang II-infused mice after two week Ang II infusion. (B-C) Albumin and creatinine concentrations were measured in 24-hour collected mouse urine of the WT and IL-6-deficient mice on day 7 (B) and day 14 (C) with or without Ang II infusion. Data are expressed as mean ± s.e.m. (n = 6). *P < 0.05 versus WT mice. **P < 0.05 versus Ang II-infused WT mice.
IL-6 contributes to progression of renal fibrosis in Ang II-infused mice
One of the major features associated with CKD is renal fibrosis. To determine the critical role of elevated IL-6 in the progression of renal fibrosis, histological studies were conducted to characterize the renal fibrosis in each group of mice described above on day 14 of Ang II-infusion. Analysis of hematoxylin-eosin stained sections from Ang II-infused mice revealed extensive renal damage (Figure 3A). The majority of the glomeruli seen in these mice demonstrated decreased Bowman’s space, decreased capillary lumen and mesangial hypercellularity (Figure 3A, Top Panel). Masson’s Trichrome staining showed significant fibrosis in both glomeruli and interstitial areas between tubules (Figure 3A, Lower Panel). Quantitative image analysis showed significantly increased collagen staining in kidneys of Ang II-infused mice (Figure 3B). Consistent with histological studies, total collagen measurements of the kidneys of Ang II-infused mice were significantly elevated (Figure 3C). In contrast, the Ang II-induced renal fibrosis, collagen staining and total collagen content was significantly reduced in IL-6-deficient mice (Figure 3A-C). Taken together, these results demonstrate that increased IL-6 contributes to Ang II-induced renal fibrosis.
Figure 3. IL-6 promotes chronic renal fibrosis in Ang II-infused mice.

(A) H&E and Trichrome staining showed that Ang II-infused mice displayed kidney damage and fibrosis on day 14 of Ang II infusion. Genetic deletion of IL-6 significantly reduced vascular damage and fibrosis. Scale bars=200μm. (B) Quantitative image analyses showed Ang II-infused mice exhibited increased collagen staining in the kidney. Genetic deletion of IL-6 significantly decreased collagen staining in the kidney of Ang II-infused mice. (C) Collagen content in the kidney was significantly increased in Ang II-infused mice. Genetic deletion of IL-6 significantly decreased collagen content in the kidneys of Ang II-infused mice. Data are expressed as mean ± s.e.m. (n=6). *P < 0.05 versus WT mice. **P < 0.05 versus Ang II-infused WT mice.
Ang II-induced hypertension is dependent on IL-6 and occurs prior to proteinuria and kidney fibrosis
In addition to renal injury and fibrosis, we also monitored hypertension (a key feature associated with CKD) in the Ang II-infused mice. We found that in wild type mice systolic blood pressure began to increase by day 3, increased significantly by day 7, reached a peak by day 10 and continuously maintained high blood pressure through the end of two week Ang II-infusion (Figure 4A). Thus, these findings demonstrate that Ang II-induced hypertension occurred prior to proteinuria and subsequent renal fibrosis which were significantly induced in wild type mice after 14 days of Ang II-infusion (Figure 2B). Next, we found that genetic deletion of IL-6 in mice led to a significant reduction of Ang II-induced hypertension (Figure 4A). Consistent with tail cuff measurements, we showed that IL-6 deficiency significantly attenuated Ang II-induced intracarotid mean arterial pressure in anesthetized mice on day 14 of infusion with Ang II (Figure 4B). Taken together, these results provide direct evidence that increased IL-6 plays an important role in Ang II-induced hypertension and the associated proteinuria and progression to renal fibrosis.
Figure 4. Ang II-induced hypertension is dependent on IL-6 in mice.

(A-B) Systolic blood pressure was measured at different time points in wild type (WT) and IL-6-deficient mice with or without Ang II infusion. (B) Intracarotid mean arterial blood pressure was monitored in WT and IL-6-deficient mice with or without Ang II infusion on day 14. * P<0.05 versus WT. ** P<0.05 versus Ang II infused WT mice. n=6 for each group.
