

Abstract
Vascular cognitive impairment defines alterations in cognition, ranging from subtle deficits to full-blown dementia, attributable to cerebrovascular causes. Often coexisting with Alzheimer’s disease, mixed vascular and neurodegenerative dementia has emerged as the leading cause of age-related cognitive impairment. Central to the disease mechanism is the crucial role that cerebral blood vessels play in brain health, not only for the delivery of oxygen and nutrients, but also for the trophic signaling that links inextricably the well being of neurons and glia to that of cerebrovascular cells. This review will examine how vascular damage disrupts these vital homeostatic interactions, focusing on the hemispheric white matter, a region at heightened risk for vascular damage, and on the interplay between vascular factors and Alzheimer’s disease. Finally, preventative and therapeutic prospects will be examined, highlighting the importance of midlife vascular risk factor control in the prevention of late-life dementia.
Introduction
Age related dementia, an irreversible condition resulting in progressive cognitive decline, has emerged as one of the leading health problems of our time. Advances in prevention and healthcare have increased life expectancy and produced a shift in the burden of disease worldwide. Thus, non-communicable diseases, including dementia, have been recognized for the first time as the major threat to the world population (World Health Organization, 2012). The World Health Organization estimates that 35.6 million people live with dementia, a number that is anticipated to triple by 2050 (World Health Organization, 2012). Every year 7.7 million new cases of dementia are diagnosed, imposing a tremendous burden on families, the primary caregivers, and financial cost to society. Although recent data suggest a decline in prevalence (Matthews et al., 2013), dementia remains a devastating and costly disease. In the US such cost has already surpassed that of cancer and heart diseases (Hurd et al., 2013). The realization of its paramount public health impact has led nations, including the US, to develop national plans to cope with dementia and attempt to reduce its devastating effects (National Alzheimer’s Project Act; Public Law 111-375).
Vascular dementia, a heterogeneous group of brain disorders in which cognitive impairment is attributable to cerebrovascular pathologies, is responsible for at least 20% of cases of dementia, being second only to Alzheimer’s disease (AD) (Gorelick et al., 2011). Recent clinical-pathological studies have highlighted the role of cerebrovascular disease, not only as a primary cause of cognitive impairment, but also as an adjuvant to the expression of dementia caused by other factors, including AD and other neurodegenerative pathologies (Gorelick et al., 2011; Schneider et al., 2007a; Toledo et al., 2013). At the same time, new experimental findings have revealed a previously unrecognized functional and pathogenic synergy between neurons, glia and vascular cells (Iadecola, 2010; Quaegebeur et al., 2011; Zlokovic, 2011), providing a new framework to reevaluate how alterations in cerebral blood vessels could contribute to the neuronal dysfunction underlying cognitive impairment. These advances call for a re-appraisal of the role of vascular factors in cognitive health. To this end, the major cerebrovascular causes of cognitive dysfunction will be briefly reviewed, focusing on neuropathology, emerging mechanisms and overlap with neurodegeneration.
Dementia through the ages
In Alois Alzheimer’s time (1900s), dementia was thought to be caused predominantly by “hardening of the arteries” (arteriosclerotic dementia) (Bowler, 2007; Jellinger, 2006). Vascular factors were considered a major player in dementia well into the 20th century, until, in the 1980s, the Aβ peptide was identified as the main component of parenchymal (amyloid plaque) and vascular (amyloid angiopathy) amyloid deposits, pathological hallmarks of AD (Glenner and Wong, 1984; Kang et al., 1987). Shortly after, mutations in the amyloid precursor protein (APP) gene were identified in familial forms AD (Bertram and Tanzi, 2008). Since then, the emphasis shifted from vascular dementia to AD, a process defined as the “Alzheimerization of dementia” (fig. 1) (Bowler, 2007). However, an increasing appreciation of the impact of cerebrovascular lesions on AD brought to the forefront the importance of cerebrovascular health in cognitive function (Esiri et al., 1999; Gold et al., 2007; Snowdon et al., 1997). Furthermore, community based clinical-pathological studies revealed that the largest proportion of dementia cases have mixed pathology, comprising features of AD (amyloid plaques and neurofibrillary tangles) as well as ischemic lesions (Launer et al., 2008; Schneider et al., 2009). These developments have promoted an interest to gain a better understanding of how vascular brain lesions affect cognition, and how vascular pathology and neurodegeneration interact to amplify their respective pathogenic contribution.
Figure 1.

Changing views about dementia through the years. In the early 1900s vascular factors were thought to be the main cause of dementia. Over the next several decades Alzheimer’s disease was felt to be the main cause. Clinical-pathological studies have revealed that mixed dementia, combining feature of vascular dementia and AD, is currently the most common cause of cognitive impairment in the aged.
Defining dementia on vascular bases: From arteriosclerotic dementia to vascular cognitive impairment
The concept of dementia caused by cerebrovascular pathology has evolved considerably over the years (fig. 2). For many decades vascular dementia was attributed to sclerosis of cerebral arteries leading to diffuse ischemic injury and brain atrophy (Jellinger, 2006). The first significant departure from this concept, inspired by the work of Tomlinson and colleagues (Tomlinson et al., 1970), was proposed by Hachinski and colleagues (Hachinski et al., 1974), who suggested that dementia on vascular bases was caused by multiple and discrete ischemic lesions in patients with vascular risk factors, such as hypertension (multi-infarct dementia) (figs. 2, 3). The construct of multi-infarct dementia, by attributing cognitive impairment to multiple strokes, raised the possibility that preventing cerebrovascular diseases could also prevent dementia (Hachinski et al., 1974). The introduction of brain imaging led to the realization that diffuse white matter lesions, termed leukoaraiosis (Hachinski et al., 1987), were a frequent correlate of cognitive impairment, much more common than multiple infarcts, which turned out to be a rare cause of dementia (Hulette et al., 1997) (fig. 2, 3). Genetic causes of white matter lesions were discovered, the prototypical one being the Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) (Chabriat et al., 2009). In the early 90s the criteria for diagnosis of vascular dementia were largely based on those used for AD, which emphasized memory impairment, irreversibility of the deficits, and impaired activities of daily living (Roman et al., 1993). This definition was felt to be restrictive since it did not take into due consideration cognitive deficits more commonly associated with cerebrovascular lesions, such as executive dysfunction and psychomotor slowing (supplemental table 1). Therefore, the term vascular cognitive impairment (VCI) was introduced to better reflect the full range of cognitive alterations resulting from vascular factors (Hachinski and Bowler, 1993) (fig. 2). By doing so, it was hoped that the vascular nature of the cognitive deficit could be recognized early, providing the opportunity to slow down disease progression by controlling vascular risk factors (Hachinski and Bowler, 1993). The concept of VCI has gained wide acceptance and is currently defined as “a syndrome with evidence of clinical stroke or subclinical vascular brain injury and cognitive impairment affecting at least one cognitive domain” (Gorelick et al., 2011), vascular dementia being the most severe form of VCI.
Figure 2.
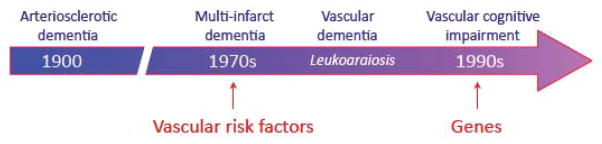
Evolution of the concept of cognitive impairment on vascular bases. Hardening of the arteries was considered the main cause in the early 1900s. The concept of multi-infarct dementia introduced the possibility of preventing dementia by controlling vascular risk factors. The introduction of brain imaging modalities (computer tomography, then magnetic resonance imaging) led to the realization that white matter disease, termed leukoaraiosis, was a major cause of cognitive impairment. In the 1990s the term VCI was introduced to broaden the spectrum of cognitive deficits caused by vascular factors. At this time, genetic causes of vascular damage causing dementia were also discovered, CADASIL being the first monogenic cause of vascular cognitive impairment, identified by M-G. Bousser and colleagues.
Figure 3.
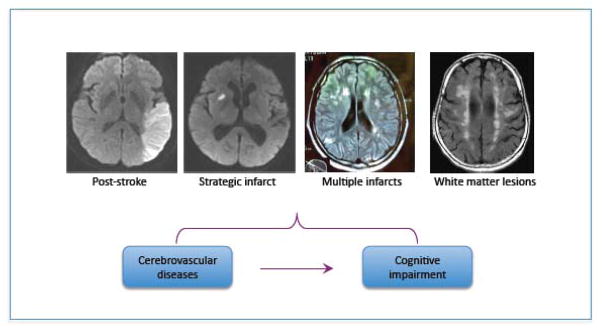
Brain lesions responsible for vascular cognitive impairment. All MRI sequences are diffusion weighted imaging, except for the white matter lesions, which is a fluid attenuated inversion recovery sequence. Images are courtesy of Dr. Hooman Kamel.
The neurovascular unit: blood flow regulation and beyond
The fundamental role that cerebral blood vessels play in the broad spectrum of pathologies underlying cognitive impairment highlights the importance of vascular structure and function in brain health. Owing to its high energy needs and lack of fuel reserves, the brain requires a continuous and well-regulated supply of blood (Iadecola, 2004). Most energy is used by neurons to fuel ionic pumps to maintain and restore the ionic gradients dissipated by synaptic activity (Harris et al., 2012). Due to fewer synapses, white matter energy usage, and consequently blood flow, is 1/3 lower of that of the gray matter (Harris and Attwell, 2012). The brain vasculature has an intimate developmental, structural and functional relationship with the brain tissue, their cellular elements forming a functional domain termed the neurovascular unit (Iadecola, 2004). Due to their intimate association with brain cells, cerebral blood vessels have unique characteristics that sets them apart from vessels in other organs (Abboud, 1981; Bevan, 1979; Quaegebeur et al., 2011). The salient structural and functional features of the cerebral circulation are briefly examined next.
The brain vascular network and neurovascular unit
The brain is supplied by arteries arising from the circle of Willis, a polygon of interconnected vessels at the base of the brain formed by the confluence of the internal carotid arteries and the basilar artery (fig. 4). The main vessels arising from the circle of Willis - the anterior middle and posterior cerebral arteries, and their branches - give rise to a rich anastomotic network on the brain surface (pial arteries and arterioles). Pial vessels are endowed with a smooth muscle cell coat, which surrounds a monolayer of endothelial cells (fig. 4). Pial arterial branches dive into the brain substance, giving rise to smaller arterioles still surrounded by an extension of the subarachnoid space filled with cerebrospinal fluid (perivascular space or Virchow-Robin space) (fig. 4). Delimited by the vascular basement membrane and the basement membrane of the glia limitans (fig. 4) (Dyrna et al., 2013), the perivascular space has emerged as critically important for the disposal of unwanted proteins and peptides, e.g., Aβ (Carare et al., 2013; Iliff et al., 2013; Laman and Weller, 2013). As intracerebral arterioles reach deeper into the brain parenchyma and become smaller (diameter <100μm), the perivascular space disappears and the vessel’s basement membrane enters in direct contact with the glial basement membrane enveloping the end-feet of astrocytes (fig. 4). In capillaries, smooth muscle cells are replaced by pericytes, contractile cells that are particularly abundant in brain vessels and are involved in the development and maintenance of the BBB (Armulik et al., 2010; Bell et al., 2010; Quaegebeur et al., 2011).
Figure 4.

Anatomy of the cerebral blood supply. A: Circle of Willis. B: The arterial supply of the deep white matter arises from branches of the ACA and the MCA. The supply of the basal ganglia white matter is provided by arteries arising directly form the circle of Willis and its proximal branches. Abbreviations: ACA: anterior cerebral artery; ICA: internal carotid artery; MCA: middle cerebral artery; PCA: posterior cerebral artery. C: Anatomy of the wall of arteries, arterioles and capillaries.
The “outside in” vascularization pattern of the brain differs from that of other major organs, like the kidney and liver that are vascularized from the “inside out”, and places key vessels regulating intracerebral blood flow, the pial arterioles, outside the brain parenchyma. Consequently, elaborate vascular signaling mechanisms coordinate changes in vascular diameter of pial arterioles on the brain’s surface with those of the intracerebral microvasculature (Bagher and Segal, 2011; Iadecola, 2004). Another consequence of this vascular arrangement is that occlusion of penetrating arterioles or venules, unlike pial vessels, cannot be effectively compensated by anastomotic branches (Blinder et al., 2013), and results in reductions in flow sufficient to produce small ischemic lesions akin to microinfarcts (Nguyen et al., 2011; Nishimura et al., 2010; Shih et al., 2013). In addition, regions of the deep white matter are supplied by long penetrating arterioles arising from the pial cortical network at the border between non-overlapping vascular territories of the anterior and middle cerebral arteries, and as such are more vulnerable to reductions in blood flow (Brown and Thore, 2011; De Reuck, 1971) (fig. 4). On the other hand, the basal ganglia and brainstem are supplied by penetrating arterioles arising directly from the circle of Willis and its proximal branches (fig. 4), rendering these vessels more susceptible to the mechanical stresses imposed by chronic hypertension or stiffening of large arteries (Scuteri et al., 2011; Sörös et al., 2013).
Neurovascular control mechanisms
The brain is endowed with vasoregulatory mechanisms that assure that it receives enough blood to support the energy needs to its cellular constituents. Consequently, neural activity, which uses most of the brain’s energy budget, is the major determinant of the dynamic regulation of CBF. The increases in blood flow induced by activation depend on the concerted action of neurons, astrocytes and vascular cells through a wide variety of molecular signals including ions, arachidonic acid metabolites, nitric oxide (NO), adenosine, neurotransmitters and neuropeptides (Drake and Iadecola, 2007). The hemodynamic changes underlying the increases in blood flow are mediated by vasoactive agents with opposing vascular actions (vasodilatation or vasoconstriction), generated by synaptic activity, astrocytes, interneurons, and afferent projections from the basal forebrain and brainstem (Cauli and Hamel, 2010; Drake and Iadecola, 2007; Kleinfeld et al., 2011). These highly coordinated signals converge on specific sites of the cerebrovascular network to shape the hemodynamic response to neural activation with a remarkable degree of spatial and temporal precision (Iadecola, 2004). Thus, the regional hemodynamic changes induced by activation are widely used to localize neuronal activity in the living brain using functional imaging (Attwell and Iadecola, 2002).
Like in other organs, endothelial cells regulate vascular tone by releasing vasoactive factors in response to chemical signals, e.g., transmitters (Andresen et al., 2006), or mechanical forces, e.g., shear stress (Ando and Yamamoto, 2013). Unlike other organs, cerebral endothelial cells in most brain regions are adjoined by intricate junctional complexes formed by occludins and claudins (tight junctions) that prevent the bidirectional exchange of hydrophilic substances between blood the brain, a key feature of the BBB (Dyrna et al., 2013). Rather, specialized transport proteins on the endothelial cell membrane control the traffic of solutes in and out or the brain. For example, GLUT1 and aminoacid transporters regulate the transfer of glucose and aminoacids into the brain, whereas “efflux transporters”, such as LRP-1, ABC transporters and others, remove drugs and metabolic by-products form the brain, including Aβ and lactate (Neuwelt et al., 2011).
Vascular smooth muscle cells, owing to their ability to constrict when intravascular pressure increases (myogenic tone), adjust vascular tone in response to changes in arterial pressure to maintain CBF relatively constant within a range of pressures (cerebrovascular autoregulation) (Cipolla, 2010). Autoregulation protects cerebral blood vessels from the wide swings in arterial pressure associated with the activities of daily living, and provides a stabile CBF baseline on which the dynamic changes induced by neurovascular coupling and endothelium are superimposed. These neurovascular control mechanisms work in concert to assure that the brain receives sufficient blood flow to meet the metabolic needs of its active cellular constituents.
Trophic coupling in the neurovascular unit
Neurons, astrocytes, oligodendrocytes, as well as vascular and perivascular cells are in state of close trophic and metabolic co-dependence that plays a defining role in brain development, function and reaction to injury. In the developing nervous system, prototypical neural guidance signals, ephrins, netrins, slit glycoproteins and semaphorins, also contribute to endothelial tip cell guidance (Carmeliet and Jain, 2011). In turn, classical angiogenic molecules, such as VEGF, participate in neurogenesis (neurovascular niche), neuronal cell migration, axon guidance, dendritogenesis, and oligodendrocyte precursor migration (Butler et al., 2010; Carmeliet and Ruiz de Almodovar, 2013; Quaegebeur et al., 2011). In the adult nervous system, neuroblasts migrate along blood vessels, a process dependent on BDNF secretion by endothelial cells (Snapyan et al., 2009). Endothelial cells have the potential to stimulate the proliferation of neuronal precursors and to stir their differentiation toward the neuronal lineage (Shen et al., 2004). Furthermore, through BDNF, insulin growth factor 2, chemokine (C-X-C motif) ligand 12, and pleiotrophin, endothelial cells support neuronal survival and protect them from injury (Dugas et al., 2008; Guo et al., 2008). Endothelial cells can also promote the proliferation and survival of oligodendrocytes (oligovascular niche) by activating the Akt/PI3 kinase pathway through BDNF and FGF (Arai and Lo, 2009). In addition to their well established interactions with neurons, astrocytes are also needed for the development and maintenance of BBB characteristics in endothelial cells (Wolburg et al., 2009), and for the reorganization of vascular networks after brain injury (Hayakawa et al., 2012). In turn, endothelial cells regulate glycolytic metabolism in astrocytes through the production of NO (Brix et al., 2012). Therefore, neurovascular cells are trophically and metabolically interdependent, such that damage to one cell type removes a vital source of support to the whole unit and has deleterious consequences also for the other cell types.
Immune trafficking and regulation
The cells of the neurovascular unit are involved in the initiation and expression of adaptive and innate immune responses of the brain. Pericytes and perivascular macrophages have the potential for antigen presentation, the first step in adaptive immunity, whereas endothelial cells and microglia are richly endowed with innate immunity receptors including CD36, toll like receptors (TLR) and the receptor for advances glycation end-products (RAGE) (Lampron et al., 2013; Park et al., 2011). The perivascular space, which drains into the subarachnoid space and then into cervical lymphnodes (Laman and Weller, 2013), is the “afferent arm” through which brain antigens reach the systemic immune system (Galea et al., 2007). The cells of the neurovascular unit also regulate the “efferent arm” of the immune system, which relies on the transfer of effector immune cells into the brain. In conditions of hypoxia-ischemia, endothelial cells express adhesion receptors, such as P-selectin, E-selectin, ICAM, VCAM, instrumental for the transfer of circulating leukocytes into the perivascular space (Iadecola and Anrather, 2011). In turn, perivascular macrophages, located in contact with the vascular basement membrane, are required for inflammatory cells, such as lymphocytes, to cross the glial basement membrane and move from the perivascular space into the brain parenchyma (Tran et al., 1998). Astrocytes express “death” ligands (CD95L) on their perivascular end feet, and control immune trafficking by triggering apoptosis of CD95+ lymphocytes attempting to enter the brain (Bechmann et al., 1999). Therefore, the neurovascular unit is an important checkpoint regulating the afferent and efferent arms of the immune system and shaping the immune responses of the brain. Vital to vascular homeostasis are circulating endothelial progenitor cells (EPC), hematopoietic stem cells involved in the maintenance and repair of endothelial cells (Hill et al., 2003). EPC development and function is controlled by CD31+ T-cells (angiogenic T-cells) through the release proangiogenic cytokines (Hur et al., 2007; Kushner et al., 2010). Thus, immune cells are also involved in the maintenance of vascular homeostasis.