Elevated IL-6 is responsible for increased fibrotic mediator gene expression in the kidneys of Ang II-infused mice
To determine whether IL-6-mediated renal fibrosis in Ang II-infused mice is associated with an induction of genes encoding fibrotic mediators, we measured gene expression profiles of the renal tissue of both wild type and IL-6-deficient mice with or without Ang II-infusion. We found that the expression of fibrotic marker genes including pro-collagen I, TGF-β and Pai-1 mRNAs were significantly increased in the kidneys of Ang II-infused mice (Figure 5A-C). In contrast, genetic deficiency in IL-6 significantly decreased the elevated profibrotic gene expression in the kidneys of Ang II-infused mice (Figure 5A-C). These results reveal that Ang II-induced renal fibrosis is associated with increased expression of fibrotic marker genes in the kidney.
Figure 5. IL-6 contributes to Ang II-induced expression of fibrotic marker genes in the mouse kidney.
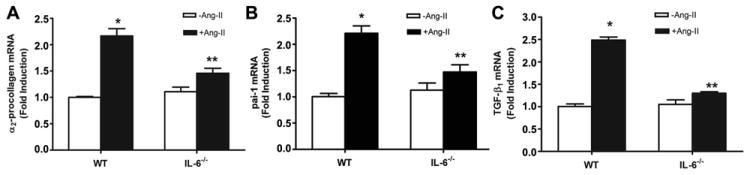
(A-C) The expression of fibrotic marker genes in kidneys of wild type (WT) and IL-6-deficient mice with or without Ang II infusion. Data are expressed as mean ± s.e.m. (n = 4-5). *P < 0.05 versus WT mice. **P < 0.05 versus WT mice with Ang II infusion.
IL-6 underlies increased preproET-1 mRNA levels and ET-1 production in the kidneys of Ang II-infused mice
Endothelin-1 (ET-1) is a 21-amino acid peptide and a key mediator of vascular tone and renal function. Elevated ET-1 signaling is associated with hypertension, proteinuria, and kidney fibrosis 28-31,32 and Ang II is known to induce its production.33, 34 However, the molecules functioning downstream of Ang II responsible for ET-1 induction and subsequent hypertension and kidney damage are largely unidentified. Because we found that IL-6 played an important role in Ang II-induced hypertension, renal injury and fibrosis, it is possible that elevated IL-6 in the kidney may underlie the induction of ET-1 by Ang II. To test this hypothesis, we first measured preproET-1 mRNA levels in the kidneys of both wild type mice and IL-6-deficient mice with or without Ang II infusion. We found that the expression of preproET-1 was significantly increased in the kidneys of Ang II-infused mice (Figure 6A). Additionally, using a sensitive ELISA, we confirmed that Ang II-induced preproET-1 mRNA led to an elevated ET-1 production in the mouse kidneys with Ang II (Figure 6B). Consistently, immunostaining revealed that ET-1 was highly expressed in endothelial cells and epithelial cells of renal tubules (Figure 6C). Image quantification indicated that ET-1 levels were significantly elevated in both glomeruli and tubules of Ang II-infused mice (Figure 6D). Strikingly, genetic deletion of IL-6 significantly decreased preproET-1 mRNA levels and ET-1 levels in the kidneys of Ang II-infused mice (Figure 6A). These results demonstrate that IL-6 is a downstream signaling molecule that contributes to Ang II-mediated induction of elevated ET-1 production in the kidney.
Figure 6. IL-6 contributes to increased ET-1 production in kidneys of Ang II-infused mice.