Cerebrovascular pathologies underlying cognitive impairment are diverse
Considering the vital importance of the cerebral blood supply for the structural and functional integrity of the brain, it is not surprising that alterations in cerebral blood vessels have a profound impact on cognitive function. The vascular alterations that cause cognitive impairment are diverse, and include systemic conditions affecting global cerebral perfusion or alterations involving cerebral blood vessels, most commonly small size arterioles or venules (fig. 5). Table 1 describes some of the most common conditions, their vascular bases, and neuropathological correlates (see (Jellinger, 2013) for a more complete list).
Figure 5.

Vascular lesions leading to VCI and their effects on the brain. See text for details. CAA: cerebral amyloid angiopathy; ATS: atherosclerosis.
Table 1.
Selected causes of cognitive impairment related to vascular factors
| Condition | Predominant association/cause | Target vessel and vascular pathology | Resulting brain lesions | Refs. |
|---|---|---|---|---|
| Hypoperfusion dementia |
|
|
|
(Jellinger, 2013; Johnston et al., 2004; Marshall et al., 2012) |
| “Strategic infarct” dementia |
|
|
|
(Jellinger, 2013) |
| Multiinfarct dementia |
|
|
|
(Thal et al., 2012) |
| White matter lesions (Leukoaraiosis) and lacunes |
|
|
|
(Black et al., 2009; Brown and Thore, 2011; Thal et al., 2012) |
| Microinfarcts |
|
|
|
(Smith et al., 2012) |
| Microbleeds and hemorrhages |
|
|
|
(Charidimou and Werring, 2012; Henskens et al., 2008) |
| CADASIL |
|
|
|
(Chabriat et al., 2009; Federico et al., 2012; Schmidt et al., 2012) |
| Cerebral amyloid angiopathy |
|
|
|
(Attems et al., 2011; Charidimou and Werring, 2012) |
| Post-stroke dementia |
|
|
|
(Leys et al., 2005) (Iadecola and Anrather, 2011) |
| Mixed AD vascular dementia |
|
|
|
(Jellinger, 2013; Thal et al., 2012) |
Large infarct: >1cm Ø; Lacunar infarct: 5–15 mm Ø; microinfarct: <1mm Ø; microbleeds: <5mm Ø; ATS: atherosclerosis; GOM: graular osmophilic material; Vascular risk factors: hypertension, diabetes, smoking, etc.
Reduced cerebral perfusion and post-stroke dementia
Reduction in global cerebral perfusion caused by cardiac arrest, arrhythmias, cardiac failure, or hypotension can produce brain dysfunction and impair cognition transiently or permanently (table 1) (Alosco et al., 2013; Justin et al., 2013; Marshall, 2012; Stefansdottir et al., 2013). High-grade stenosis or occlusion of the internal carotid arteries is associated with chronic ischemia and can lead to cognitive impairment even in the absence of ischemic lesions (Balestrini et al., 2013; Cheng et al., 2012; Johnston et al., 2004; Marshall, 2012) (fig. 5).
On the other hand, if the reduction in CBF is sustained and severe, ischemic stroke ensues (Moskowitz et al., 2010). Stroke doubles the risk for dementia (post-stroke dementia), and approximately 30% of stroke patients go on to develop cognitive dysfunction within 3 years (Allan et al., 2011; Leys et al., 2005; Pendlebury and Rothwell, 2009). The association between stroke and dementia is also observed in patients younger than 50 years, up to 50% of whom exhibit cognitive deficits after a decade (Schaapsmeerders et al., 2013). As mentioned, multiple infarcts, caused by multiple arterial occlusions over time, are well know to impair cognition (multi-infarct dementia), as can a single infarct located in a brain regions critical for cognitive function, such as frontal lobe or thalamus (table 1) (strategic infarct dementia) (fig. 3). However, ischemic strokes are often associated with many of the vascular pathologies described below, which also contribute to the total vascular burden.
Small vessel disease, leukoaraiosis and lacunar infarcts
By far, the most prevalent vascular lesions associated with VCI are related to alterations in small vessels in the hemispheric white matter (Jellinger, 2013). These microvascular alterations result in different neuropathological lesions, which can occur in isolation but, more typically, coexist in the same brain (table 1). Confluent white matter lesions, the imaging correlate of which is termed leukoaraiosis (fig. 3), and lacunes, small (<1.5 cm) white matter infarcts typically in the basal ganglia, are common occurrence in VCI and are strongly associated with cardiovascular risk factors, especially hypertension, diabetes, hyperlipidemia and smoking (Gorelick et al., 2011; Wardlaw et al., 2013b). The vascular pathologies underlying these lesions consist of atherosclerotic plaques affecting small cerebral vessels, deposition of a hyaline substance in the vascular wall (lipohyalinosis), fibrotic changes in the vessel wall resulting in stiffening and microvascular distortion (arteriolosclerosis), and total loss of integrity of the vascular wall (fibrinoid necrosis) (fig. 5)(Thal et al., 2012). Arterioles become tortuous, have thickened basement membranes and are surrounded by enlarged perivascular spaces (Brown and Thore, 2011). Capillaries are reduced in number and “string vessels”, non-functional capillaries that have lost endothelial cells and have only a basement membrane, are observed (Brown and Thore, 2011). Collagen deposits are observed in venules (venous collagenosis) (Black et al., 2009; Brown and Thore, 2011). The white matter damage resulting from these lesions consists of vacuolation, demyelination, axonal loss, and lacunar infarcts. The white matter lesions generally correspond to hyperintensities observed on MRI, which, however, can also reflect other pathological substrates (Gouw et al., 2011). The white matter lesions evolve over time by expansion of existing lesions, rather than formation of new foci (Maillard et al., 2012), resembling the patters of progression of amyloid angiopathy (Alonzo et al., 1998; Robbins et al., 2006). The expansion of the white matter lesions correlates with the evolution of the cognitive impairment (Maillard et al., 2012), new lacunes causing a steeper decline, especially in motor speed and executive functions (Jokinen et al., 2011). White matter lesions and lacunar infarcts are also present in uncommon genetic conditions resulting in VCI and vascular dementia (Federico et al., 2012; Schmidt et al., 2012). The better studied of these, CADASIL, is associated with extensive leukoaraiosis and lacunar infarcts (Chabriat et al., 2009). CADASIL vascular lesions are related to accumulation of granular osmiofilic material (GOM) in vascular and perivascular cells, which include the Notch 3 ectodomain (Yamamoto et al., 2013).
Microinfarcts and microhemorrhages
Microscopic infarcts (microinfarcts) and hemorrhages (microbleeds) (fig. 5) are independent predictors of cognitive dysfunction, but are commonly associated with other vascular pathologies, such as leukoaraiosis, lacunar infarcts, large infarcts, and hemorrhage (Smith et al., 2012; van Norden et al., 2013), as well as CADASIL and AD (table 1). Microinfarcts are sharply delineated lesions consisting of pallor, necrosis, cavitation and inflammation (astrocytosis, microgliosis and macrophage infiltration) (Thal et al., 2012), caused by the small vessel pathology described above (table 1). Microbleeds are microscopic areas of blood extravasation from leaky arterioles, which are restricted to the perivascular space and do not disrupt the brain parenchyma (De Reuck, 2012). Observed in 17% of demented patients (Cordonnier and van der Flier, 2011), cortical microbleeds are frequently associated with cerebral amyloid angiopathy (CAA), whereas microbleeds in deep regions tend to be associated with white matter disease secondary to vascular risk factors (De Reuck, 2012; Park et al., 2013a).
Cerebral amyloid angiopathy
It is well known that deposits of Aβ in cerebral blood vessels or CAA are associated with vascular cognitive impairment. Although inherited forms of CAA have been described, CAA is most prevalent in AD, being present in over 90% of cases (Attems et al., 2011; Charidimou et al., 2012). CAA is also observed in demented (50–60%) and non-demented (20–40%) elderly people (Attems et al., 2011; Charidimou et al., 2012). The major risk factor for CAA is advanced age, and cardiovascular risk factors seem to play a lesser role (Charidimou et al., 2012). CAA is a major cause of microbleeds and large hemorrhages, typically located in the cortex (lobar hemorrhages) (Auriel and Greenberg, 2012). The amyloid accumulation occurs in the media and the adventitia of cerebral vessels, leading to degeneration of smooth muscle cells and pericytes (Thal et al., 2012). In extreme cases, the vascular wall develops fibrinoid necrosis and the vessels assumes a characteristic double barrel appearance (Thal et al., 2012).
Mixed lesions
Overlap of AD neuropathology (amyloid plaques and neurofibrillary tangles) with cerebrovascular lesions is observed in up to 50% of cases of dementia (Jellinger, 2013). These lesions include atherosclerosis of the circle of Willis and its branches, leukoaraiosis and lacunar infarcts, microbleeds, microinfarcts, and CAA (Benedictus et al., 2013; Charidimou et al., 2012; Honig et al., 2005; Jellinger, 2013; Richardson et al., 2012; Roher et al., 2004; Toledo et al., 2013; Yarchoan et al., 2012). Ischemic lesions in regions between arterial territories (watershed infarcts) have also been reported in AD, implicating hypoperfusion and CAA in their mechanisms (Miklossy, 2003; Suter et al., 2002). Vascular lesions are also present in other age related neurodegenerative diseases, such as synucleinopathies, hippocampal sclerosis and frontotemporal lobar degeneration linked to tau or TDP-43, but the coexistence with AD is the most frequent (Toledo et al., 2013).
How do vascular factors cause cognitive impairment?
As reviewed above, VCI can stem from a wide variety of cardiovascular and cerebrovascular pathologies, but it has been difficult to pin down the contribution of each condition to cognitive dysfunction because of the coexistence of the different lesions and overlap with neurodegenerative pathology (Gorelick et al., 2011). Reductions in global cerebral perfusion, such as those caused by heart diseases or carotid artery stenosis/occlusion, if below a critical threshold, can impair cognition independently of brain lesions (Marshall et al., 2012). Reductions in CBF by 40–50% are associated with suppression of brain activity and cognitive dysfunction, which are reversible upon re-establishing normal CBF levels (Marshall et al., 1999; Marshall, 2012; Tatemichi et al., 1995). As for the other pathologies underlying VCI, there is a general correlation between the total burden of vascular pathology and cognitive deficits (Gelber et al., 2012; Gorelick et al., 2011; Inzitari et al., 2009). A caveat is that, due to confounding factors, such as overlap with AD, differences in educational level (see below), and microscopic pathology not seen by in vivo imaging, the exact parameters of the relationship have been hard to define (Black et al., 2009; Brickman et al., 2011). However, there is general consensus that cognitive impairment results from the brain dysfunction caused by cumulative tissue damage (Gorelick et al., 2011), as originally proposed by Tomlinson et al. for large cerebral infarcts (Tomlinson et al., 1970).
In addition to gray matter damage, disruption of the white matter can have profound effects on the precision and fidelity of the information transfer underlying brain function and cognitive health (Nave, 2010a). Fast-conducting myelinated white matter tracts are responsible for long range connectivity, interhemispheric synchronization and neurotrophic effects through spike timing dependent plasticity and axonal transport (Dan and Poo, 2004; Nave, 2010a; Stone and Tesche, 2013). Indeed, white matter lesions affect brain structure and function broadly, and are associated with reductions in frontal lobe glucose utilization (Decarli et al., 1995; Haight et al., 2013; Tullberg et al., 2004), global reduction in cortical blood flow (Appelman et al., 2008; Chen et al., 2013a; Dam et al., 2007; Kobari et al., 1990), disruption of brain connectivity (Lawrence et al., 2013; Sun et al., 2011) and cerebral atrophy (Appelman et al., 2009). In addition, since myelination of previously naked fibers participates in neuroplasticity and skilled motor learning (Fields, 2010; Richardson et al., 2011), myelin damage could also compromise these important functions and contribute to cognitive impairment.
Risk factors for vascular cognitive impairments
Ascertaining the genetic and modifiable risk factors of VCI is problematic due to the multiplicity of underlying pathologies, coexistence with cardiovascular diseases, and the frequent overlap with AD and other neurodegenerative diseases (Gorelick et al., 2011). Therefore, the risk attributable to specific factors remains unclear, although the recent development of biomarkers for in vivo AD diagnosis (Hampel et al., 2012) promises to alleviate this problem.
Advanced age is a powerful risk factor for VCI, and the prevalence and incidence of cognitive impairment increases exponentially after age 65 (Gorelick et al., 2011). The level of education, a surrogate marker of cognitive reserves (Stern, 2012), is an important determinant of the expression of VCI, such that for a given level of neuropathology higher education is associated with less cognitive deficits (Zieren et al., 2013). However, the education level does not influence the rate of progression of VCI and no longer has an impact in advanced disease (Elbaz et al., 2013; Zieren et al., 2013). Although education could account for individual differences in the susceptibility to cognitive impairment given comparable burdens of disease, socioeconomic status, coexisting chronic diseases, ethnicity, and pre-morbid intellectual capacity are important confounders (Gorelick et al., 2011).
Vascular risk factors, including hypertension, diabetes, hyperlipidemia, smoking, atrial fibrillation, and hyperhomocystinemia, increase the risk of dementia independently of the associated increase in stroke risk (Sahathevan et al., 2012). Furthermore, the metabolic syndrome, including insulin resistance, hypertension and dyslipidemia, has been associated with lower cognitive performance (Yates et al., 2012). However, recurrent stroke is one of the strongest predictors of dementia onset (Pendlebury and Rothwell, 2009). Remarkably, in VCI as in AD, the increase in risk afforded by vascular risk factors is observed decades later, a finding that may explain why some studies did not find a cognitive improvement with risk factor control later in life (Sahathevan et al., 2012).
A host of rare genetic mutations are associated with VCI (Federico et al., 2012; Schmidt et al., 2012). The most common of these is the CADASIL syndrome caused by a frame shift mutation of Notch-3 that either creates or eliminates a cystein residue (Chabriat et al., 2009). Other hereditary cerebral vasculopathies include familial CAAs caused by mutations or duplications of APP (Auriel and Greenberg, 2012; Rannikmäe et al., 2013), the cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) caused by mutation of the TGFβ repressor HTRA1, the autosomal dominant retinal vasculopathy with cerebral leukodystrophy caused by frameshift deletions in the exonuclease TREX1, and mutations of the COL4A1 gene encoding the type IV collagen alpha 1 chain (Federico et al., 2012; Gorelick et al., 2011; Lanfranconi and Markus, 2010). The ApoE 4 allele is a well-established susceptibility gene for increased cardiovascular risk and Alzheimer disease (Verghese et al., 2011). The ApoE4 allele is associated with increased risk of CAA, whereas both ApoE2 and 4 increase the risk of lobar hemorrhages (Charidimou et al., 2012). Nevertheless, a strong link between ApoE and sporadic VCI has not been established (Lee and Kim, 2013; Yu et al., 2013). Studies of candidate genes have revealed weak associations with genes involved in the renin-angiotensin system, endothelial nitric oxide synthase, oxidative stress, lipid metabolism and inflammation, but have not been replicated (Fornage et al., 2011; Lee and Kim, 2013; Markus, 2008). GWAS of vascular dementia have shown small effect of SNPs in the androgen receptor gene locus (Schrijvers et al., 2012), a finding not observed in all ethnic groups (Lee and Kim, 2013). The diversity of pathologies underlying VCI and the overlap with AD complicate the interpretation of these studies. Linkage studies in patients with white matter lesions on MRI have discovered several loci (Schmidt et al., 2012), but no specific gene has been identified and the findings await replication and validation (Lee and Kim, 2013; Markus, 2008).
Pathogenic mechanisms responsible for white matter injury
Although as described in the previous section severe ischemia resulting from arterial occlusion can lead to brain damage and VCI, e.g., multi-infarct dementia, cognitive dysfunction is most often associated with more subtle vascular alterations targeting predominantly the deep hemispheric white matter (fig. 5). Here we examine the major pathogenic mechanisms leading to white matter damage, inferred either from brain imaging and post-mortem studies in humans, or animal models (fig. 6).
Figure 6.

Potential mechanisms of the blood vessel damage induced by vascular risk factors. Endothelial dysfunction, impairment of autoregulation and dysfunction of neurovascular coupling, partly mediated by oxidative stress and NO deficit, reduce CBF resulting in hypoperfusion and tissue hypoxia. In addition to hypoperfusion, a critical consequence of endothelial dysfunction is increased BBB permeability, which leads to extravasation of plasma proteins, including fibrinogen, into the brain. Fibrinogen activates CD11b and TLR leading to production of ROS, proinflammatory cytokines and MMPs from activated microglia, reactive astrocytes and OPCs. Inflammation, in turn, aggravates the BBB breakdown and induces expression of adhesion molecules in endothelial cells, contributing to leukocyte and platelet adhesion and microvascular occlusion.
Hypoperfusion and hypoxia
Owing to their location at the distal border between different vascular territories (De Reuck, 1971) (fig 4) and to the susceptibility of their vasculature to risk factors (Brown and Thore, 2011), deep white matter tracts are particularly vulnerable to vascular insufficiency. Even in healthy individuals, hypercapnia, a potent vasodilator, does not increase, but reduces, CBF in the periventricular white matter, suggesting that vasodilatation of upstream vessels diverts blood flow to other regions (intracerebral steal) (Mandell et al., 2008). This finding highlights the hemodynamic precariousness of the periventricular white matter, even in the absence of vascular damage.
Increasing evidence suggests that the white matter cerebral blood supply is compromised in VCI (fig. 6). Resting flow is reduced in areas of leukoaraiosis and vascular reactivity attenuated (Kobari et al., 1990; Makedonov et al., 2013; Markus et al., 2000; 1994; Marstrand et al., 2002; O’Sullivan et al., 2002; Yao et al., 1992). In patients with VCI risk factors, like hypertension and diabetes, the ability of neural activity to increase blood flow in brain or retina is compromised (Delles et al., 2004; Jennings et al., 2005; Sorond et al., 2011). Cerebrovascular autoregulation is impaired, increasing the susceptibility of the white matter to damage during fluctuation in blood pressure (Matsushita et al., 1994). Interestingly, CBF alterations have also been described in normal appearing white matter (O’Sullivan et al., 2002), suggesting that the flow reduction precedes and, as such, may contribute to the white matter damage. Indeed, in the general population, lower global CBF and lower cerebrovascular reactivity to hypercapnia is associated with a greater volume of white matter lesions (Bakker et al., 1999; Vernooij et al., 2008). The CBF reduction is observed prior to the onset of dementia (Ruitenberg et al., 2005). Due to their hemodynamic vulnerability, deep white matter regions are marginally perfused, and, in the presence of vascular risk factors, their vessels may be unable to adapt CBF to the metabolic needs of the tissue. Consistent with this hypothesis, post-mortem studies have shown that areas of leukoaraiosis are chronically hypoxic, as indicated by the expression of hypoxia inducible factors and related hypoxia-inducible genes (Fernando et al., 2006; Rosenberg et al., 2001).
In addition to local factors affecting white matter microvessels, broader-acting systemic factors are also involved. White matter lesions and lacunar strokes are associated with increases in circulating levels of the NO synthase inhibitor asymmetric dimethylarginine (ADMA) (Notsu et al., 2009; Pikula et al., 2009; Rufa et al., 2008). ADMA may contribute to the impairment of NO-dependent vasodilatation in peripheral and cerebral arteries (Chen et al., 2006; Knottnerus et al., 2009; Pretnar-Oblak et al., 2006; Stevenson et al., 2010). Furthermore, stiffness of large vessels and increased pulsatility are associated with reduced white matter CBF and are strong predictors of leukoaraiosis and lacunes (Brisset et al., 2013; Tarumi et al., 2011; Webb et al., 2012), independently of vascular risk factors (Kearney-Schwartz et al., 2009). These findings implicate loss of large artery elasticity and increased pulsatile stress on microvessels, especially those branching directly from the circle of Willis, in the microvascular damage underlying white matter lesions (Scuteri et al., 2011). Similar microvascular changes occur also in other organs, suggesting that small vessel disease in brain may be the manifestation of a systemic vasculopathy (Thompson and Hakim, 2009).
Increased blood-brain barrier permeability
Reflecting another aspect of endothelial dysfunction, alterations in BBB permeability are also associated with leukoaraiosis and lacunar stroke (Wardlaw et al., 2013a; Yang and Rosenberg, 2011). Several lines of evidence indicate that the BBB is disrupted in the course of the disease. First, the plasma protein albumin is increased in the CSF of patient with VCI, reflecting BBB breakdown (Candelario-Jalil et al., 2011). Second, plasma proteins, including complement, fibrinogen, albumin and immunoglobulins are detected in astrocytes in white matter lesions (Akiguchi et al., 1998; Alafuzoff et al., 1985; Simpson et al., 2007; Tomimoto et al., 1996). Third, the permeability to MRI tracers is increased in white matter lesions (Hanyu et al., 2002; Taheri et al., 2011; Wardlaw et al., 2009) and in normal appearing white matter (Topakian et al., 2010). The latter finding suggests that the BBB disruption could precede white matter injury and contribute to its development. BBB leakiness in white matter was found in lacunar strokes, but not cortical strokes (Wardlaw et al., 2008), raising the possibility of a specific association with small vessel disease of the deep white matter.
Several factors could contribute to the BBB disruption (Rosenberg, 2012). Hypoxia-ischemia, which has been demonstrated in white matter lesions, is well known to damage endothelial cells leading to increased BBB leakage in vitro (Ahmad et al., 2012). In vivo, hypoperfusion produced by bilateral carotid stenosis in rat increases BBB permeability (Ueno et al., 2002). In a similar model, the BBB alteration was found to be due to MMP9 production by oligodendrocyte precursors, which are increased in ischemic white matter injury in rodent models (Seo et al., 2013) and in patients with VCI (Candelario-Jalil et al., 2011). In stroke prone spontaneously hypertensive rats, which have a strong vascular risk factor profile, a high salt diet induces fast-developing vasculopathy with BBB leakage that leads to ischemic injury in the absence of arterial occlusions (Schreiber et al., 2013). This finding indicates that chronic BBB disruption has the potential of induce ischemic damage. Indeed, vascular risk factors, and the associated oxidative stress and vascular inflammation also alter BBB permeability and could play a role.
Oxidative stress and inflammation
Pathological studies have shown markers of oxidative stress (isoprostanes) and inflammation (cytokines and adhesion molecules) in the damaged white matter associated with VCI (Back et al., 2011; Candelario-Jalil et al., 2011; Fernando et al., 2006). Furthermore, microglial activation and reactive astrocytes are also present in the lesions (Akiguchi et al., 1998; Simpson et al., 2007; Tomimoto et al., 1996) (fig. 6). Markers of endothelial activation, hemostasis, inflammation and oxidative stress are also upregulated in blood, consistent with more widespread effects in the systemic circulation (Gallacher et al., 2010; Knottnerus et al., 2010; Markus et al., 2005; Rouhl et al., 2012a; Shibata et al., 2004; Xu et al., 2010) (fig. 6). The mechanisms of these responses have not been fully elucidated, but several factors may play a role. Cerebral hypoperfusion is associated with white matter inflammation and oxidative stress in rodent models (Dong et al., 2011; Huang et al., 2010; Ihara et al., 2001; Juma et al., 2011; Masumura et al., 2001; Reimer et al., 2011; Yoshizaki et al., 2008), indicating that hypoxia-ischemia is sufficient to trigger these responses. Vascular risk factors for VCI, such as hypertension, insulin resistance and diabetes, lead to vascular oxidative stress and inflammation, both in animal models and in humans (Cohen and Tong, 2010; Iadecola and Davisson, 2008; Yates et al., 2012), which, in turn, impair the factors regulating the cerebral circulation (Faraci, 2011). Thus, functional hyperemia and endothelium dependent responses are attenuated in models of aging, hypertension, and diabetes (Ergul, 2011; Kazama et al., 2004; Park et al., 2007), whereas the ability of the vessels to adjust cerebral perfusion in response to changes in blood pressure (autoregulation) is blunted in patients with diabetes or hypertension (Kim et al., 2008b; Novak et al., 2003). Such neurovascular dysfunction would aggravate the CBF reduction in critically perfused deep white matter regions and contribute to the white matter damage. Accordingly, scavenging of free radicals or approaches to suppress inflammation counteract white matter damage and behavioral deficits in rodent models of cerebral hypoperfusion (Dong et al., 2011; Kim et al., 2008a; Maki et al., 2011; Ueno et al., 2009; Wakita et al., 2008; Wang et al., 2010; Washida et al., 2010; Zhang et al., 2011). NADPH oxidase, a multiunit enzyme particularly enriched in cerebral blood vessels (Miller et al., 2005), has emerges as an important source in vascular oxidative stress in aging, hypertension, hyperlipidemia and diabetes (Faraci, 2011), and inhibition or genetic inactivation of this enzyme has been shown to ameliorate the vascular dysfunction (Drummond et al., 2011). Extravasation of plasma proteins, due to the BBB alterations, is also likely to play a role, since fibrinogen, immunoglobulins, and complement are potent activators of inflammation and free radical production (Crehan et al., 2013; Davalos and Akassoglou, 2012; Yoshida et al., 2002). In particular, fibrinogen extravasation activates inflammatory pathway through its interaction with integrin (CD11b/CD18) and non-integrin receptors (TLRs), leading to activation of microglia and astrocytes (Davalos and Akassoglou, 2012; Davalos et al., 2012) (fig. 6). As discussed next, inflammation and oxidative stress have also deleterious effects on the trophic interaction among the cells of the neurovascular unit.
Trophic uncoupling
ROS and inflammation suppress the prosurvival action of endothelial cells on neurons by reducing BDNF levels, an effect mediated by impairing integrin linked kinase signaling (Guo et al., 2008). In models of diabetes, advanced glycation end-products lead to MMP9 secretion by endothelial cells and cleavage of the ectodomain of the BDNF receptor TRKB on neurons, reducing neurotrophic signaling (Navaratna et al., 2013). Owing to their trophic support of vascular cells, dysfunction and damage to neurons and glia is associated with endothelial cell atrophy and microvascular rarefaction (Brown and Thore, 2011). Systemic factor also play a role in the mechanisms of trophic uncoupling. EPC are reduced by stroke risk factors (Hill et al., 2003) and predict cardiovascular morbidity and mortality (Werner et al., 2005). EPC are reduced in age associated white matter lesions, the reduction correlating with lesion burden (Jickling et al., 2009). In addition, EPC may be less functionally competent in patients with vascular risk factors and stroke. For example, the ability of colony forming units, a subset of EPC, to form vascular tubes in a matrigel assay is impaired patients with large artery atherosclerosis or lacunar stroke (Chu et al., 2008). Interestingly, EPC colony forming units are also reduced in AD patients, in whom the magnitude of the reduction correlates with the degree of cognitive impairment (Lee et al., 2009). Angiogenic T-cells are reduced in patients with vascular risk factors (Hur et al., 2007; Weil et al., 2011), and in hypertensive patients with small vessels disease (Rouhl et al., 2012b). Furthermore, angiogenic T-cells migration in vitro is positively correlated with preservation of endothelium-dependent vasodilatation in patients with cardiovascular risk factors (Weil et al., 2011), highlighting their protective role in vascular function. These findings, raise the possibility that vascular risk factors suppress the production of angiogenic T-cells, reduce the repair potential of EPC, and contribute to the microvascular degeneration underlying leukoaraiosis and lacunar stroke. Accordingly, capillary density is reduced not only at lesioned sites, but also in normal appearing white matter in patients with VCI (Brown et al., 2007). Vessels devoid of endothelium (string vessels) are often observed, reflecting a failure of endothelial repair, possibly due to EPC dysfunction or loss of neuron and/or glial-derived growth factors.
Lesions of white matter tracts also lead to distant effects resulting from loss of trophic support at their site of termination. Leukoaraiosis is associated with focal cortical thinning especially in frontal cortex, a finding correlated with executive dysfunction (Seo et al., 2012). Focal cortical thinning was also observed in a prospective study of patients with CADASIL subsequent to a subcortical infarct (Duering et al., 2012), indicating a causal link between white matter lesions and cortical atrophy. These processes are likely to play a role in the progressive cerebral atrophy observed in patients with leukoaraiosis, who experience a brain volume loss of 1% per year, twice that of age matches controls (Nitkunan et al., 2011). However, it has not been established whether white matter lesions cause the atrophy independently of age and other risk factors (Appelman et al., 2009; 2010). Trophic interactions are also critically involved in the demyelination and remyelination associated with leukoaraiosis, which are examined next.
Demyelination and remyelination
One of the consequences of the oxidative and proinflammatory environment induced by hypoperfusion and BBB breakdown is damage to the myelin sheet and demyelination. Myelination allows axons to conduct 100 times faster, and reduce energy expenditures by restricting the depolarization of the axonal membrane to the Na+ channel rich Ranvier nodes (Nave, 2010b). Some of the energy saving afforded by myelination is offset by the cost of maintaining the resting potential of oligodendrocytes, which is estimated to be high (Harris and Attwell, 2012). Loss of myelin has important consequences for the white matter tracts. In addition to the brain dysfunction caused by slowing down the transmission of axon potentials, demyelination threatens the integrity of the axons and leads to axonal loss (Franklin and Ffrench-Constant, 2008; Matute and Ransom, 2012). Several factors contribute to the demise of the axons. Oligodendrocytes release growth factors, such as IGF-1 and glial cell-derived neurotrophic factor that support the survival of axons (Wilkins et al., 2003). Thus, loss of myelin deprives the axons of trophic support and increases their vulnerability. In addition, demyelination exposes the axons to the deleterious effects of cytokine and free radicals in the hypoxic white matter, which may impair axonal energy production leading to failure of the Na+/K+ ATPase. The resulting accumulation of intracellular Na+ reverses the operation of the Na+/Ca2+ exchanger, resulting in intracellular Ca2+ accumulation (Matute and Ransom, 2012; Stys et al., 1992). Furthermore, the adaptive upregulation of voltage-dependent Na+ channels (VNa+) in the denuded internodal axoplasm, attempting to preserve impulse propagation in demyelinated axons, leads to Na+ entry and aggravates the energy deficit and Ca2+ overload. Upregulation of VNa+1.2 channels increases the activity of the Na+/K+ ATPase, stressing further the energy budget of the marginally perfused white matter (Trapp and Stys, 2009). In turn, excess intracellular Ca2+ activates protease dependent processes that lead to microtubule fragmentation and perturbation of axonal flow (Franklin and Ffrench-Constant, 2008; Matute and Ransom, 2012).
Attempts to remyelinate are present in the damaged white matter in leukoaraiosis (Jonsson et al., 2012). Oligodendrocytes are responsible for the formation and maintenance of the myelin sheet. A large pool of oligodendrocyte progenitor cells (OPC) is present in the brain, which goes through several stages of development before becoming mature and competent to lay down myelin (Fancy et al., 2011a). However, in demyelinating diseases, including leukoaraiosis, axons fail to fully remyelinate (Franklin and Ffrench-Constant, 2008). Several factors are thought to be responsible (fig. 7). First, OPC in the late stage of development are particularly susceptible to injury in conditions of chronic hypoxia and oxidative stress existing in the ischemic white matter (Back et al., 2011; 2002; Fernando et al., 2006; French et al., 2009). Oligodendrocytes are also susceptible to damage caused by extracellular ATP, which increases in hypoxia-ischemia, through activation of the P2X7 purinergic receptors (Domercq et al., 2010). Second, withdrawal of trophic support from damaged endothelial cells and astrocytes could reduce the vitality of the OPC pool and contribute to their demise in the hypoxic environment of the vulnerable white matter (Arai and Lo, 2010). Third, failure to remyelinate could be related to an arrest in OPC maturation. OPC are abundant in areas of leukoaraiosis, which are enriched with hyluronan (HA), a high molecular weight glycosaminoglycan produced by reactive astrocytes and other cells (Back et al., 2011). HA is a component of the matrix and is involved in neurodevelopment promoting neuronal migration (Sherman and Back, 2008). In white matter lesions, HA is degraded by the hyaluronidase PH20 and its cleavage products inhibit the maturation of OPC into oligodendrocytes capable of myelination (Preston et al., 2013) (fig. 7). Dysregulation of the Wnt signaling pathway could also play a role in the OPC developmental arrest (Fancy et al., 2011b). In addition, OPC produce MMP9, which, as seen in the previous sections, promotes BBB breakdown perpetuating the cytotoxic milieu underlying demyelination (Seo et al., 2013).
Figure 7.
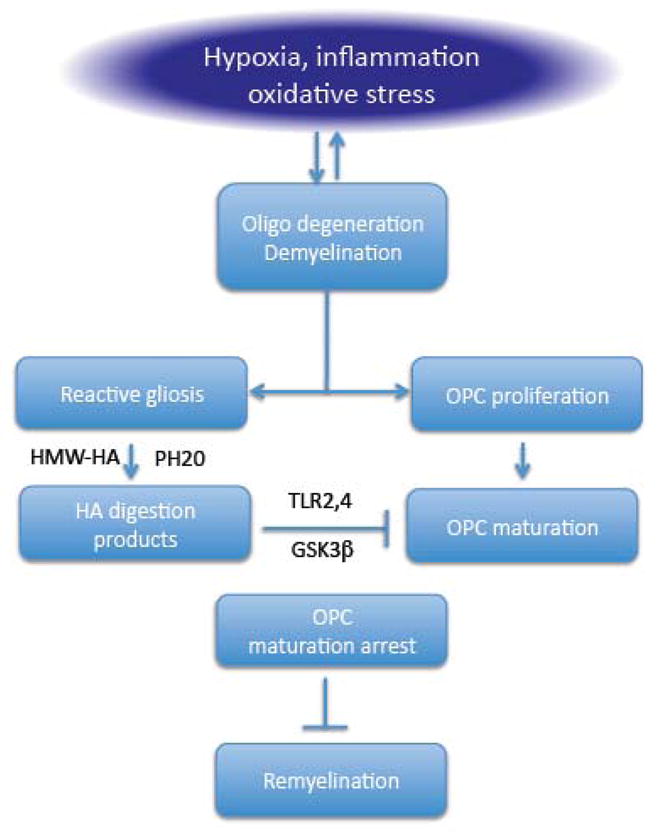
Potential mechanisms of failure to remyelinate the damaged white matter. Inflammation, oxidative stress and hypoxia induced demyelination. OPC proliferate to attempt remyelination. High molecular weight hyaluronic acid (HMW-HA) produced by reactive astrocytes is cleaved by the hyaluronidase PH20 generating digestion products that inhibit OPC maturation through mechanisms involving TLR2 and 4 and GSK3β. The resulting OPC maturation arrest prevents efficient remyelination.
Putting it all together
The evidence reviewed above suggests a convergence of pathogenic factors on cerebral blood vessels, which in turn leads to white matter damage (figs. 6, 7). Oxidative stress-induced endothelial dysfunction caused by risk factors is most likely an early event leading to white matter damage. Endothelial dysfunction has two major pathogenic consequences: reductions in resting CBF in the marginally perfused white matter, and alterations in the permeability of the BBB. In turn, hypoperfusion and BBB disruption lead to additional oxidative stress by inducing tissue hypoxia and by extravasation of plasma proteins, such as fibrinogen. Tissue edema resulting from increased BBB permeability may exacerbate these alterations by compressing blood vessels and reducing CBF further. Tissue hypoxia and oxidative stress activate inflammatory pathways through NFκb-dependent transcription, leading to production of cytokines and adhesion molecules in vascular cells, reactive astrocytes and activated microglia. Hypoxia, inflammation and oxidative stress damage oligodendrocytes and leads to trophic uncoupling in the neurovascular unit, which, in turn, contribute to the damage to vascular cells and oligodendrocytes. Oligodendrocyte damage, oxidative stress and inflammation lead to demyelination and attempted remyelination through OPC proliferation. Developmental arrest of OPC, due to HA degradation products, leads to accumulation of these cells which secrete MMP9 and worsen the BBB impairment (fig. 7). Once demyelination occurs, the increased energy requirement of the denuded axons aggravates the hypoxic stress of the tissue, leading to a vicious circle that perpetuates these pathogenic processes and exacerbates the tissue damage.
Is hypoperfusion involved also in inherited and autoimmune white matter diseases?