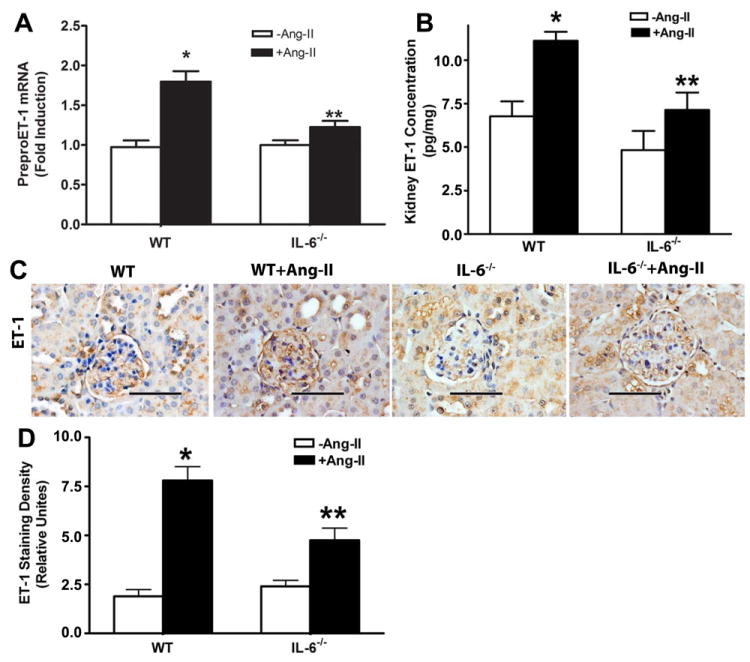
Wild type (WT) and IL-6-deficient mice were infused with Ang II for 2 weeks. (A) The expression of preproET-1 mRNA in the kidneys of wild type (WT) and IL-6-deficient mice with or without Ang II infusion. (B) ET-1 levels in the kidneys of WT and IL-6 deficient mice with or without Ang II infusion. (C) Immunohistochemistry study of ET-1 expression in the kidneys of WT and Il-6-deficient mice with or without Ang II infusion. Scale bar=400 μM. (D) Quantitative imagine analysis of ET-1 expression levels in the kidneys from WT and IL-6-deficient mice with or without Ang II infusion. The average densities (± s.e.m.) of 25 areas per kidney were determined. n = 15-48 kidneys for each category.
*p<0.05 compared to WT without Ang II infusion, ** p<0.05 compared to WT with Ang II infusion.
IL-6 contributes to Ang II-induced expression of profibrotic marker genes and the preproET-1 gene in cultured mouse kidney explants
To determine if IL-6 is a potential mediator of Ang II-induced fibrosis and preproET-1 gene expression in the kidney, we performed experiments using kidney organ cultures. Specifically, we isolated kidneys from wild type mice and IL-6-deficient mice and incubated renal explants in the presence or absence of Ang II for 24 hours. We found that Ang II directly induced Pai-1, pro-collagen I, TGF-β and preproET-1 mRNA levels (Figure 7A-D). Consistent with in vivo findings, we found that genetic deletion of IL-6 significantly abolished the Ang II-induced pro-collagen I, Pai-1, TGF-β and preproET-1 mRNA production in the kidney cultures (Figure 7A-D). Consistent with these genetic studies, we found that blockade with neutralizing antibodies for IL-6 or gp130 (co-receptor for IL-6 receptor activation), but not isotype control antibodies, significantly attenuated Ang II-induced fibrotic gene expression and preproET-1 mRNA levels in the cultured kidney explants isolated from wild-type mice (Figure 7A-D). Taken together, the studies demonstrate that the IL-6 directly contributes to Ang II-induced expression of profibrotic genes and the preproET-1 gene in cultured mouse kidneys.
Figure 7. Direct effects of IL-6 on Ang II-induced fibrotic and preproET-1 gene expression in cultured mouse kidney explants.
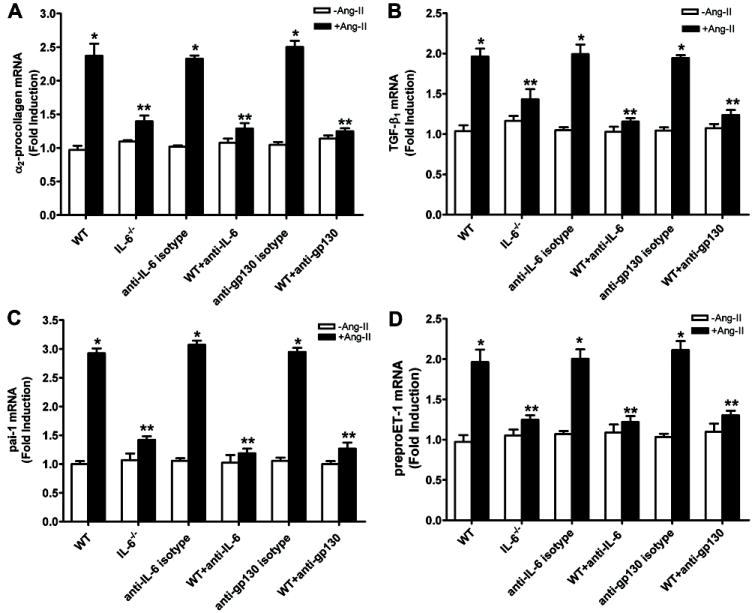
(A) Analysis of α2-procollagen (A), TGF-β (B), Pai-1 (C) and preproET-1 (D) by quantitative RT-PCR in cultured kidney explants isolated from wild type (WT) and IL-6-deficient mice in response to Ang II treatment. Some of the kidney explants from WT mice were treated with or without Ang II in the presence of absence of neutralizing antibodies to IL-6, gp130 (IL-6 co-receptor) or isotype control antibody. Data are expressed as mean ± s.e.m. *P < 0.05 versus without Ang II treatment. **P < 0.05 versus WT treated with Ang II (n = 4-6).