There is emerging evidence that reduced cerebral perfusion may contribute to other diseases characterized by white matter damage. Multiple sclerosis (MS) is the prototypical neuroinflammatory disease in which demyelination is thought to be related to a T-cell mediated autoimmune attack on myelin (McFarland and Martin, 2007). However, in MS patients CBF is reduced in the normal appearing white matter (Law et al., 2004), as well as in the gray matter (D’haeseleer et al., 2011). In contrast, in active lesions displaying BBB disruption CBF is increased, consistent with vasodilatation caused by inflammation (D’haeseleer et al., 2011). The reduction in CBF in the normal white matter could be caused by a primary vascular dysfunction pathogenically linked to the disease process, or could be secondary to loss of white matter elsewhere, due to distal Wallerian degeneration, or reduced synaptic activity (De Keyser et al., 2008). Studies in which CBF measurements in the normal appearing white matter were coupled to diffusion tensor imaging, revealed that the reductions in CBF are associated with restricted diffusion and not with increased fractional anisotropy, as anticipated if the CBF changes were secondary to Wallerian degeneration (Saindane et al., 2007). Although the possibility that the reduction in CBF is secondary to reduced local synaptic activity has not been ruled out, the fact that the hypoperfusion is normalized by an endothelin receptor antagonist suggest a primary vascular cause (D’haeseleer et al., 2013). Consistent with the hypoperfusion hypothesis, HIF-1α and dependent genes are upregulated in normal appearing white matter (Graumann et al., 2003).
Reductions in white matter CBF has also been found X-linked adrenoleukodystrophy (ALD), a disease caused by mutations in ABCD1, which encodes a peroxisomal membrane transporter protein, leading to accumulation of very long chain fatty acids in brain, spinal cord and adrenal glands (Moser et al., 2000). In its infantile form, the disease starts between 4 and 8 years of age and is characterized by a progressive cognitive decline associated with rampant inflammatory demyelination of the white matter (Moser et al., 2000). BBB alterations predict disease progression (Melhem et al., 2000). Cerebral blood volume, assessed by susceptibility contrast MRI (Musolino et al., 2012), or CBF, assessed by single photon emission tomography (Suhaili et al., 1994), is reduced in the normal appearing and abnormal white matter. The mechanisms of the white matter hypoperfusion remain to be defined. Reductions in CBF prior to white matter damage were also observed in a patient with Alexander disease, a rare childhood disease caused by a dominant mutation of the GFAP gene (Ito et al., 2009).
It is noteworthy that, despite fundamental differences in their pathogenesis, inherited and autoimmune diseases of the white matter exhibit cerebrovascular alterations before pathology develops, just like in white matter disease caused by vascular factors. Thus, hypoperfusion and BBB disruption seem necessary correlates of the process leading to white matter damage independently of the primary disease cause. Collectively, these observations highlight the importance of neurovascular factors in maintaining white matter health.
Overlap between vascular and neurodegenerative dementia
The realization that most cases of dementia have mixed pathological features has raised the intriguing possibility that vascular factors play role in AD and other neurodegenerative diseases. As discussed in the section on “Mixed lesions”, AD brains have a wide variety of vascular lesions, suggesting a potential pathogenic interaction between vascular factors and AD. However, since cerebrovascular diseases and AD are common in the aged, the coexistence of the two pathologies could simply be coincidental (Hachinski, 2011). The overall effect on cognition would results from the combined burden of vascular and neurodegenerative pathology, according to an additive model. Alternatively, vascular disease could promote AD and vice-versa, resulting in a reciprocal interaction amplifying their pathogenic effects. The cognitive impact of vascular and AD neuropathology depends on the severity of the AD pathology and location of the vascular lesions (Gold et al., 2007). In advanced cases of AD, vascular lesions do not seem to have a major influence on the progression of the cognitive deficits, suggesting the AD pathology is the major driver of the cognitive dysfunction (Chui et al., 2006; Jellinger, 2001). On the other hand, in older individuals with moderate AD pathology subcortical vascular lesions are a major determinant of the expression of the dementia (Esiri et al., 1999; Schneider et al., 2007b; Snowdon et al., 1997).
Cerebrovascular factors and AD
Cerebrovascular function is reduced in patients with early AD or at risk for AD (Claassen et al., 2009; Gao et al., 2013; Luckhaus et al., 2008; Mentis et al., 1996; Niedermeyer, 2006; Ruitenberg et al., 2005; Sabayan et al., 2012; Tanaka et al., 2002), implicating reduced cerebral perfusion in the pathobiology of the disease (Iadecola, 2004). Conversely, some studies (Jendroska et al., 1995; Ly et al., 2012), but not others (Aho et al., 2006; Mastaglia et al., 2003), have reported increased amyloid deposition in stroke patients, implicating that ischemia promotes AD pathology. Furthermore, AD and cerebrovascular diseases may have common risk factors, such as hypertension, insulin resistance, diabetes, obesity, hyperhomocystinemia, hyperlipidemia, etc. (Craft, 2009; Fillit et al., 2008; Honjo et al., 2012; Purnell et al., 2009). However, the correlation was most evident when the risk factors were considered together and not individually (Chui et al., 2012). Furthermore, the correlation was strongest for vascular dementia and weakest for AD, suggesting that vascular risk factors may independently increase the likelihood of dementia without exacerbating AD pathology (Chui et al., 2012). In contrast, studies that have prospectively evaluated representative patients cohorts with confirmation of the clinical diagnosis at autopsy failed to establish a link between the burden of AD pathology and vascular risk factors (Chui et al., 2012). It is, therefore, conceivable that in cases in which AD was diagnosed clinically there might have been a component of vascular pathology. New imaging and CSF biomarkers for the in vivo diagnosis of AD may provide additional insights into whether vascular factors are pathogenically linked to AD (Chui et al., 2012; Haight et al., 2013; Purnell et al., 2009).
Vascular effects of Aβ
Mounting evidence that Aβ has powerful vascular effects also suggests a link between AD and vascular disease. Aβ4 constrict isolated cerebral and systemic blood vessels (Niwa et al., 2001; Paris et al., 2003; Thomas et al., 1996), whereas application of Aβ4 to the exposed cerebral cortex of mice reduces CBF and impairs the increase in CBF induced by endothelium-dependent vasodilators and functional hyperemia (Niwa et al., 2000a; 2000b). Similarly, functional hyperemia, endothelium-dependent responses and autoregulation are profoundly impaired in young mice overexpressing mutated forms of APP, in which brain Aβ is elevated, but there are no plaques, behavioral alterations, or reductions in resting glucose utilization (Niwa et al., 2000b; 2002; Tong et al., 2012). These data suggest that the cerebrovascular effects of Aβ are not attributable to CAA or amyloid plaques, and are not a consequence of neuronal energy hypometabolism. APP-overexpressing mice have increased brain damage following occlusion of the middle cerebral artery (Koistinaho et al., 2002; Zhang et al., 1997), an effect in part related to poor collateral circulation due to vascular dysregulation (Zhang et al., 1997). The vascular alterations induced by Aβ are abrogated by overexpression of the ROS scavenging enzyme superoxide dismutase or deficiency of the NADPH oxidase subunit NOX2 (Iadecola et al., 1999; Park et al., 2005; 2008), implicating ROS produced by the enzyme NADPH oxidase in the vascular dysfunction. The mechanisms of NADPH oxidase activation involve the Aβ-binding scavenger receptor CD36 (Park et al., 2011). Aged APP mice deficient in CD36 are protected from cerebrovascular alterations and behavioral deficits, effects associated with reduced CAA compared to controls, but no reduction of amyloid plaques (Park et al., 2013b). Thus, CD36, which is located in vascular and perivascular cells, may contribute to the accumulation of Aβ in cerebral blood vessels.
Aβ production and clearance
Hypoperfusion and hypoxia caused by vascular insufficiency may also facilitate Aβ production by activating the APP cleavage enzyme β-secretase (Kitaguchi et al., 2009; Sun et al., 2006; Tesco et al., 2007; Wen et al., 2004a). Cerebral ischemia promotes amyloid plaque formation (Garcia-Alloza et al., 2011; Kitaguchi et al., 2009; Okamoto et al., 2012), and tau phosphorylation (Koike et al., 2010; Wen et al., 2007; 2004b). The vascular effects of Aβ may also impair the clearance of the peptide, a key factor in brain Aβ accumulation in sporadic AD (Mawuenyega et al., 2010). The vascular pathway is estimated to be a major route of removal of Aβ from the brain (Castellano et al., 2012; Shibata et al., 2000). Brain Aβ is transported along the perivascular pathway draining into the cervical lymphnodes (Carare et al., 2013; Iliff et al., 2013). In addition, Aβ is cleared from the brain through a transvascular transport system involving LRP-1 (Shibata et al., 2000), a protein that acts in concert with P-glycoprotein, ApoE, ApoJ, and α2-macroglobulin to regulate brain Aβ homeostasis (Zlokovic, 2008). Interestingly, ApoE4, a major genetic risk factor for AD, leads to BBB disruption through a proinflammatory pathway involving cyclophilin A in pericytes (Bell et al., 2012). Activation of this pathway causes MMP-9-mediated degradation of endothelial tight junctions and basement membrane proteins, as shown in human ApoE4 targeted replacement mice (Bell et al., 2012). ApoE4 positive individuals may develop a similar age-dependent BBB breakdown prior to cognitive decline (Halliday et al., 2013). In patients with vascular risk factors, such as hypertension, sedentary life style, or ApoE4 genotype, there is a greater tendency for amyloid accumulation (Head et al., 2012; Rodrigue et al., 2013), whereas amyloid accumulation is reduced in patients who exercise regularly (Liang et al., 2010). Experimental studies indicate that this clearance mechanism is altered in the presence of vascular dysfunction and damage, contributing to parenchymal and vascular Aβ accumulation (Deane et al., 2004; Park et al., 2013b). In particular, suppression of LRP1 in vascular smooth muscle cells due to upregulation of serum response factor and myocardin, is a key factor in the clearance impairment (Bell et al., 2009). Collectively, these observations suggest a link between cerebrovascular health and brain Aβ clearance.
These lines of evidence suggest that AD is frequently associated with cerebral macro- and micro-vascular pathology, which can contribute to the expression of the dementia. Vascular risk factors can increase amyloid accumulation and the risk of clinically defined AD. The vasoactivity of Aβ and the influence of cerebral perfusion on APP processing and Aβ clearance suggest that cerebral blood vessels can have a role the accumulation of Aβ in the brain parenchyma and cerebral blood vessels. Preliminary evidence suggests that control of vascular risk factors reduces vascular lesions in AD (Richard et al., 2010), and may delay disease progression (Deschaintre et al., 2009), at least early in the disease course (Richard et al., 2010). Although replication in representative cohorts in which AD is confirmed pathologically or with biomarkers is needed, these observations provide initial evidence that improving vascular health may also help in AD.
Prospects for prevention and therapy
The development of treatments for VCI has been hampered by the lack of a suitable animal model recapitulating the multifaceted features of the disease (Gorelick et al., 2011). Although several animal models have been developed (Hainsworth et al., 2012; Lee et al., 2012), the most widely used has been white matter damage produced by chronic forebrain ischemia (Ihara and Tomimoto, 2011). These models have demonstrated that counteracting some of the pathogenic factors, including chronic ischemia, inflammation and oxidative stress, reduce white matter damage and/or behavioral deficits (Dong et al., 2011; Ihara and Tomimoto, 2011; Maki et al., 2011). Other approaches have attempted to promote remyelination by stimulating the survival and differentiation of OPC (Miyamoto et al., 2010). Despite these positive results in models of hypoperfusion-induced white matter damage, there are no FDA approved treatments for VCI and vascular dementia (Butler and Radhakrishnan, 2012). Treatment with antioxidants, anti-inflammatory agents or agents increasing cerebral perfusion have not led to consistent results (Butler and Radhakrishnan, 2012). Some agents, like the neurotrophic factor cerebrolysin, showed a modest cognitive improvement, but the evidence is not sufficiently strong to justify clinical use (Chen et al., 2013b). Clinical trials are currently exploring other agents, including cholinergic stimulants (choline alphoscerate), vasodilators (udenafil), inhibitor of platelet aggregation (cilostazol) and delta-9-tetrahydrocannabinol (a complete list can be found at www.clinicaltrials.gov).
On the other hand, increasing evidence indicates that the risk of VCI and vascular dementia can be reduced by preventive measures. A study in the UK population suggests that the prevalence of dementia may be decreasing, a finding interpreted to reflect the beneficial effects of controlling blood pressure and other risk factors (Matthews et al., 2013). Indeed, rigorous blood pressure control reduces white matter damage and staves off cognitive decline (Sharp et al., 2011; Sörös et al., 2013). Physical and mental activity, social engagement, and a diet rich in antioxidants or polyunsaturated fatty acids reduce dementia risk (Aarsland et al., 2010; Akinyemi et al., 2013; Middleton and Yaffe, 2009; Verdelho et al., 2012). Therefore, controlling vascular risk factors and promoting a healthy diet, exercise and mental activity are promising strategies to reduce VCI. This hypothesis is supported by a study indicating that weight control, a healthy diet, nonsmoking, physical activity, and keeping total cholesterol, blood pressure, and fasting glucose at goal levels are associated with better cognitive performance later in life (Reis et al., 2013). However, most of the evidence is based on observational studies, which have not been confirmed by randomized clinical trials of risk factor modification, stressing the need for further large scale studies (Dichgans and Zietemann, 2012; Middleton and Yaffe, 2009).
Conclusions
VCI and vascular dementia are major contributors to age-relate dementing illnesses and comprise a heterogeneous group of cognitive disorders attributable to vascular causes. Vascular pathology is also an integral part of AD and other late-life neurodegenerative conditions associated with dementia, and play a defining role in the expression of the cognitive dysfunction. Despite the diversity of the underlying brain pathology, the vascular alterations have a similar pathogenic bases, resulting from hypoperfusion, oxidative stress and inflammation, which in turn lead to endothelial damage, BBB breakdown, activation of innate immunity and disruption of trophic coupling between vascular and brain cells. The hemispheric white matter, which is particularly susceptible to the deleterious effects of vascular risk factors, is a major target of these vascular alterations. The resulting demyelination and axonal loss plays a role in the broad functional brain changes underlying cognitive impairment and in the associated cerebral atrophy. This chain of events highlights the critical role that vascular cells play in the maintenance of the health of neurons, glia and myelin.
However, several fundamental questions remain to be addressed. The predilection of the vascular pathology for the deep hemispheric white matter, a remarkable constant in conditions as diverse as CADASIL and sporadic white matter disease, remains incompletely understood. Although its peculiar vascular topology and precarious blood supply are likely to play a role, the cellular and molecular bases determining the characteristic vascular lesions remain to be defined. In particular, how aging and vascular risk factors interact with the vascular wall to induce vascular lesions preferentially in the white matter remains unclear. The relative contribution of hypoperfusion, BBB damage and oxidative stress to vascular and parenchymal damage remain to be defined. Furthermore, what determines the pathological diversity, e.g., lacunes, microinfarcts, microhemorrhages, etc., and spatial localization of the brain lesions resulting from similar vascular pathology remain unexplained. A better understanding of ischemic demyelination and abortive remyelination is needed. Fundamental questions concerning the interaction of AD pathology with vascular pathology also remains unanswered. Studies elucidating the vascular biology of the white matter and the interaction with risk factors and AD pathology would be needed to shed light on some of these issues and provide better insight into potential therapeutic targets. These mechanistic studies can benefit from the increasing availability of cell specific conditional genetic models, viral-based gene delivery methods, and novel approaches for targeted cell replacement/modification in the brain, e.g., (Goldman et al., 2012).
Developing treatments for VCI remains a challenge. In addition to the lack of predictive animal models to guide target selection, the heterogeneity of the underlying pathology represents a therapeutic challenge. The role of hypoperfusion, BBB disruption, oxidative stress and inflammation is well established in animal models of white matter damage, but therapies based on these pathogenic mechanisms have not been successful. Although it has been difficult to prove that these approaches achieved the intended effect on cerebral perfusion, ROS production and inflammation in the white matter at risk, other considerations make the development of treatments particularly challenging. For example, the long preclinical phase of dementia is problematic, since, in VCI as in AD, initiating therapy when patients become symptomatic may be too late. Furthermore, due to frequent overlap with AD, the diagnosis of VCI can be challenging, complicating the choice of the best therapeutic approach (Wang et al., 2012). Novel imaging modalities, including amyloid and tau imaging, as well as high resolution MRI, will go a long way in addressing some of these challenges, and will make possible to characterize the pathology in vivo with an unprecedented spatial, temporal and morphological accuracy. At the same time, these approaches offer the prospect of developing new biomarkers that will be critical for identifying patients at risk, staging the progression of the disease, and assessing therapeutic efficacy.
Considering that mixed dementia is the most common cause of dementia in the elderly, it has become increasingly important to harmonize basic science, translational, and clinical approaches in AD and vascular dementia. Thus, the impact of both pathologies should be considered, independently of whether their contribution is additive or synergistic. In the absence of effective therapies, promoting and maintaining vascular health seems critical to prevent both the vascular and neurodegenerative components of the disease and is probably the best possible course of action at the present.