IL-6 is essential for Ang II-induced preproET-1 mRNA production in cultured human endothelial cells
We found that IL-6 is significantly elevated in the kidneys of CKD patients (Figure 1) and that elevated IL-6 contributes to ET-1 production in Ang II-infused mice. Furthermore, ET-1 expression was significantly elevated in the endothelial cells of the capillary lumens of kidneys of Ang II-infused mice (Figure 6B), suggesting that endothelial cells are a major cell type responsible for Ang II-induced ET-1 production. To determine if Ang II can induce ET-1 production in endothelial cells we selected human microvascular endothelial cells (HMECs) as a model in vitro system. We found that Ang II was capable of inducing IL-6 gene expression in cultured HMECs (Figure 8A). Importantly, we found that Ang II-induced preproET-1 mRNA expression was significantly attenuated by either anti-IL-6 or anti-gp130 (IL-6 coreceptor) neutralizing antibodies but not by isotype control antibodies (Figure 8B). Thus, we provide human evidence that Ang II can directly induce IL-6 expression and that elevated IL-6 is essential for Ang II-induced preproET-1 mRNA production in cultured human endothelial cells.
Figure. 8. IL-6 functions via its receptor responsible for Ang II-induced preproET-1 mRNA elevation in culture human endothelial cells.
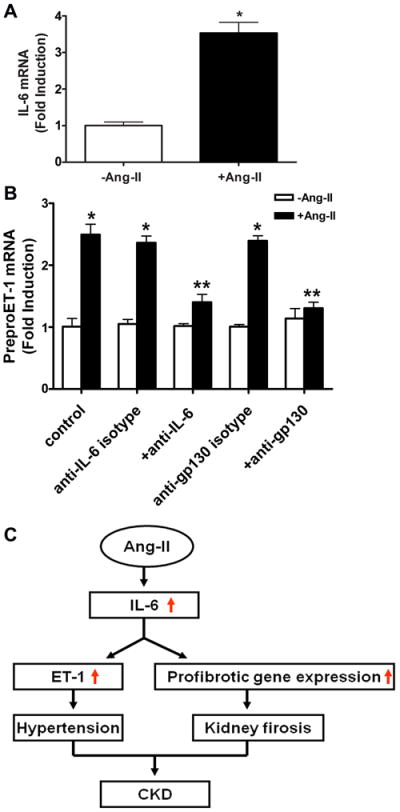
(A) Ang II increased the expression of IL-6 mRNA in cultured human microvascular endothelial cells (HMECs). (B) Ang II-mediated induction of preproET-1 mRNA in HMECs was inhibited by neutralizing antibodies to IL-6 or gp130 but not isotype control antibodies. *P < 0.05 versus without Ang II treatment. **P < 0.05 versus with Ang II treatment. (n= 4-6). (C) A model depicting the potential role of elevated IL-6 signaling in Ang II-induced CKD. Ang II stimulates increased production of IL-6. In addition to its role in hypertension, increased IL-6 may play an important pathogenic role in CKD, by directly stimulating fibrotic gene expression and preproET-1 mRNA levels via IL-6 receptor activation.