Supplementary Material
Acknowledgments
We gratefully acknowledge the support from the NIH (NINDS: NS37853, NHLBI: HL96571) and the Alzheimer’s Association (ZEN-11-202707). Dr. Giuseppe Faraco provided invaluable help with the figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aarsland D, Aarsland D, Sardahaee FS, Sardahaee FS, Anderssen S, Anderssen S, Ballard C, Ballard C Alzheimer’s Society Systematic Review group, Alzheimer’s Society Systematic Review group. Is physical activity a potential preventive factor for vascular dementia? A systematic review. Aging Ment Health. 2010;14:386–395. doi: 10.1080/13607860903586136. [DOI] [PubMed] [Google Scholar]
- Abboud FM. Special characteristics of the cerebral circulation. Fed Proc. 1981;40:2296–2300. [PubMed] [Google Scholar]
- Ahmad AlA, Gassmann M, Ogunshola OO. Involvement of oxidative stress in hypoxia-induced blood-brain barrier breakdown. Microvasc Res. 2012;84:222–225. doi: 10.1016/j.mvr.2012.05.008. [DOI] [PubMed] [Google Scholar]
- Aho L, Jolkkonen J, Alafuzoff I. Beta-amyloid aggregation in human brains with cerebrovascular lesions. Stroke. 2006;37:2940–2945. doi: 10.1161/01.STR.0000248777.44128.93. [DOI] [PubMed] [Google Scholar]
- Akiguchi I, Tomimoto H, Suenaga T, Wakita H, Budka H. Blood-brain barrier dysfunction in Binswanger’s disease; an immunohistochemical study. Acta Neuropathol. 1998;95:78–84. doi: 10.1007/s004010050768. [DOI] [PubMed] [Google Scholar]
- Akinyemi R, Mukaetova-Ladinska E, Attems J, Attems J, Ihara M, Kalaria RN. Vascular Risk Factors and Neurodegeneration in Ageing related Dementias: Alzheimer’s Disease and Vascular Dementia. Curr Alzheimer Res. 2013;10:642–653. doi: 10.2174/15672050113109990037. [DOI] [PubMed] [Google Scholar]
- Alafuzoff I, Adolfsson R, Grundke-Iqbal I, Winblad B. Perivascular deposits of serum proteins in cerebral cortex in vascular dementia. Acta Neuropathol. 1985;66:292–298. doi: 10.1007/BF00690961. [DOI] [PubMed] [Google Scholar]
- Allan LM, Rowan EN, Firbank MJ, Thomas AJ, Parry SW, Polvikoski TM, O’Brien JT, Kalaria RN. Long term incidence of dementia, predictors of mortality and pathological diagnosis in older stroke survivors. Brain. 2011;134:3716–3727. doi: 10.1093/brain/awr273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonzo N, Hyman B, Rebeck G, Greenberg S. Progression of cerebral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels. J Neuropathol Exp Neurol. 1998;57:353–9. doi: 10.1097/00005072-199804000-00008. [DOI] [PubMed] [Google Scholar]
- Alosco ML, Brickman AM, Spitznagel MB, Garcia SL, Narkhede A, Griffith EY, Raz N, Cohen R, Sweet LH, Colbert LH, Josephson R, Hughes J, Rosneck J, Gunstad J. Cerebral Perfusion is Associated With White Matter Hyperintensities in Older Adults With Heart Failure. Congest Heart Fail. 2013;19:E29–E34. doi: 10.1111/chf.12025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando J, Yamamoto K. Flow detection and calcium signalling in vascular endothelial cells. Cardiovasc Res. 2013;99:260–268. doi: 10.1093/cvr/cvt084. [DOI] [PubMed] [Google Scholar]
- Andresen J, Shafi N, Bryan R. Endothelial influences on cerebrovascular tone. J Appl Physiol. 2006;100:318–327. doi: 10.1152/japplphysiol.00937.2005. [DOI] [PubMed] [Google Scholar]
- Appelman APA, Exalto LG, van der Graaf Y, Biessels GJ, Mali WPTM, Geerlings MI. White matter lesions and brain atrophy: more than shared risk factors? A systematic review. Cerebrovasc Dis. 2009;28:227–242. doi: 10.1159/000226774. [DOI] [PubMed] [Google Scholar]
- Appelman APA, van der Graaf Y, Vincken KL, Tiehuis AM, Witkamp TD, Mali WPTM, Geerlings MI SMART Study Group. Total cerebral blood flow, white matter lesions and brain atrophy: the SMART-MR study. J Cereb Blood Flow Metab. 2008;28:633–639. doi: 10.1038/sj.jcbfm.9600563. [DOI] [PubMed] [Google Scholar]
- Appelman APA, Vincken KL, van der Graaf Y, Vlek ALM, Witkamp TD, Mali WPTM, Geerlings MI SMART Study Group. White matter lesions and lacunar infarcts are independently and differently associated with brain atrophy: the SMART-MR study. Cerebrovasc Dis. 2010;29:28–35. doi: 10.1159/000255971. [DOI] [PubMed] [Google Scholar]
- Arai K, Lo EH. An oligovascular niche: cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J Neurosci. 2009;29:4351–4355. doi: 10.1523/JNEUROSCI.0035-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K, Lo EH. Astrocytes protect oligodendrocyte precursor cells via MEK/ERK and PI3K/Akt signaling. J Neurosci Res. 2010;88:758–763. doi: 10.1002/jnr.22256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- Attems J, Jellinger K, Thal DR, Van Nostrand W. Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol. 2011;37:75–93. doi: 10.1111/j.1365-2990.2010.01137.x. [DOI] [PubMed] [Google Scholar]
- Attwell D, Iadecola C. The neural basis of functional brain imaging signals. Trends Neurosci. 2002;25:621–625. doi: 10.1016/s0166-2236(02)02264-6. [DOI] [PubMed] [Google Scholar]
- Auriel E, Greenberg SM. The pathophysiology and clinical presentation of cerebral amyloid angiopathy. Curr Atheroscler Rep. 2012;14:343–350. doi: 10.1007/s11883-012-0254-z. [DOI] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Kroenke CD, Sherman LS, Lawrence G, Gong X, Taber EN, Sonnen JA, Larson EB, Montine TJ. White matter lesions defined by diffusion tensor imaging in older adults. Ann Neurol. 2011;70:465–476. doi: 10.1002/ana.22484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagher P, Segal SS. Regulation of blood flow in the microcirculation: role of conducted vasodilation. Acta Physiol (Oxf) 2011;202:271–284. doi: 10.1111/j.1748-1716.2010.02244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker SL, de Leeuw FE, de Groot JC, Hofman A, Koudstaal PJ, Breteler MM. Cerebral vasomotor reactivity and cerebral white matter lesions in the elderly. Neurology. 1999;52:578–583. doi: 10.1212/wnl.52.3.578. [DOI] [PubMed] [Google Scholar]
- Balestrini S, Perozzi C, Altamura C, Vernieri F, Luzzi S, Bartolini M, Provinciali L, Silvestrini M. Severe carotid stenosis and impaired cerebral hemodynamics can influence cognitive deterioration. Neurology. 2013;80:2145–2150. doi: 10.1212/WNL.0b013e318295d71a. [DOI] [PubMed] [Google Scholar]
- Bechmann I, Mor G, Nilsen J, Eliza M, Nitsch R, Naftolin F. FasL (CD95L, Apo1L) is expressed in the normal rat and human brain: evidence for the existence of an immunological brain barrier. Glia. 1999;27:62–74. doi: 10.1002/(sici)1098-1136(199907)27:1<62::aid-glia7>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Bell R, Deane R, Chow N, Long X, Sagare A, Singh I, Streb J, Guo H, Rubio A, Van Nostrand W, Miano J, Zokovic B. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat Cell Biol. 2009;11:143–153. doi: 10.1038/ncb1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Sagare AP, Singh I, Larue B, Deane R, Zlokovic BV. Pericytes Control Key Neurovascular Functions and Neuronal Phenotype in the Adult Brain and during Brain Aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk B, Zlokovic B. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedictus MR, Goos JDC, Binnewijzend MAA, Muller M, Barkhof F, Scheltens P, Prins ND, van der Flier WM. Specific risk factors for microbleeds and white matter hyperintensities in Alzheimer’s disease. Neurobiol Aging. 2013;34:2488–2494. doi: 10.1016/j.neurobiolaging.2013.04.023. [DOI] [PubMed] [Google Scholar]
- Bertram L, Tanzi R. Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci. 2008;9:768–778. doi: 10.1038/nrn2494. [DOI] [PubMed] [Google Scholar]
- Bevan JA. Sites of transition between functional systemic and cerebral arteries of rabbits occur at embryological junctional sites. Science. 1979;204:635–637. doi: 10.1126/science.432670. [DOI] [PubMed] [Google Scholar]
- Black S, Gao F, Bilbao J. Understanding white matter disease: imaging-pathological correlations in vascular cognitive impairment. Stroke. 2009;40:S48–S52. doi: 10.1161/STROKEAHA.108.537704. [DOI] [PubMed] [Google Scholar]
- Blinder P, Tsai PS, Kaufhold JP, Knutsen PM, Suhl H, Kleinfeld D. The cortical angiome: an interconnected vascular network with noncolumnar patterns of blood flow. Nat Neurosci. 2013;16:889–897. doi: 10.1038/nn.3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowler J. Modern concept of vascular cognitive impairment. Br Med Bull. 2007;83:291–305. doi: 10.1093/bmb/ldm021. [DOI] [PubMed] [Google Scholar]
- Brickman AM, Siedlecki KL, Muraskin J, Manly JJ, Luchsinger JA, Yeung LK, Brown TR, DeCarli C, Stern Y. White matter hyperintensities and cognition: testing the reserve hypothesis. Neurobiol Aging. 2011;32:1588–1598. doi: 10.1016/j.neurobiolaging.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brisset M, Boutouyrie P, Pico F, Zhu Y, Zureik M, Schilling S, Dufouil C, Mazoyer B, Laurent S, Tzourio C, Debette S. Large-vessel correlates of cerebral small-vessel disease. Neurology. 2013;80:662–669. doi: 10.1212/WNL.0b013e318281ccc2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brix B, Mesters JR, Pellerin L, Jöhren O. Endothelial cell-derived nitric oxide enhances aerobic glycolysis in astrocytes via HIF-1α-mediated target gene activation. J Neurosci. 2012;32:9727–9735. doi: 10.1523/JNEUROSCI.0879-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown WR, Thore CR. Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol Appl Neurobiol. 2011;37:56–74. doi: 10.1111/j.1365-2990.2010.01139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown W, Moody D, Thore C, Challa V, Anstrom J. Vascular dementia in leukoaraiosis may be a consequence of capillary loss not only in the lesions, but in normal-appearing white matter and cortex as well. J Neurol Sci. 2007;257:62–66. doi: 10.1016/j.jns.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JM, Kobayashi H, Rafii S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer. 2010;10:138–146. doi: 10.1038/nrc2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler R, Radhakrishnan R. Dementia. Clin Evid (Online) Sep. 2012;12:1–27. [PMC free article] [PubMed] [Google Scholar]
- Candelario-Jalil E, Thompson J, Taheri S, Grossetete M, Adair JC, Edmonds E, Prestopnik J, Wills J, Rosenberg GA. Matrix metalloproteinases are associated with increased blood-brain barrier opening in vascular cognitive impairment. Stroke. 2011;42:1345–1350. doi: 10.1161/STROKEAHA.110.600825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carare RO, Hawkes CA, Jeffrey M, Kalaria RN, Weller RO. Cerebral amyloid angiopathy, Prion angiopathy, CADASIL and the spectrum of Protein Elimination-Failure Angiopathies (PEFA) in neurodegenerative disease with a focus on therapy. Neuropathol Appl Neurobiol. 2013;39:593–611. doi: 10.1111/nan.12042. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Ruiz de Almodovar C. VEGF ligands and receptors: implications in neurodevelopment and neurodegeneration. Cell Mol Life Sci. 2013;70:1763–1778. doi: 10.1007/s00018-013-1283-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Deane R, Gottesdiener AJ, Verghese PB, Stewart FR, West T, Paoletti AC, Kasper TR, DeMattos RB, Zlokovic BV, Holtzman DM. Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Aβ clearance in a mouse model of β-amyloidosis. Proc Natl Acad Sci USA. 2012;109:15502–15507. doi: 10.1073/pnas.1206446109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauli B, Hamel E. Revisiting the role of neurons in neurovascular coupling. Front Neuroenergetics. 2010;2:1–9. doi: 10.3389/fnene.2010.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG. Cadasil. Lancet Neurol. 2009;8:643–653. doi: 10.1016/S1474-4422(09)70127-9. [DOI] [PubMed] [Google Scholar]
- Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatr. 2012;83:124–137. doi: 10.1136/jnnp-2011-301308. [DOI] [PubMed] [Google Scholar]
- Chen JJ, Rosas HD, Salat DH. The relationship between cortical blood flow and sub-cortical white-matter health across the adult age span. PLoS ONE. 2013a;8:e56733. doi: 10.1371/journal.pone.0056733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Yang M, Guo J, Zhou M, Zhu C, He L. Cerebrolysin for vascular dementia. Cochrane Database Syst Rev. 2013b doi: 10.1002/14651858.CD008900.pub2. [DOI] [PubMed] [Google Scholar]
- Chen PL, Wang PY, Sheu WH, Chen YT, Ho YP, Hu HH, Hsu HY. Changes of brachial flow-mediated vasodilation in different ischemic stroke subtypes. Neurology. 2006;67:1056–1058. doi: 10.1212/01.wnl.0000237526.32692.67. [DOI] [PubMed] [Google Scholar]
- Cheng HL, Lin CJ, Soong BW, Wang PN, Chang FC, Wu YT, Chou KH, Lin CP, Tu PC, Lee IH. Impairments in cognitive function and brain connectivity in severe asymptomatic carotid stenosis. Stroke. 2012;43:2567–2573. doi: 10.1161/STROKEAHA.111.645614. [DOI] [PubMed] [Google Scholar]
- Chu K, Jung KH, Lee ST, Park HK, Sinn DI, Kim JM, Kim DH, Kim JH, Kim SJ, Song EC, Kim M, Lee SK, Roh JK. Circulating endothelial progenitor cells as a new marker of endothelial dysfunction or repair in acute stroke. Stroke. 2008;39:1441–1447. doi: 10.1161/STROKEAHA.107.499236. [DOI] [PubMed] [Google Scholar]
- Chui HC, Zarow C, Mack WJ, Ellis WG, Zheng L, Jagust WJ, Mungas D, Reed BR, Kramer JH, Decarli CC, Weiner MW, Vinters HV. Cognitive impact of subcortical vascular and Alzheimer’s disease pathology. Ann Neurol. 2006;60:677–687. doi: 10.1002/ana.21009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chui HC, Zheng L, Reed BR, Vinters HV, Mack WJ. Vascular risk factors and Alzheimer’s disease: are these risk factors for plaques and tangles or for concomitant vascular pathology that increases the likelihood of dementia? An evidence-based review. Alzheimers Res Ther. 2012;4:1. doi: 10.1186/alzrt98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolla M. The Cerebral Circulation. 2010 doi: 10.4199/C00005ED1V01Y200912ISP002. [DOI] [Google Scholar]
- Claassen JAHR, Diaz-Arrastia R, Martin-Cook K, Levine BD, Zhang R. Altered cerebral hemodynamics in early Alzheimer disease: a pilot study using transcranial Doppler. J Alzheimers Dis. 2009;17:621–629. doi: 10.3233/JAD-2009-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen RA, Tong X. Vascular oxidative stress: the common link in hypertensive and diabetic vascular disease. J Cardiovasc Pharmacol. 2010;55:308–316. doi: 10.1097/fjc.0b013e3181d89670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordonnier C, van der Flier WM. Brain microbleeds and Alzheimer’s disease: innocent observation or key player? Brain. 2011;134:335–344. doi: 10.1093/brain/awq321. [DOI] [PubMed] [Google Scholar]
- Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol. 2009;66:300–305. doi: 10.1001/archneurol.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crehan H, Hardy J, Pocock J. Blockage of CR1 prevents activation of rodent microglia. Neurobiol Dis. 2013;54:139–149. doi: 10.1016/j.nbd.2013.02.003. [DOI] [PubMed] [Google Scholar]
- D’haeseleer M, Beelen R, Fierens Y, Cambron M, Vanbinst AM, Verborgh C, Demey J, De Keyser J. Cerebral hypoperfusion in multiple sclerosis is reversible and mediated by endothelin-1. Proc Natl Acad Sci USA. 2013;110:5654–5658. doi: 10.1073/pnas.1222560110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’haeseleer M, Cambron M, Vanopdenbosch L, De Keyser J. Vascular aspects of multiple sclerosis. Lancet Neurol. 2011;10:657–666. doi: 10.1016/S1474-4422(11)70105-3. [DOI] [PubMed] [Google Scholar]
- Dam ten VH, van den Heuvel DMJ, de Craen AJM, Bollen ELEM, Murray HM, Westendorp RGJ, Blauw GJ, van Buchem MA. Decline in total cerebral blood flow is linked with increase in periventricular but not deep white matter hyperintensities. Radiology. 2007;243:198–203. doi: 10.1148/radiol.2431052111. [DOI] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity of neural circuits. Neuron. 2004;44:23–30. doi: 10.1016/j.neuron.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Davalos D, Akassoglou K. Fibrinogen as a key regulator of inflammation in disease. Semin Immunopathol. 2012;34:43–62. doi: 10.1007/s00281-011-0290-8. [DOI] [PubMed] [Google Scholar]
- Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, Deerinck TJ, Smirnoff DS, Bedard C, Hakozaki H, Gonias Murray S, Ling JB, Lassmann H, Degen JL, Ellisman MH, Akassoglou K. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun. 2012;3:1227. doi: 10.1038/ncomms2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keyser J, Steen C, Mostert JP, Koch MW. Hypoperfusion of the cerebral white matter in multiple sclerosis: possible mechanisms and pathophysiological significance. J Cereb Blood Flow Metab. 2008;28:1645–1651. doi: 10.1038/jcbfm.2008.72. [DOI] [PubMed] [Google Scholar]
- De Reuck J. The human periventricular arterial blood supply and the anatomy of cerebral infarctions. Eur Neurol. 1971;5:321–334. doi: 10.1159/000114088. [DOI] [PubMed] [Google Scholar]
- De Reuck JL. Histopathological stainings and definitions of vascular disruptions in the elderly brain. Exp Gerontol. 2012;47:834–837. doi: 10.1016/j.exger.2012.03.012. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu H, Spijkers P, Guo H, Song X, Lenting P, Van Nostrand W, Zlokovic B. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Decarli C, Murphy DG, Tranh M, Grady CL, Haxby JV, Gillette JA, Salerno JA, Gonzales-Aviles A, Horwitz B, Rapoport SI. The effect of white matter hyperintensity volume on brain structure, cognitive performance, and cerebral metabolism of glucose in 51 healthy adults. Neurology. 1995;45:2077–2084. doi: 10.1212/wnl.45.11.2077. [DOI] [PubMed] [Google Scholar]