Discussion
In this study, we showed that IL-6 is elevated in the kidneys of CKD patients and its level is further elevated in CKD patients with hypertension. We provided in vivo mouse evidence that IL-6 is an important cytokine that contributes to hypertension and multiple features of CKD including proteinuria and renal fibrosis in Ang II infused-mice. Additional studies show that IL-6 functions downstream of Ang II and contributes to upregulating the expression of multiple profibrotic genes (α2-procollagen, TGF-β and Pai-1) and the preproendothelin-1 gene in the mouse kidney. Thus, in addition to the role of IL-6 in Ang II-induced hypertension, we have provided both in vivo and in vitro evidence that IL-6 contributes to Ang II induced gene expression in the kidney. Overall, both the mouse and human studies reported here provide strong evidence that Ang II stimulates increased IL-6 production and that, in addition to its role in hypertension, increased IL-6 may play an important pathogenic role in CKD by inducing fibrotic gene expression and ET-1 gene expression in the kidney.
The increased inflammatory response associated with CKD is speculated to contribute to the pathogenesis of the disease6, 9. For example, multiple in vitro studies have demonstrated that increased IL-6 production may lead to mesangial cell proliferation, leukocyte proliferation and infiltration, epithelial cell apoptosis and endothelial cell damage that are relevant to the pathophysiology of the disease35-37. However, the direct cause of the increased IL-6 production is unknown and the pathogenic role of this cytokine in CKD is undetermined. The renin angiotensin system is elevated in CKD patients2, 38, 39 and infusion of Ang II into multiple species leads to hypertension, renal injury and progression to renal fibrosis.26, 27 Here, using an Ang II-infusion mouse model of hypertension and CKD, we have shown that Ang II leads to increased IL-6 production in the mouse kidney. Next, we found that genetic deletion of IL-6 significantly attenuates Ang II-induced hypertension and multiple features associated with CKD including, proteinuria, renal injury and kidney fibrosis. In agreement with our mouse studies, we found that IL-6 levels were also elevated in the kidneys of CKD patients and that IL-6 levels were additionally elevated in the kidneys of CKD patients with hypertension. Finally, we provide evidence that Ang II can directly induce IL-6 expression in human endothelial cells and in mouse kidney explants. Thus, we have shown here for the first time that Ang II is a key mediator to induce IL-6 production in the kidney and that elevated IL-6 contributes to the enhanced expression of specific genes in the kidney that may directly contribute to the pathophysiology of CKD and progression to renal fibrosis. Consistent with earlier studies17, we showed that hypertension preceded a significant increase in proteinuria, consistent with the view that hypertension contributes to kidney injury and fibrosis. However, our results also raise the possibility of a direct effect of Ang II induced IL-6 production in the kidney contributing directly to renal injury and fibrosis. Future studies are needed to delineate the contributions of hypertension and IL-6 mediated gene expression on renal injury and fibrosis.
Although earlier studies showed that IL-6 deficiency attenuated Ang II-induced hypertension,17-19 the pathogenic mechanisms underlying these effects have not been clearly identified. Here, we showed that Ang II-induced preproET-1 mRNA expression and ET-1 protein levels in mouse kidney were significantly reduced in IL-6 deficient mice. Similarly, we found that Ang II-induced preproET-1 gene expression was significantly attenuated by IL-6 and gp-130 neutralizing antibodies in cultured human microvascular endothelial cells. Thus, both in vivo mouse studies and in vitro human studies revealed a previously unrecognized role for IL-6 in Ang II-induced ET-1 production. ET-1 is a downstream mediator known to be responsible for Ang II-induced hypertension33, 34. Thereby, our findings lead to a novel hypothesis that IL-6-mediated ET-1 elevation in the kidney contributes to Ang II-induced hypertension and renal injury seen in CKD. Our studies are supported by earlier findings showing that IL-6 deficiency attenuated Ang II-induced hypertension in non-pregnant mice17-19 and knocking down IL-6 by RNA interference ameliorated cold-induced hypertension in non-pregnant rats20. Consistently, earlier studies using pregnant rats showed that chronic infusion of IL-6 led to hypertension and reduced renal function21. However, these effects mediated by IL-6 infusion were only seen in the pregnant rats but not in virgin rats21 and did not involve induction of ET-1 in the kidneys of pregnant rats,21 suggesting that IL-6 induced-preeclamptic features in the rats are pregnancy-dependent and ET-1 independent. Nevertheless, our studies in mice provide strong genetic evidence that IL-6 is a key cytokine stimulating ET-1 production from kidney and contributes to Ang II-induced hypertension and kidney injury.