- Delles C, Michelson G, Harazny J, Oehmer S, Hilgers KF, Schmieder RE. Impaired endothelial function of the retinal vasculature in hypertensive patients. Stroke. 2004;35:1289–1293. doi: 10.1161/01.STR.0000126597.11534.3b. [DOI] [PubMed] [Google Scholar]
- Deschaintre Y, Richard F, Leys D, Pasquier F. Treatment of vascular risk factors is associated with slower decline in Alzheimer disease. Neurology. 2009;73:674–680. doi: 10.1212/WNL.0b013e3181b59bf3. [DOI] [PubMed] [Google Scholar]
- Dichgans M, Zietemann V. Prevention of vascular cognitive impairment. Stroke. 2012;43:3137–3146. doi: 10.1161/STROKEAHA.112.651778. [DOI] [PubMed] [Google Scholar]
- Domercq M, Perez-Samartin A, Aparicio D, Alberdi E, Pampliega O, Matute C. P2X7 receptors mediate ischemic damage to oligodendrocytes. Glia. 2010;58:730–740. doi: 10.1002/glia.20958. [DOI] [PubMed] [Google Scholar]
- Dong YF, Kataoka K, Toyama K, Sueta D, Koibuchi N, Yamamoto E, Yata K, Tomimoto H, Ogawa H, Kim-Mitsuyama S. Attenuation of brain damage and cognitive impairment by direct renin inhibition in mice with chronic cerebral hypoperfusion. Hypertension. 2011;58:635–642. doi: 10.1161/HYPERTENSIONAHA.111.173534. [DOI] [PubMed] [Google Scholar]
- Drake CT, Iadecola C. The role of neuronal signaling in controlling cerebral blood flow. Brain Lang. 2007;102:141–152. doi: 10.1016/j.bandl.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duering M, Righart R, Csanadi E, Jouvent E, Hervé D, Chabriat H, Dichgans M. Incident subcortical infarcts induce focal thinning in connected cortical regions. Neurology. 2012;79:2025–2028. doi: 10.1212/WNL.0b013e3182749f39. [DOI] [PubMed] [Google Scholar]
- Dugas JC, Mandemakers W, Rogers M, Ibrahim A, Daneman R, Barres BA. A novel purification method for CNS projection neurons leads to the identification of brain vascular cells as a source of trophic support for corticospinal motor neurons. J Neurosci. 2008;28:8294–8305. doi: 10.1523/JNEUROSCI.2010-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyrna F, Hanske S, Krueger M, Bechmann I. The Blood-Brain Barrier. J Neuroimmune Pharmacol. 2013;8:763–773. doi: 10.1007/s11481-013-9473-5. [DOI] [PubMed] [Google Scholar]
- Elbaz A, Vicente-Vytopilova P, Tavernier B, Sabia S, Dumurgier J, Mazoyer B, Singh-Manoux A, Tzourio C. Motor function in the elderly: Evidence for the reserve hypothesis. Neurology. 2013;81:417–426. doi: 10.1212/WNL.0b013e31829d8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergul A. Endothelin-1 and diabetic complications: Focus on the vasculature. Pharmacol Res. 2011 doi: 10.1016/j.phrs.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esiri M, Nagy Z, Smith M, Barnetson L, Smith A. Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer’s disease. Lancet. 1999;354:919–20. doi: 10.1016/S0140-6736(99)02355-7. [DOI] [PubMed] [Google Scholar]
- Fancy SPJ, Chan JR, Baranzini SE, Franklin RJM, Rowitch DH. Myelin regeneration: a recapitulation of development? Annu Rev Neurosci. 2011a;34:21–43. doi: 10.1146/annurev-neuro-061010-113629. [DOI] [PubMed] [Google Scholar]
- Fancy SPJ, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, Bruce CC, Otero JJ, Huang EJ, Nusse R, Franklin RJM, Rowitch DH. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat Neurosci. 2011;14:1009–1016. doi: 10.1038/nn.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraci FM. Protecting Against Vascular Disease in Brain. Am J Physiol Heart Circ Physiol. 2011;300:H1566–H1582. doi: 10.1152/ajpheart.01310.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federico A, Di Donato I, Bianchi S, Di Palma C, Taglia I, Dotti MT. Hereditary cerebral small vessel diseases: a review. J Neurol Sci. 2012;322:25–30. doi: 10.1016/j.jns.2012.07.041. [DOI] [PubMed] [Google Scholar]
- Fernando MS, Simpson JE, Matthews F, Brayne C, Lewis CE, Barber R, Kalaria RN, Forster G, Esteves F, Wharton SB, Shaw PJ, O’Brien JT, Ince PG MRC Cognitive Function and Ageing Neuropathology Study Group . White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke. 2006;37:1391–1398. doi: 10.1161/01.STR.0000221308.94473.14. [DOI] [PubMed] [Google Scholar]
- Fields RD. Neuroscience. Change in the brain’s white matter. Science. 2010;330:768–769. doi: 10.1126/science.1199139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillit H, Nash DT, Rundek T, Zuckerman A. Cardiovascular risk factors and dementia. Am J Geriatr Pharmacother. 2008;6:100–118. doi: 10.1016/j.amjopharm.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Fornage M, Debette S, Bis JC, Schmidt H, Ikram MA, Dufouil C, Sigurdsson S, Lumley T, DeStefano AL, Fazekas F, Vrooman HA, Shibata DK, Maillard P, Zijdenbos A, Smith AV, Gudnason H, de Boer R, Cushman M, Mazoyer B, Heiss G, Vernooij MW, Enzinger C, Glazer NL, Beiser A, Knopman DS, Cavalieri M, Niessen WJ, Harris TB, Petrovic K, Lopez OL, Au R, Lambert J-C, Hofman A, Gottesman RF, Garcia M, Heckbert SR, Atwood LD, Catellier DJ, Uitterlinden AJ, Yang Q, Smith NL, Aspelund T, Romero JR, Rice K, Taylor KD, Nalls MA, Rotter JI, Sharrett R, van Duijn CM, Amouyel P, Wolf PA, Gudnason V, van der Lugt A, Boerwinkle E, Psaty BM, Seshadri S, Tzourio C, Breteler MB, Mosley TH, Schmidt R, Longstreth WT, DeCarli C, Launer LJ. Genome-wide association studies of cerebral white matter lesion burden: the CHARGE consortium. Ann Neurol. 2011;69:928–939. doi: 10.1002/ana.22403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJM, Ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 2008;9:839–855. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- French HM, Reid M, Mamontov P, Simmons RA, Grinspan JB. Oxidative stress disrupts oligodendrocyte maturation. J Neurosci Res. 2009;87:3076–3087. doi: 10.1002/jnr.22139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. 2007;28:12–18. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Gallacher J, Bayer A, Lowe G, Fish M, Pickering J, Pedro S, Dunstan F, White J, Yarnell J, Ben-Shlomo Y. Is sticky blood bad for the brain?: Hemostatic and inflammatory systems and dementia in the Caerphilly Prospective Study. Arterioscler Thromb Vasc Biol. 2010;30:599–604. doi: 10.1161/ATVBAHA.109.197368. [DOI] [PubMed] [Google Scholar]
- Gao YZ, Zhang JJ, Liu H, Wu GY, Xiong L, Shu M. Regional cerebral blood flow and cerebrovascular reactivity in Alzheimer’s disease and vascular dementia assessed by arterial spinlabeling magnetic resonance imaging. Current Neurovascular Research. 2013;10:49–53. doi: 10.2174/156720213804806016. [DOI] [PubMed] [Google Scholar]
- Garcia-Alloza M, Gregory J, Kuchibhotla KV, Fine S, Wei Y, Ayata C, Frosch MP, Greenberg SM, Bacskai BJ. Cerebrovascular lesions induce transient β-amyloid deposition. Brain. 2011;134:3697–3707. doi: 10.1093/brain/awr300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelber RP, Launer LJ, White LR. The Honolulu-Asia Aging Study: epidemiologic and neuropathologic research on cognitive impairment. Curr Alzheimer Res. 2012;9:664–672. doi: 10.2174/156720512801322618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner G, Wong C. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Gold G, Giannakopoulos P, Herrmann FR, Bouras C, Kövari E. Identification of Alzheimer and vascular lesion thresholds for mixed dementia. Brain. 2007;130:2830–2836. doi: 10.1093/brain/awm228. [DOI] [PubMed] [Google Scholar]
- Goldman SA, Nedergaard M, Windrem MS. Glial progenitor cell-based treatment and modeling of neurological disease. Science. 2012;338:491–495. doi: 10.1126/science.1218071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelick PB, Scuteri A, Black SE, DeCarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Román GC, Sellke FW, Seshadri S. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke. 2011;42:2672–2713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouw AA, Seewann A, van der Flier WM, Barkhof F, Rozemuller AM, Scheltens P, Geurts JJG. Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatr. 2011;82:126–135. doi: 10.1136/jnnp.2009.204685. [DOI] [PubMed] [Google Scholar]
- Graumann U, Reynolds R, Steck AJ, Schaeren-Wiemers N. Molecular changes in normal appearing white matter in multiple sclerosis are characteristic of neuroprotective mechanisms against hypoxic insult. Brain Pathol. 2003;13:554–573. doi: 10.1111/j.1750-3639.2003.tb00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Kim W, Lok J, Lee S, Besancon E, Luo B, Stins M, Wang X, Dedhar S, Lo E. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci U S A. 2008;105:7582–7587. doi: 10.1073/pnas.0801105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachinski V, Bowler J. Vascular dementia. Neurology. 1993;43:2159–2160. doi: 10.1212/wnl.43.10.2159-a. [DOI] [PubMed] [Google Scholar]
- Hachinski VC, Potter P, Merskey H. Leuko-araiosis. Arch Neurol. 1987;44:21–23. doi: 10.1001/archneur.1987.00520130013009. [DOI] [PubMed] [Google Scholar]
- Hachinski V, Lassen N, Marshall J. Multi-infarct dementia. A cause of mental deterioration in the elderly. Lancet. 1974;2:207–10. doi: 10.1016/s0140-6736(74)91496-2. [DOI] [PubMed] [Google Scholar]
- Hachinski V. Stroke and Alzheimer disease: fellow travelers or partners in crime? Arch Neurol. 2011;68:797–798. doi: 10.1001/archneurol.2011.118. [DOI] [PubMed] [Google Scholar]
- Haight TJ, Landau SM, Carmichael O, Schwarz C, DeCarli C, Jagust WJ for the Alzheimer’s Disease Neuroimaging Initiative. Dissociable Effects of Alzheimer Disease and White Matter Hyperintensities on Brain Metabolism. JAMA Neurol. 2013:1–8. doi: 10.1001/jamaneurol.2013.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainsworth AH, Brittain JF, Khatun H. Pre-clinical models of human cerebral small vessel disease: utility for clinical application. J Neurol Sci. 2012;322:237–240. doi: 10.1016/j.jns.2012.05.046. [DOI] [PubMed] [Google Scholar]
- Halliday MR, Pomara N, Sagare AP, Mack WJ, Frangione B, Zlokovic BV. Relationship between cyclophilin a levels and matrix metalloproteinase 9 activity in cerebrospinal fluid of cognitively normal apolipoprotein e4 carriers and blood-brain barrier breakdown. JAMA Neurol. 2013;70:1198–1200. doi: 10.1001/jamaneurol.2013.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel H, Lista S, Khachaturian ZS. Development of biomarkers to chart all Alzheimer’s disease stages: the royal road to cutting the therapeutic Gordian Knot. Alzheimers Dement. 2012;8:312–336. doi: 10.1016/j.jalz.2012.05.2116. [DOI] [PubMed] [Google Scholar]
- Hanyu H, Asano T, Tanaka Y, Iwamoto T, Takasaki M, Abe K. Increased blood-brain barrier permeability in white matter lesions of Binswanger’s disease evaluated by contrast-enhanced MRI. Dement Geriatr Cogn Disord. 2002;14:1–6. doi: 10.1159/000058326. [DOI] [PubMed] [Google Scholar]
- Harris JJ, Attwell D. The energetics of CNS white matter. J Neurosci. 2012;32:356–371. doi: 10.1523/JNEUROSCI.3430-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. 2012;75:762–777. doi: 10.1016/j.neuron.2012.08.019. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Pham LDD, Katusic ZS, Arai K, Lo EH. Astrocytic high-mobility group box 1 promotes endothelial progenitor cell-mediated neurovascular remodeling during stroke recovery. Proc Natl Acad Sci USA. 2012;109:7505–7510. doi: 10.1073/pnas.1121146109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head D, Bugg JM, Goate AM, Fagan AM, Mintun MA, Benzinger T, Holtzman DM, Morris JC. Exercise Engagement as a Moderator of the Effects of APOE Genotype on Amyloid Deposition. Arch Neurol. 2012;69:636–643. doi: 10.1001/archneurol.2011.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JM, Zalos G, Halcox JPJ, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- Honig L, Kukull W, Mayeux R. Atherosclerosis and AD: analysis of data from the US National Alzheimer’s Coordinating Center. Neurology. 2005;64:494–500. doi: 10.1212/01.WNL.0000150886.50187.30. [DOI] [PubMed] [Google Scholar]
- Honjo K, Black SE, Verhoeff NPLG. Alzheimer’s disease, cerebrovascular disease, and the β-amyloid cascade. Can J Neurol Sci. 2012;39:712–728. doi: 10.1017/s0317167100015547. [DOI] [PubMed] [Google Scholar]
- Huang Y, Zhang W, Lin L, Feng J, Chen F, Wei W, Zhao X, Guo W, Li J, Yin W, Li L. Is endothelial dysfunction of cerebral small vessel responsible for white matter lesions after chronic cerebral hypoperfusion in rats? J Neurol Sci. 2010;299:72–80. doi: 10.1016/j.jns.2010.08.035. [DOI] [PubMed] [Google Scholar]
- Hulette C, Nochlin D, McKeel D, Morris JC, Mirra SS, Sumi SM, Heyman A. Clinical-neuropathologic findings in multi-infarct dementia: a report of six autopsied cases. Neurology. 1997;48:668–672. doi: 10.1212/wnl.48.3.668. [DOI] [PubMed] [Google Scholar]
- Hur J, Yang HM, Yoon CH, Lee CS, Park KW, Kim JH, Kim TY, Kim JY, Kang HJ, Chae IH, Oh B-H, Park Y-B, Kim H-S. Identification of a novel role of T cells in postnatal vasculogenesis: characterization of endothelial progenitor cell colonies. Circulation. 2007;116:1671–1682. doi: 10.1161/CIRCULATIONAHA.107.694778. [DOI] [PubMed] [Google Scholar]
- Hurd MD, Martorell P, Delavande A, Mullen KJ, Langa KM. Monetary costs of dementia in the United States. N Engl J Med. 2013;368:1326–1334. doi: 10.1056/NEJMsa1204629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao KK, Carlson GA. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci. 1999;2:157–161. doi: 10.1038/5715. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Iadecola C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol. 2010;120:287–296. doi: 10.1007/s00401-010-0718-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Davisson RL. Hypertension and cerebrovascular dysfunction. Cell Metab. 2008;7:476–484. doi: 10.1016/j.cmet.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihara M, Tomimoto H, Kinoshita M, Oh J, Noda M, Wakita H, Akiguchi I, Shibasaki H. Chronic cerebral hypoperfusion induces MMP-2 but not MMP-9 expression in the microglia and vascular endothelium of white matter. J Cereb Blood Flow Metab. 2001;21:828–834. doi: 10.1097/00004647-200107000-00008. [DOI] [PubMed] [Google Scholar]
- Ihara M, Tomimoto H. Lessons from a mouse model characterizing features of vascular cognitive impairment with white matter changes. J Aging Res. 2011;2011:978761. doi: 10.4061/2011/978761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Lee H, Yu M, Feng T, Logan J, Nedergaard M, Benveniste H. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J CLin Invest. 2013;123:1299–1309. doi: 10.1172/JCI67677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inzitari ID, Pracucci G, Poggesi A, Carlucci G, Barkhof F, Chabriat H, Erkinjuntti T, Fazekas F, Ferro JM, Hennerici M the LADIS study group. Changes in white matter as determinant of global functional decline in older independent outpatients: three year follow-up of LADIS (leukoaraiosis and disability) study cohort. Bmj. 2009;339:b2477. doi: 10.1136/bmj.b2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Takeuchi S, Hasuo K, Morooka M, Kubota K, Matsuda H. Cerebral blood flow on ECD SPECT in a patient with adult onset Alexander disease. Clin Nucl Med. 2009;34:931–933. doi: 10.1097/RLU.0b013e3181bed1ef. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Small concomitant cerebrovascular lesions are not important for cognitive decline in severe Alzheimer disease. Arch Neurol. 2001;58:520–521. doi: 10.1001/archneur.58.3.520. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Alzheimer 100--highlights in the history of Alzheimer research. J Neural Transm. 2006;113:1603–1623. doi: 10.1007/s00702-006-0578-3. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Pathology and pathogenesis of vascular cognitive impairment-a critical update. Front Aging Neurosci. 2013;5:1–19. doi: 10.3389/fnagi.2013.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jendroska K, Poewe W, Daniel SE, Pluess J, Iwerssen-Schmidt H, Paulsen J, Barthel S, Schelosky L, Cervós-Navarro J, DeArmond SJ. Ischemic stress induces deposition of amyloid beta immunoreactivity in human brain. Acta Neuropathol. 1995;90:461–466. doi: 10.1007/BF00294806. [DOI] [PubMed] [Google Scholar]
- Jennings J, Muldoon M, Ryan C, Price J, Greer P, Sutton-Tyrrell K, van der Veen F, Meltzer C. Reduced cerebral blood flow response and compensation among patients with untreated hypertension. Neurology. 2005;64:1358–1365. doi: 10.1212/01.WNL.0000158283.28251.3C. [DOI] [PubMed] [Google Scholar]
- Jickling G, Salam A, Mohammad A, Hussain MS, Scozzafava J, Nasser AM, Jeerakathil T, Shuaib A, Camicioli R. Circulating endothelial progenitor cells and age-related white matter changes. Stroke. 2009;40:3191–3196. doi: 10.1161/STROKEAHA.109.554527. [DOI] [PubMed] [Google Scholar]
- Johnston SC, O’Meara ES, Manolio TA, Lefkowitz D, O’Leary DH, Goldstein S, Carlson MC, Fried LP, Longstreth WT. Cognitive impairment and decline are associated with carotid artery disease in patients without clinically evident cerebrovascular disease. Ann Intern Med. 2004;140:237–247. doi: 10.7326/0003-4819-140-4-200402170-00005. [DOI] [PubMed] [Google Scholar]
- Jokinen H, Gouw AA, Madureira S, Ylikoski R, van Straaten ECW, van der Flier WM, Barkhof F, Scheltens P, Fazekas F, Schmidt R the LADIS study group. Incident lacunes influence cognitive decline: the LADIS study. Neurology. 2011;76:1872–1878. doi: 10.1212/WNL.0b013e31821d752f. [DOI] [PubMed] [Google Scholar]
- Jonsson M, Zetterberg H, Rolstad S, Edman A, Gouw AA, Bjerke M, Lind K, Blennow K, Pantoni L, Inzitari D, Wallin A. Low cerebrospinal fluid sulfatide predicts progression of white matter lesions: The LADIS study. Dement Geriatr Cogn Disord. 2012;34:61–67. doi: 10.1159/000341576. [DOI] [PubMed] [Google Scholar]
- Juma WM, Lira A, Marzuk A, Marzuk Z, Hakim AM, Thompson CS. C-reactive protein expression in a rodent model of chronic cerebral hypoperfusion. Brain Res. 2011;1414:85–93. doi: 10.1016/j.brainres.2011.07.047. [DOI] [PubMed] [Google Scholar]
- Justin BN, Turek M, Hakim AM. Heart disease as a risk factor for dementia. Clin Epidemiol. 2013;5:135–145. doi: 10.2147/CLEP.S30621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Lemaire H, Unterbeck A, Salbaum J, Masters C, Grzeschik K, Multhaup G, Beyreuther K, Muller H. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- Kazama K, Anrather J, Zhou P, Girouard H, Frys K, Milner TA, Iadecola C. Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase-derived radicals. Circ Res. 2004;95:1019–1026. doi: 10.1161/01.RES.0000148637.85595.c5. [DOI] [PubMed] [Google Scholar]
- Kearney-Schwartz A, Rossignol P, Bracard S, Felblinger J, Fay R, Boivin JM, Lecompte T, Lacolley P, Benetos A, Zannad F. Vascular structure and function is correlated to cognitive performance and white matter hyperintensities in older hypertensive patients with subjective memory complaints. Stroke. 2009;40:1229–1236. doi: 10.1161/STROKEAHA.108.532853. [DOI] [PubMed] [Google Scholar]
- Kim JS, Yun I, Choi YB, Lee KS, Kim YI. Ramipril protects from free radical induced white matter damage in chronic hypoperfusion in the rat. J Clin Neurosci. 2008a;15:174–178. doi: 10.1016/j.jocn.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Kim YS, Immink RV, Stok WJ, Karemaker JM, Secher NH, van Lieshout JJ. Dynamic cerebral autoregulatory capacity is affected early in Type 2 diabetes. Clin Sci. 2008b;115:255–262. doi: 10.1042/CS20070458. [DOI] [PubMed] [Google Scholar]
- Kitaguchi H, Tomimoto H, Ihara M, Shibata M, Uemura K, Kalaria RN, Kihara T, Asada-Utsugi M, Kinoshita A, Takahashi R. Chronic cerebral hypoperfusion accelerates amyloid beta deposition in APPSwInd transgenic mice. Brain Res. 2009;1294:202–210. doi: 10.1016/j.brainres.2009.07.078. [DOI] [PubMed] [Google Scholar]
- Kleinfeld D, Blinder P, Drew PJ, Driscoll JD, Muller A, Tsai PS, Shih AY. A guide to delineate the logic of neurovascular signaling in the brain. Front Neuroenergetics. 2011;3:1–9. doi: 10.3389/fnene.2011.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knottnerus ILH, Ten Cate H, Lodder J, Kessels F, van Oostenbrugge RJ. Endothelial dysfunction in lacunar stroke: a systematic review. Cerebrovasc Dis. 2009;27:519–526. doi: 10.1159/000212672. [DOI] [PubMed] [Google Scholar]
- Knottnerus ILH, Govers-Riemslag JWP, Hamulyak K, Rouhl RPW, Staals J, Spronk HMH, van Oerle R, van Raak EPM, Lodder J, Ten Cate H, van Oostenbrugge RJ. Endothelial activation in lacunar stroke subtypes. Stroke. 2010;41:1617–1622. doi: 10.1161/STROKEAHA.109.576223. [DOI] [PubMed] [Google Scholar]
- Kobari M, Meyer JS, Ichijo M, Oravez WT. Leukoaraiosis: correlation of MR and CT findings with blood flow, atrophy, and cognition. AJNR Am J Neuroradiol. 1990;11:273–281. [PMC free article] [PubMed] [Google Scholar]
- Koike MA, Green KN, Blurton-Jones M, LaFerla FM. Oligemic Hypoperfusion Differentially Affects Tau and Amyloid-{beta} Am J Pathol. 2010;177:300–310. doi: 10.2353/ajpath.2010.090750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Kettunen M, Goldsteins G, Keinanen R, Salminen A, Ort M, Bures J, Liu D, Kauppinen R, Higgins L, Koistinaho J. Beta-amyloid precursor protein transgenic mice that harbor diffuse A beta deposits but do not form plaques show increased ischemic vulnerability: role of inflammation. Proc Natl Acad Sci U S A. 2002;99:1610–5. doi: 10.1073/pnas.032670899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner EJ, Weil BR, MacEneaney OJ, Morgan RG, Mestek ML, Van Guilder GP, Diehl KJ, Stauffer BL, DeSouza CA. Human aging and CD31+ T-cell number, migration, apoptotic susceptibility, and telomere length. J Appl Physiol. 2010;109:1756–1761. doi: 10.1152/japplphysiol.00601.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laman JD, Weller RO. Drainage of Cells and Soluble Antigen from the CNS to Regional Lymph Nodes. J Neuroimmune Pharmacol. 2013;8:840–856. doi: 10.1007/s11481-013-9470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampron A, ElAli A, Rivest S. Innate immunity in the CNS: redefining the relationship between the CNS and Its environment. Neuron. 2013;78:214–232. doi: 10.1016/j.neuron.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Lanfranconi S, Markus HS. COL4A1 mutations as a monogenic cause of cerebral small vessel disease: a systematic review. Stroke. 2010;41:e513–e518. doi: 10.1161/STROKEAHA.110.581918. [DOI] [PubMed] [Google Scholar]
- Launer LJ, Petrovitch H, Ross GW, Markesbery W, White LR. AD brain pathology: vascular origins? Results from the HAAS autopsy study. Neurobiol Aging. 2008;29:1587–1590. doi: 10.1016/j.neurobiolaging.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law M, Saindane AM, Ge Y, Babb JS, Johnson G, Mannon LJ, Herbert J, Grossman RI. Microvascular abnormality in relapsing-remitting multiple sclerosis: perfusion MR imaging findings in normal-appearing white matter. Radiology. 2004;231:645–652. doi: 10.1148/radiol.2313030996. [DOI] [PubMed] [Google Scholar]
- Lawrence AJ, Patel B, Morris RG, Mackinnon AD, Rich PM, Barrick TR, Markus HS. Mechanisms of Cognitive Impairment in Cerebral Small Vessel Disease: Multimodal MRI Results from the St George’s Cognition and Neuroimaging in Stroke (SCANS) Study. PLoS ONE. 2013;8:e61014. doi: 10.1371/journal.pone.0061014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Kim Y. Complex genetic susceptibility to vascular dementia and an evidence for its underlying genetic factors associated with memory and associative learning. Gene. 2013;516:152–157. doi: 10.1016/j.gene.2012.12.032. [DOI] [PubMed] [Google Scholar]
- Lee JH, Bacskai BJ, Ayata C. Genetic animal models of cerebral vasculopathies. Prog Mol Biol Transl Sci. 2012;105:25–55. doi: 10.1016/B978-0-12-394596-9.00002-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ST, Chu K, Jung KH, Park HK, Kim DH, Bahn JJ, Kim JH, Oh MJ, Lee SK, Kim M, Roh JK. Reduced circulating angiogenic cells in Alzheimer disease. Neurology. 2009;72:1858–1863. doi: 10.1212/WNL.0b013e3181a711f4. [DOI] [PubMed] [Google Scholar]
- Leys D, Hénon H, Mackowiak-Cordoliani MA, Pasquier F. Poststroke dementia. Lancet Neurol. 2005;4:752–759. doi: 10.1016/S1474-4422(05)70221-0. [DOI] [PubMed] [Google Scholar]
- Liang KY, Mintun MA, Fagan AM, Goate AM, Bugg JM, Holtzman DM, Morris JC, Head D. Exercise and Alzheimer’s disease biomarkers in cognitively normal older adults. Ann Neurol. 2010;68:311–318. doi: 10.1002/ana.22096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckhaus C, Flüb MO, Wittsack HJ, Grass-Kapanke B, Jänner M, Khalili-Amiri R, Friedrich W, Supprian T, Gaebel W, Mödder U, Cohnen M. Detection of changed regional cerebral blood flow in mild cognitive impairment and early Alzheimer’s dementia by perfusion-weighted magnetic resonance imaging. Neuroimage. 2008;40:495–503. doi: 10.1016/j.neuroimage.2007.11.053. [DOI] [PubMed] [Google Scholar]
- Ly JV, Rowe CC, Villemagne VL, Zavala JA, Ma H, Sahathevan R, O’Keefe G, Gong SJ, Gunawan R, Churilov L, Sounder T, Ackerman U, Tochon-Danguy H, Donnan GA. Subacute ischemic stroke is associated with focal 11C PiB positron emission tomography retention but not with global neocortical Aβ deposition. Stroke. 2012;43:1341–1346. doi: 10.1161/STROKEAHA.111.636266. [DOI] [PubMed] [Google Scholar]
- Maillard P, Carmichael O, Fletcher E, Reed B, Mungas D, DeCarli C. Coevolution of white matter hyperintensities and cognition in the elderly. Neurology. 2012;79:442–448. doi: 10.1212/WNL.0b013e3182617136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makedonov I, Black SE, MacIntosh BJ. Cerebral small vessel disease in aging and Alzheimer’s disease: a comparative study using MRI and SPECT. Eur J Neurol. 2013;20:243–250. doi: 10.1111/j.1468-1331.2012.03785.x. [DOI] [PubMed] [Google Scholar]
- Maki T, Ihara M, Fujita Y, Nambu T, Miyashita K, Yamada M, Washida K, Nishio K, Ito H, Harada H, Yokoi H, Arai H, Itoh H, Nakao K, Takahashi R, Tomimoto H. Angiogenic and vasoprotective effects of adrenomedullin on prevention of cognitive decline after chronic cerebral hypoperfusion in mice. Stroke. 2011;42:1122–1128. doi: 10.1161/STROKEAHA.110.603399. [DOI] [PubMed] [Google Scholar]
- Mandell D, Han J, Poublanc J, Crawley A, Kassner A, Fisher J, Mikulis D. Selective reduction of blood flow to white matter during hypercapnia corresponds with leukoaraiosis. Stroke. 2008;39:1993–1998. doi: 10.1161/STROKEAHA.107.501692. [DOI] [PubMed] [Google Scholar]
- Markus HS, Lythgoe DJ, Ostegaard L, O’Sullivan M, Williams SC. Reduced cerebral blood flow in white matter in ischaemic leukoaraiosis demonstrated using quantitative exogenous contrast based perfusion MRI. J Neurol Neurosurg Psychiatr. 2000;69:48–53. doi: 10.1136/jnnp.69.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus H, Vallance P, Brown M. Differential effect of three cyclooxygenase inhibitors on human cerebral blood flow velocity and carbon dioxide reactivity. Stroke. 1994;25:1760–1764. doi: 10.1161/01.str.25.9.1760. [DOI] [PubMed] [Google Scholar]
- Markus HS. Genes, endothelial function and cerebral small vessel disease in man. Exp Physiol. 2008;93:121–127. doi: 10.1113/expphysiol.2007.038752. [DOI] [PubMed] [Google Scholar]
- Markus HS, Hunt B, Palmer K, Enzinger C, Schmidt H, Schmidt R. Markers of endothelial and hemostatic activation and progression of cerebral white matter hyperintensities: longitudinal results of the Austrian Stroke Prevention Study. Stroke. 2005;36:1410–1414. doi: 10.1161/01.STR.0000169924.60783.d4. [DOI] [PubMed] [Google Scholar]
- Marshall RS, Festa JR, Cheung YK, Chen R, Pavol MA, Derdeyn CP, Clarke WR, Videen TO, Grubb RL, Adams HP, Powers WJ, Lazar RM. Cerebral hemodynamics and cognitive impairment: baseline data from the RECON trial. Neurology. 2012;78:250–255. doi: 10.1212/WNL.0b013e31824365d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall RS, Lazar RM, Mohr JP, Pile-Spellman J, Hacein-Bey L, Duong DH, Joshi S, Chen X, Levin B, Young WL. Higher cerebral function and hemispheric blood flow during awake carotid artery balloon test occlusions. J Neurol Neurosurg Psychiatr. 1999;66:734–738. doi: 10.1136/jnnp.66.6.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall RS. Effects of altered cerebral hemodynamics on cognitive function. J Alzheimers Dis. 2012;32:633–642. doi: 10.3233/JAD-2012-120949. [DOI] [PubMed] [Google Scholar]
- Marstrand JR, Garde E, Rostrup E, Ring P, Rosenbaum S, Mortensen EL, Larsson HBW. Cerebral perfusion and cerebrovascular reactivity are reduced in white matter hyperintensities. Stroke. 2002;33:972–976. doi: 10.1161/01.str.0000012808.81667.4b. [DOI] [PubMed] [Google Scholar]
- Mastaglia FL, Byrnes ML, Johnsen RD, Kakulas BA. Prevalence of cerebral vascular amyloid-beta deposition and stroke in an aging Australian population: a postmortem study. J Clin Neurosci. 2003;10:186–189. doi: 10.1016/s0967-5868(02)00317-x. [DOI] [PubMed] [Google Scholar]
- Masumura M, Hata R, Nagai Y, Sawada T. Oligodendroglial cell death with DNA fragmentation in the white matter under chronic cerebral hypoperfusion: comparison between normotensive and spontaneously hypertensive rats. Neurosci Res. 2001;39:401–412. doi: 10.1016/s0168-0102(01)00195-x. [DOI] [PubMed] [Google Scholar]
- Matsushita K, Kuriyama Y, Nagatsuka K, Nakamura M, Sawada T, Omae T. Periventricular white matter lucency and cerebral blood flow autoregulation in hypertensive patients. Hypertension. 1994;23:565–568. doi: 10.1161/01.hyp.23.5.565. [DOI] [PubMed] [Google Scholar]
- Matthews FE, Arthur A, Barnes LE, Bond J, Jagger C, Robinson L, Brayne C on behalf of the Medical Research Council Cognitive Function and Ageing Collaboration. A two-decade comparison of prevalence of dementia in individuals aged 65 years and older from three geographical areas of England: results of the Cognitive Function and Ageing Study I and II. Lancet. 2013 doi: 10.1016/S0140-6736(13)61570-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute C, Ransom BR. Roles of white matter in central nervous system pathophysiologies. ASN Neuro. 2012;4:89–101. doi: 10.1042/AN20110060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8:913–919. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- Melhem ER, Loes DJ, Georgiades CS, Raymond GV, Moser HW. X-linked adrenoleukodystrophy: the role of contrast-enhanced MR imaging in predicting disease progression. AJNR Am J Neuroradiol. 2000;21:839–844. [PMC free article] [PubMed] [Google Scholar]
- Mentis M, Horwitz B, Grady C, Alexander G, VanMeter J, Maisog J, Pietrini P, Schapiro M, Rapoport S. Visual cortical dysfunction in Alzheimer’s disease evaluated with a temporally graded ‘stress test’ during PET. Am Psychiatry. 1996;153:32–40. doi: 10.1176/ajp.153.1.32. [DOI] [PubMed] [Google Scholar]
- Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol. 2009;66:1210–1215. doi: 10.1001/archneurol.2009.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklossy J. Cerebral hypoperfusion induces cortical watershed microinfarcts which may further aggravate cognitive decline in Alzheimer’s disease. Neurol Res. 2003;25:605–610. doi: 10.1179/016164103101202048. [DOI] [PubMed] [Google Scholar]
- Miller A, Drummond G, Schmidt H, Sobey C. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ Res. 2005;97:1055–1062. doi: 10.1161/01.RES.0000189301.10217.87. [DOI] [PubMed] [Google Scholar]
- Miyamoto N, Tanaka R, Shimura H, Watanabe T, Mori H, Onodera M, Mochizuki H, Hattori N, Urabe T. Phosphodiesterase III inhibition promotes differentiation and survival of oligodendrocyte progenitors and enhances regeneration of ischemic white matter lesions in the adult mammalian brain. J Cereb Blood Flow Metab. 2010;30:299–310. doi: 10.1038/jcbfm.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser HW, Loes DJ, Melhem ER, Raymond GV, Bezman L, Cox CS, Lu SE. X-Linked adrenoleukodystrophy: overview and prognosis as a function of age and brain magnetic resonance imaging abnormality. A study involving 372 patients. Neuropediatrics. 2000;31:227–239. doi: 10.1055/s-2000-9236. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musolino PL, Rapalino O, Caruso P, Caviness VS, Eichler FS. Hypoperfusion predicts lesion progression in cerebral X-linked adrenoleukodystrophy. Brain. 2012;135:2676–2683. doi: 10.1093/brain/aws206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navaratna D, Fan X, Leung W, Lok J, Guo S, Xing C, Wang X, Lo EH. Cerebrovascular degradation of TRKB by MMP9 in the diabetic brain. J CLin Invest. 2013;123:3373–3377. doi: 10.1172/JCI65767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave KA. Myelination and support of axonal integrity by glia. Nature. 2010a;468:244–252. doi: 10.1038/nature09614. [DOI] [PubMed] [Google Scholar]
- Nave KA. Myelination and the trophic support of long axons. Nat Rev Neurosci. 2010b;11:275–283. doi: 10.1038/nrn2797. [DOI] [PubMed] [Google Scholar]
- Neuwelt EA, Bauer B, Fahlke C, Fricker G, Iadecola C, Janigro D, Leybaert L, Molnár Z, O’Donnell ME, Povlishock JT, Saunders NR, Sharp F, Stanimirovic D, Watts RJ, Drewes LR. Engaging neuroscience to advance translational research in brain barrier biology. Nat Rev Neurosci. 2011;12:169–182. doi: 10.1038/nrn2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen J, Nishimura N, Fetcho RN, Iadecola C, Schaffer CB. Occlusion of cortical ascending venules causes blood flow decreases, reversals in flow direction, and vessel dilation in upstream capillaries. J Cereb Blood Flow Metab. 2011;31:2243–2254. doi: 10.1038/jcbfm.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedermeyer E. Alzheimer Disease: Caused by Primary Deficiency of the Cerebral Blood Flow. Clin EEG Neurosci. 2006;37:175–177. doi: 10.1177/155005940603700303. [DOI] [PubMed] [Google Scholar]
- Nishimura N, Rosidi NL, Iadecola C, Schaffer CB. Limitations of collateral flow after occlusion of a single cortical penetrating arteriole. J Cereb Blood Flow Metab. 2010;30:1914–1927. doi: 10.1038/jcbfm.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitkunan A, Lanfranconi S, Charlton RA, Barrick TR, Markus HS. Brain atrophy and cerebral small vessel disease: a prospective follow-up study. Stroke. 2011;42:133–138. doi: 10.1161/STROKEAHA.110.594267. [DOI] [PubMed] [Google Scholar]
- Niwa K, Carlson GA, Iadecola C. Exogenous A beta1-40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. J Cereb Blood Flow Metab. 2000a;20:1659–1668. doi: 10.1097/00004647-200012000-00005. [DOI] [PubMed] [Google Scholar]