In addition to hypertension and kidney injury, renal fibrosis is a common progressive feature seen in CKD. Without interference, it will eventually lead to CRF. Thus, understanding the molecular basis of progression of renal fibrosis in CKD is extremely importantly. Using both intact animals and isolated kidney cultures, we provide new evidence for mechanisms that may underlie Ang II-induced renal fibrosis. Our in vivo and in vitro studies reveal an important role for IL-6 in Ang II-mediated induction of multiple fibrotic genes in mouse kidney. Overall, our findings show that IL-6 is an important downstream mediator of Ang II signaling and contributes not only to hypertension but also to Ang II-mediated induction of gene expression in the kidney. Specifically, IL-6 contributes to Ang II mediated induction of multiple fibrotic genes and ET-1 production in the kidneys, and in this way may contribute to renal injury and renal fibrosis (Figure 8C) The research reported here provides new insight regarding the pathogenesis of CKD and provides a foundation for future clinical trials to treat CKD, a life-threatening disorder with limited therapeutic options.
Perspectives
Taken together, our studies identified Ang II as a candidate responsible for the elevated expression of IL-6 in the kidneys of patients with CKD. Both human and mouse studies demonstrated that this inflammatory cytokine plays an important role in the pathogenesis of Ang II-induced hypertension and CKD and its progression. Of significant importance, genetic deletion or pharmacological neutralization of IL-6 reduces hypertension and renal impairment in Ang II-infused mice. In addition, Ang II-induced ET-1 production is significantly inhibited by blockade of IL-6 and its co-receptor anti-gp130 in human endothelial cells. Taken together, these findings support a central role of IL-6 in Ang II-induced hypertension and draw attention to a potentially direct contribution of IL-6 to renal dysfunction and subsequent development of renal fibrosis resulting from the induction of profibrotic genes and increased production of ET-1. These findings have revealed novel therapeutic possibilities by targeting these signaling pathways.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by National Institute of Health Grants DK077748 (to YX), DK083559 (to YX), RC4HD067977 (to YX and REK), American Heart Association Grant 10GRNT3760081 (to YX) and China Scholarship Council 2009637520 (to WRZ).
Footnotes
Disclosures
None.
References
- 1.Klahr S. Heart disease. Vol. 3. Hagerstown, Md: 2001. Progression of chronic renal disease; pp. 205–209. [DOI] [PubMed] [Google Scholar]
- 2.Khosla N, Kalaitzidis R, Bakris GL. The kidney, hypertension, and remaining challenges. The Medical clinics of North America. 2009;93:697–715. doi: 10.1016/j.mcna.2009.02.001. Table of Contents. [DOI] [PubMed] [Google Scholar]
- 3.Bidani AK, Griffin KA. Pathophysiology of hypertensive renal damage: Implications for therapy. Hypertension. 2004;44:595–601. doi: 10.1161/01.HYP.0000145180.38707.84. [DOI] [PubMed] [Google Scholar]
- 4.Ritz E. Hypertension and kidney disease. Clinical nephrology. 2010;74(Suppl 1):S39–43. [PubMed] [Google Scholar]
- 5.Kuroki A, Akizawa T. management of chronic kidney disease--preventing the progression of renal disease. Nippon rinsho. 2008;66:1735–1740. [PubMed] [Google Scholar]
- 6.Carrero JJ, Stenvinkel P. Inflammation in end-stage renal disease--what have we learned in 10 years? Seminars in dialysis. 2010;23:498–509. doi: 10.1111/j.1525-139X.2010.00784.x. [DOI] [PubMed] [Google Scholar]
- 7.Nangaku M, Fujita T. Activation of the renin-angiotensin system and chronic hypoxia of the kidney. Hypertens Res. 2008;31:175–184. doi: 10.1291/hypres.31.175. [DOI] [PubMed] [Google Scholar]
- 8.Leoncini G, Viazzi F, Pontremoli R. Chronic kidney disease and albuminuria in arterial hypertension. Current hypertension reports. 2010;12:335–341. doi: 10.1007/s11906-010-0141-3. [DOI] [PubMed] [Google Scholar]
- 9.Horl WH. Hypertension in end-stage renal disease: Different measures and their prognostic significance. Nephrol Dial Transplant. 2010;25:3161–3166. doi: 10.1093/ndt/gfq428. [DOI] [PubMed] [Google Scholar]
- 10.Campese VM, Park J. The kidney and hypertension: Over 70 years of research. Journal of nephrology. 2006;19:691–698. [PubMed] [Google Scholar]
- 11.Schiffrin EL, Lipman ML, Mann JF. Chronic kidney disease: Effects on the cardiovascular system. Circulation. 2007;116:85–97. doi: 10.1161/CIRCULATIONAHA.106.678342. [DOI] [PubMed] [Google Scholar]
- 12.Hackam DG, Spence JD, Garg AX, Textor SC. Role of renin-angiotensin system blockade in atherosclerotic renal artery stenosis and renovascular hypertension. Hypertension. 2007;50:998–1003. doi: 10.1161/HYPERTENSIONAHA.107.097345. [DOI] [PubMed] [Google Scholar]
- 13.Pecoits-Filho R, Heimburger O, Barany P, Suliman M, Fehrman-Ekholm I, Lindholm B, Stenvinkel P. Associations between circulating inflammatory markers and residual renal function in crf patients. Am J Kidney Dis. 2003;41:1212–1218. doi: 10.1016/s0272-6386(03)00353-6. [DOI] [PubMed] [Google Scholar]
- 14.Honda H, Qureshi AR, Heimburger O, Barany P, Wang K, Pecoits-Filho R, Stenvinkel P, Lindholm B. Serum albumin, c-reactive protein, interleukin 6, and fetuin a as predictors of malnutrition, cardiovascular disease, and mortality in patients with esrd. Am J Kidney Dis. 2006;47:139–148. doi: 10.1053/j.ajkd.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 15.Pecoits-Filho R, Lindholm B, Axelsson J, Stenvinkel P. Update on interleukin-6 and its role in chronic renal failure. Nephrol Dial Transplant. 2003;18:1042–1045. doi: 10.1093/ndt/gfg111. [DOI] [PubMed] [Google Scholar]
- 16.Stenvinkel P, Barany P, Heimburger O, Pecoits-Filho R, Lindholm B. Mortality, malnutrition, and atherosclerosis in esrd: What is the role of interleukin-6? Kidney international. 2002:103–108. doi: 10.1046/j.1523-1755.61.s80.19.x. [DOI] [PubMed] [Google Scholar]
- 17.Lee DL, Sturgis LC, Labazi H, Osborne JB, Jr, Fleming C, Pollock JS, Manhiani M, Imig JD, Brands MW. Angiotensin ii hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol. 2006;290:H935–940. doi: 10.1152/ajpheart.00708.2005. [DOI] [PubMed] [Google Scholar]
- 18.Coles B, Fielding CA, Rose-John S, Scheller J, Jones SA, O’Donnell VB. Classic interleukin-6 receptor signaling and interleukin-6 trans-signaling differentially control angiotensin ii-dependent hypertension, cardiac signal transducer and activator of transcription-3 activation, and vascular hypertrophy in vivo. The American journal of pathology. 2007;171:315–325. doi: 10.2353/ajpath.2007.061078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brands MW, Banes-Berceli AK, Inscho EW, Al-Azawi H, Allen AJ, Labazi H. Interleukin 6 knockout prevents angiotensin ii hypertension: Role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension. 2010;56:879–884. doi: 10.1161/HYPERTENSIONAHA.110.158071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crosswhite P, Sun Z. Ribonucleic acid interference knockdown of interleukin 6 attenuates cold-induced hypertension. Hypertension. 2010;55:1484–1491. doi: 10.1161/HYPERTENSIONAHA.109.146902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gadonski G, LaMarca BB, Sullivan E, Bennett W, Chandler D, Granger JP. Hypertension produced by reductions in uterine perfusion in the pregnant rat: Role of interleukin 6. Hypertension. 2006;48:711–716. doi: 10.1161/01.HYP.0000238442.33463.94. [DOI] [PubMed] [Google Scholar]
- 22.Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bluethmann H, Kohler G. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 1994;368:339–342. doi: 10.1038/368339a0. [DOI] [PubMed] [Google Scholar]
- 23.Zhou CC, Ahmad S, Mi T, Xia L, Abbasi S, Hewett PW, Sun C, Ahmed A, Kellems RE, Xia Y. Angiotensin ii induces soluble fms-like tyrosine kinase-1 release via calcineurin signaling pathway in pregnancy. Circulation research. 2007;100:88–95. doi: 10.1161/01.RES.0000254703.11154.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou CC, Irani RA, Zhang Y, Blackwell SC, Mi T, Wen J, Shelat H, Geng YJ, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibody-mediated tumor necrosis factor-alpha induction contributes to increased soluble endoglin production in preeclampsia. Circulation. 2010;121:436–444. doi: 10.1161/CIRCULATIONAHA.109.902890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou CC, Zhang Y, Irani RA, Zhang H, Mi T, Popek EJ, Hicks MJ, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nature medicine. 2008;14:855–862. doi: 10.1038/nm.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolf G. Angiotensin ii as a renal growth factor. Contributions to nephrology. 2001:92–110. doi: 10.1159/000060159. [DOI] [PubMed] [Google Scholar]
- 27.Kim S, Iwao H. Molecular and cellular mechanisms of angiotensin ii-mediated cardiovascular and renal diseases. Pharmacological reviews. 2000;52:11–34. [PubMed] [Google Scholar]
- 28.Simonson M, Osanai T, Wann S, Baldi E, Mene P, Nakazato Y, Kester M, Thomas C, Dunn M. Effects of endothelin on cultured human and rat glomerular mesangial cells. Contributions to nephrology. 1991;95:1–11. doi: 10.1159/000420633. [DOI] [PubMed] [Google Scholar]
- 29.Simonson MS, Dunn MJ. Endothelin peptides: A possible role in glomerular inflammation. Laboratory investigation; a journal of technical methods and pathology. 1991;64:1–4. [PubMed] [Google Scholar]
- 30.Khimji AK, Rockey DC. Endothelin--biology and disease. Cellular signalling. 2010;22:1615–1625. doi: 10.1016/j.cellsig.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 31.Kohan DE. Endothelin, hypertension and chronic kidney disease: New insights. Current opinion in nephrology and hypertension. 2010;19:134–139. doi: 10.1097/MNH.0b013e328335f91f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suga S. molecular biology in regulation of kidney functions: Endothelin. Nippon rinsho. 2006;64(Suppl 2):209–212. [PubMed] [Google Scholar]
- 33.Ge Y, Huang Y, Kohan DE. Role of the renin-angiotensin-aldosterone system in collecting duct-derived endothelin-1 regulation of blood pressure. Canadian journal of physiology and pharmacology. 2008;86:329–336. doi: 10.1139/Y08-028. [DOI] [PubMed] [Google Scholar]
- 34.Schiffrin EL. The angiotensin-endothelin relationship: Does it play a role in cardiovascular and renal pathophysiology? Journal of hypertension. 2003;21:2245–2247. doi: 10.1097/01.hjh.0000098171.36890.e2. [DOI] [PubMed] [Google Scholar]
- 35.Grassl C, Luckow B, Schlondorff D, Dendorfer U. Transcriptional regulation of the interleukin-6 gene in mesangial cells. J Am Soc Nephrol. 1999;10:1466–1477. doi: 10.1681/ASN.V1071466. [DOI] [PubMed] [Google Scholar]
- 36.Chen WP, Chen A, Lin CY. In vitro effects of interleukins on human mesangial cells: Implications for glomerulonephritis. The Journal of pathology. 1995;175:327–337. doi: 10.1002/path.1711750311. [DOI] [PubMed] [Google Scholar]
- 37.Coleman DL, Ruef C. Interleukin-6: An autocrine regulator of mesangial cell growth. Kidney Int. 1992;41:604–606. doi: 10.1038/ki.1992.91. [DOI] [PubMed] [Google Scholar]
- 38.Covic A, Gusbeth-Tatomir P. The role of the renin-angiotensin-aldosterone system in renal artery stenosis, renovascular hypertension, and ischemic nephropathy: Diagnostic implications. Progress in cardiovascular diseases. 2009;52:204–208. doi: 10.1016/j.pcad.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 39.Benavente D, Chue CD, Ferro CJ. The importance of renin-angiotensin blockade in patients with cardio-renal disease. Journal of renal care. 36(Suppl 1):97–105. doi: 10.1111/j.1755-6686.2010.00166.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.