- Niwa K, Porter VA, Kazama K, Cornfield D, Carlson GA, Iadecola C. A beta-peptides enhance vasoconstriction in cerebral circulation. Am J Physiol Heart Circ Physiol. 2001;281:H2417–H2424. doi: 10.1152/ajpheart.2001.281.6.H2417. [DOI] [PubMed] [Google Scholar]
- Niwa K, Younkin L, Ebeling C, Turner SK, Westaway D, Younkin S, Ashe KH, Carlson GA, Iadecola C. Abeta 1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci U S A. 2000b;97:9735–9740. doi: 10.1073/pnas.97.17.9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa K, Kazama K, Younkin SG, Carlson GA, Iadecola C. Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol Dis. 2002;9:61–68. doi: 10.1006/nbdi.2001.0460. [DOI] [PubMed] [Google Scholar]
- Notsu Y, Nabika T, Bokura H, Suyama Y, Kobayashi S, Yamaguchi S, Masuda J. Evaluation of asymmetric dimethylarginine and homocysteine in microangiopathy-related cerebral damage. Am J Hypertens. 2009;22:257–262. doi: 10.1038/ajh.2008.346. [DOI] [PubMed] [Google Scholar]
- Novak V, Chowdhary A, Farrar B, Nagaraja H, Braun J, Kanard R, Novak P, Slivka A. Altered cerebral vasoregulation in hypertension and stroke. Neurology. 2003;60:1657–1663. doi: 10.1212/01.wnl.0000068023.14587.06. [DOI] [PubMed] [Google Scholar]
- O’Sullivan M, Lythgoe DJ, Pereira AC, Summers PE, Jarosz JM, Williams SCR, Markus HS. Patterns of cerebral blood flow reduction in patients with ischemic leukoaraiosis. Neurology. 2002;59:321–326. doi: 10.1212/wnl.59.3.321. [DOI] [PubMed] [Google Scholar]
- Okamoto Y, Yamamoto T, Kalaria RN, Senzaki H, Maki T, Hase Y, Kitamura A, Washida K, Yamada M, Ito H, Tomimoto H, Takahashi R, Ihara M. Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol. 2012;123:381–394. doi: 10.1007/s00401-011-0925-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris D, Humphrey J, Quadros A, Patel N, Crescentini R, Crawford F, Mullan M. Vasoactive effects of A beta in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer’s disease: role of inflammation. Neurol Res. 2003;25:642–651. doi: 10.1179/016164103101201940. [DOI] [PubMed] [Google Scholar]
- Park JH, Seo SW, Kim C, Kim GH, Noh HJ, Kim ST, Kwak KC, Yoon U, Lee JM, Lee JW, Shin JS, Kim CH, No Y, Cho H, Kim HJ, Yoon CW, Oh SJ, Kim JS, Choe YS, Lee KH, Lee KH, Ewers M, Weiner WW, Werring DJ, Na DL. Pathogenesis of cerebral microbleeds: In vivo imaging of amyloid and subcortical ischemic small vessel disease in 226 individuals with cognitive impairment. Ann Neurol. 2013;73:584–593. doi: 10.1002/ana.23845. [DOI] [PubMed] [Google Scholar]
- Park L, Anrather J, Girouard H, Zhou P, Iadecola C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab. 2007;27:1908–1918. doi: 10.1038/sj.jcbfm.9600491. [DOI] [PubMed] [Google Scholar]
- Park L, Anrather J, Zhou P, Frys K, Pitstick R, Younkin S, Carlson GA, Iadecola C. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J Neurosci. 2005;25:1769–1777. doi: 10.1523/JNEUROSCI.5207-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Wang G, Zhou P, Zhou J, Pitstick R, Previti ML, Younkin L, Younkin SG, Van Nostrand WE, Cho S, Anrather J, Carlson GA, Iadecola C. Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid-beta. Proc Natl Acad Sci USA. 2011;108:5063–5068. doi: 10.1073/pnas.1015413108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Zhou J, Zhou P, Pistick R, Jamal ElS, Younkin L, Pierce J, Arreguin A, Anrather J, Younkin SG, Carlson GA, McEwen BS, Iadecola C. Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc Natl Acad Sci USA. 2013;110:3089–3094. doi: 10.1073/pnas.1300021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Younkin L, Younkin S, Carlson G, McEwen BS, Iadecola C. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci USA. 2008;105:1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendlebury ST, Rothwell PM. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol. 2009;8:1006–1018. doi: 10.1016/S1474-4422(09)70236-4. [DOI] [PubMed] [Google Scholar]
- Pikula A, Böger RH, Beiser AS, Maas R, DeCarli C, Schwedhelm E, Himali JJ, Schulze F, Au R, Kelly-Hayes M, Kase CS, Vasan R, Wolf PW, Seshadri S. Association of plasma ADMA levels with MRI markers of vascular brain injury: Framingham offspring study. Stroke. 2009;40:2959–2964. doi: 10.1161/STROKEAHA.109.557116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston M, Gong X, Su W, Matsumoto SG, Banine F, Winkler C, Foster S, Xing R, Struve J, Dean J, Baggenstoss B, Weigel PH, Montine TJ, Back ASA, Sherman LS. Digestion products of the PH20 hyaluronidase inhibit remyelination. Ann Neurol. 2013;73:266–280. doi: 10.1002/ana.23788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pretnar-Oblak J, Sabovic M, Sebestjen M, Pogacnik T, Zaletel M. Influence of atorvastatin treatment on L-arginine cerebrovascular reactivity and flow-mediated dilatation in patients with lacunar infarctions. Stroke. 2006;37:2540–2545. doi: 10.1161/01.STR.0000239659.99112.fb. [DOI] [PubMed] [Google Scholar]
- Purnell C, Gao S, Callahan CM, Hendrie HC. Cardiovascular risk factors and incident Alzheimer disease: a systematic review of the literature. Alzheimer Dis Assoc Disord. 2009;23:1–10. doi: 10.1097/WAD.0b013e318187541c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaegebeur A, Lange C, Carmeliet P. The neurovascular link in health and disease: molecular mechanisms and therapeutic implications. Neuron. 2011;71:406–424. doi: 10.1016/j.neuron.2011.07.013. [DOI] [PubMed] [Google Scholar]
- Rannikmäe K, Samarasekera N, Martînez-Gonzâlez NA, Al-Shahi Salman R, Sudlow CLM. Genetics of cerebral amyloid angiopathy: systematic review and meta-analysis. J Neurol Neurosurg Psychiatr. 2013;84:901–908. doi: 10.1136/jnnp-2012-303898. [DOI] [PubMed] [Google Scholar]
- Reimer MM, McQueen J, Searcy L, Scullion G, Zonta B, Desmazieres A, Holland PR, Smith J, Gliddon C, Wood ER, Herzyk P, Brophy PJ, McCulloch J, Horsburgh K. Rapid disruption of axon-glial integrity in response to mild cerebral hypoperfusion. J Neurosci. 2011;31:18185–18194. doi: 10.1523/JNEUROSCI.4936-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis JP, Loria CM, Launer LJ, Sidney S, Liu K, Jacobs DR, Zhu N, Lloyd-Jones DM, He K, Yaffe K. Cardiovascular health through young adulthood and cognitive functioning in midlife. Ann Neurol. 2013;73:170–179. doi: 10.1002/ana.23836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard E, Gouw AA, Scheltens P, van Gool WA. Vascular care in patients with Alzheimer disease with cerebrovascular lesions slows progression of white matter lesions on MRI: the evaluation of vascular care in Alzheimer’s disease (EVA) study. Stroke. 2010;41:554–556. doi: 10.1161/STROKEAHA.109.571281. [DOI] [PubMed] [Google Scholar]
- Richardson K, Stephan BCM, Ince PG, Brayne C, Matthews FE, Esiri MM. The neuropathology of vascular disease in the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS) Curr Alzheimer Res. 2012;9:687–696. doi: 10.2174/156720512801322654. [DOI] [PubMed] [Google Scholar]
- Richardson WD, Young KM, Tripathi RB, McKenzie I. NG2-glia as multipotent neural stem cells: fact or fantasy? Neuron. 2011;70:661–673. doi: 10.1016/j.neuron.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins EM, Betensky RA, Domnitz SB, Purcell SM, Garcia-Alloza M, Greenberg C, Rebeck GW, Hyman BT, Greenberg SM, Frosch MP, Bacskai BJ. Kinetics of cerebral amyloid angiopathy progression in a transgenic mouse model of Alzheimer disease. J Neurosci. 2006;26:365–371. doi: 10.1523/JNEUROSCI.3854-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigue KM, Rieck JR, Kennedy KM, Devous MD, Diaz-Arrastia R, Park DC. Risk Factors for β-Amyloid Deposition in Healthy Aging: Vascular and Genetic Effects. JAMA Neurol. 2013:1–7. doi: 10.1001/jamaneurol.2013.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roher A, Esh C, Rahman A, Kokjohn T, Beach T. Atherosclerosis of cerebral arteries in Alzheimer disease. Stroke. 2004;35:2623–2627. doi: 10.1161/01.STR.0000143317.70478.b3. [DOI] [PubMed] [Google Scholar]
- Roman G, Takemici T, Erkinjuntti T, Cummings J, Masdeu J, Garcia J, Amaducci L, Orgogozo J, Brun A, Hofman A, Moody, Moody D, O’Brien M, Yamaguchi T, Grafman J, Drayer B, Bennett D, Fisher M, Ogata J, Kokmen E, Bermejo F, Wolf P, Gorelick P, Bock K, Pajeau A, Bell M, Decarli C, Culebras A, Korczyn A, Bogousslavsky J, Hartmann A, Scheinberg P, O’Brien M, Yamaguchi T, Grafman J, Drayer D, Bennett D, Fisher M, Ogata J, Kokmen E, Bermejo F, Wolf P, Gorelick P, Bock K, Pajeau A, Bell M, Decarli C, Culebras A, Korczyn A, Bogousslavsky J, Hartmann A, Scheinberg P. Vascular dementia: diagnostic criteria for research studies: report of the NINDS-AIREN international workshop. Neurology. 1993;43:250–260. doi: 10.1212/wnl.43.2.250. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Sullivan N, Esiri MM. White matter damage is associated with matrix metalloproteinases in vascular dementia. Stroke. 2001;32:1162–1168. doi: 10.1161/01.str.32.5.1162. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA. Neurological diseases in relation to the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32:1139–1151. doi: 10.1038/jcbfm.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouhl RPW, Damoiseaux JGMC, Lodder J, Theunissen ROMFIH, Knottnerus ILH, Staals J, Henskens LHG, Kroon AA, de Leeuw PW, Tervaert JWC, van Oostenbrugge RJ. Vascular inflammation in cerebral small vessel disease. Neurobiol Aging. 2012;33:1800–1806. doi: 10.1016/j.neurobiolaging.2011.04.008. [DOI] [PubMed] [Google Scholar]
- Rouhl RPW, Mertens AECS, van Oostenbrugge RJ, Damoiseaux JGMC, Debrus-Palmans LL, Henskens LHG, Kroon AA, de Leeuw PW, Lodder J, Tervaert JWC. Angiogenic T-cells and putative endothelial progenitor cells in hypertension-related cerebral small vessel disease. Stroke. 2012b;43:256–258. doi: 10.1161/STROKEAHA.111.632208. [DOI] [PubMed] [Google Scholar]
- Rufa A, Blardi P, De Lalla A, Cevenini G, De Stefano N, Zicari E, Auteri A, Federico A, Dotti MT. Plasma levels of asymmetric dimethylarginine in cerebral autosomal dominant arteriopathy with subcortical infarct and leukoencephalopathy. Cerebrovasc Dis. 2008;26:636–640. doi: 10.1159/000166840. [DOI] [PubMed] [Google Scholar]
- Ruitenberg A, den Heijer T, Bakker SLM, van Swieten JC, Koudstaal PJ, Hofman A, Breteler MMB. Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann Neurol. 2005;57:789–794. doi: 10.1002/ana.20493. [DOI] [PubMed] [Google Scholar]
- Sabayan B, Jansen S, Oleksik AM, van Osch MJP, van Buchem MA, van Vliet P, de Craen AJM, Westendorp RGJ. Cerebrovascular hemodynamics in Alzheimer’s disease and vascular dementia: a meta-analysis of transcranial Doppler studies. Ageing Res Rev. 2012;11:271–277. doi: 10.1016/j.arr.2011.12.009. [DOI] [PubMed] [Google Scholar]
- Sahathevan R, Brodtmann A, Donnan GA. Dementia, stroke, and vascular risk factors; a review. Int J Stroke. 2012;7:61–73. doi: 10.1111/j.1747-4949.2011.00731.x. [DOI] [PubMed] [Google Scholar]
- Saindane AM, Law M, Ge Y, Johnson G, Babb JS, Grossman RI. Correlation of diffusion tensor and dynamic perfusion MR imaging metrics in normal-appearing corpus callosum: support for primary hypoperfusion in multiple sclerosis. AJNR Am J Neuroradiol. 2007;28:767–772. [PMC free article] [PubMed] [Google Scholar]
- Schaapsmeerders P, Maaijwee NAM, van Dijk EJ, Rutten-Jacobs LCA, Arntz RM, Schoonderwaldt HC, Dorresteijn LDA, Kessels RPC, de Leeuw FE. Long-term cognitive impairment after first-ever ischemic stroke in young adults. Stroke. 2013;44:1621–1628. doi: 10.1161/STROKEAHA.111.000792. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Freudenberger P, Seiler S, Schmidt R. Genetics of subcortical vascular dementia. Exp Gerontol. 2012;47:873–877. doi: 10.1016/j.exger.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007a;69:2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009;66:200–208. doi: 10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JA, Boyle PA, Arvanitakis Z, Bienias JL, Bennett DA. Subcortical infarcts, Alzheimer’s disease pathology, and memory function in older persons. Ann Neurol. 2007b;62:59–66. doi: 10.1002/ana.21142. [DOI] [PubMed] [Google Scholar]
- Schreiber S, Bueche CZ, Garz C, Braun H. Blood brain barrier breakdown as the starting point of cerebral small vessel disease? - New insights from a rat model. Exp Transl Stroke Med. 2013;5:4. doi: 10.1186/2040-7378-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrijvers EMC, Schürmann B, Koudstaal PJ, van den Bussche H, van Duijn CM, Hentschel F, Heun R, Hofman A, Jessen F, Kölsch H, Kornhuber J, Peters O, Rivadeneira F, Rüther E, Uitterlinden AG, Riedel-Heller S, Dichgans M, Wiltfang J, Maier W, Breteler MMB, Ikram MA. Genome-wide association study of vascular dementia. Stroke. 2012;43:315–319. doi: 10.1161/STROKEAHA.111.628768. [DOI] [PubMed] [Google Scholar]
- Scuteri A, Nilsson PM, Tzourio C, Redon J, Laurent S. Microvascular brain damage with aging and hypertension: pathophysiological consideration and clinical implications. J Hypertens. 2011;29:1469–1477. doi: 10.1097/HJH.0b013e328347cc17. [DOI] [PubMed] [Google Scholar]
- Seo JH, Miyamoto N, Hayakawa K, Pham L-DD, Maki T, Ayata C, Kim K-W, Lo EH, Arai K. Oligodendrocyte precursors induce early blood-brain barrier opening after white matter injury. J CLin Invest. 2013;123:782–786. doi: 10.1172/JCI65863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo SW, Lee JM, Im K, Park JS, Kim SH, Kim ST, Ahn HJ, Chin J, Cheong HK, Weiner MW, Na DL. Cortical thinning related to periventricular and deep white matter hyperintensities. Neurobiol Aging. 2012;33:1156–1167. doi: 10.1016/j.neurobiolaging.2010.12.003. [DOI] [PubMed] [Google Scholar]
- Sharp SI, Aarsland D, Day S, Sønnesyn H, Ballard C Alzheimer’s Society Vascular Dementia Systematic Review Group. Hypertension is a potential risk factor for vascular dementia: systematic review. Int J Geriatr Psychiatry. 2011;26:661–669. doi: 10.1002/gps.2572. [DOI] [PubMed] [Google Scholar]
- Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, Vincent P, Pumiglia K, Temple S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–1340. doi: 10.1126/science.1095505. [DOI] [PubMed] [Google Scholar]
- Sherman LS, Back SA. A “GAG” reflex prevents repair of the damaged CNS. Trends Neurosci. 2008;31:44–52. doi: 10.1016/j.tins.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Shibata H, Nabika T, Moriyama H, Masuda J, Kobayashi S. Correlation of NO metabolites and 8-iso-prostaglandin F2a with periventricular hyperintensity severity. Arterioscler Thromb Vasc Biol. 2004;24:1659–1663. doi: 10.1161/01.ATV.0000137415.67349.3c. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic B. Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J CLin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Blinder P, Tsai PS, Friedman B, Stanley G, Lyden PD, Kleinfeld D. The smallest stroke: occlusion of one penetrating vessel leads to infarction and a cognitive deficit. Nat Neurosci. 2013;16:55–63. doi: 10.1038/nn.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JE, Fernando MS, Clark L, Ince PG, Matthews F, Forster G, O’Brien JT, Barber R, Kalaria RN, Brayne C MRC Cognitive Function and Ageing Neuropathology Study Group . White matter lesions in an unselected cohort of the elderly: astrocytic, microglial and oligodendrocyte precursor cell responses. Neuropathol Appl Neurobiol. 2007;33:410–419. doi: 10.1111/j.1365-2990.2007.00828.x. [DOI] [PubMed] [Google Scholar]
- Smith EE, Schneider JA, Wardlaw JM, Greenberg SM. Cerebral microinfarcts: the invisible lesions. Lancet Neurol. 2012;11:272–282. doi: 10.1016/S1474-4422(11)70307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapyan M, Lemasson M, Brill MS, Blais M, Massouh M, Ninkovic J, Gravel C, Berthod F, Götz M, Barker PA, Parent A, Saghatelyan A. Vasculature guides migrating neuronal precursors in the adult mammalian forebrain via brain-derived neurotrophic factor signaling. J Neurosci. 2009;29:4172–4188. doi: 10.1523/JNEUROSCI.4956-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snowdon D, Greiner L, Mortimer J, Riley K, Greiner P, Markesbery W. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. Jama. 1997;277:813–817. [PubMed] [Google Scholar]
- Sorond FA, Kiely DK, Galica A, Moscufo N, Serrador JM, Iloputaife I, Egorova S, Dell’Oglio E, Meier DS, Newton E, Milberg WP, Guttmann CRG, Lipsitz LA. Neurovascular coupling is impaired in slow walkers: the MOBILIZE Boston Study. Ann Neurol. 2011;70:213–220. doi: 10.1002/ana.22433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sörös P, Whitehead S, Spence JD, Hachinski V. Antihypertensive treatment can prevent stroke and cognitive decline. Nat Rev Neurol. 2013;9:174–178. doi: 10.1038/nrneurol.2012.255. [DOI] [PubMed] [Google Scholar]
- Stefansdottir H, Arnar DO, Aspelund T, Sigurdsson S, Jonsdottir MK, Hjaltason H, Launer LJ, Gudnason V. Atrial fibrillation is associated with reduced brain volume and cognitive function independent of cerebral infarcts. Stroke. 2013;44:1020–1025. doi: 10.1161/STROKEAHA.12.679381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012;11:1006–1012. doi: 10.1016/S1474-4422(12)70191-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson SF, Doubal FN, Shuler K, Wardlaw JM. A systematic review of dynamic cerebral and peripheral endothelial function in lacunar stroke versus controls. Stroke. 2010;41:e434–e442. doi: 10.1161/STROKEAHA.109.569855. [DOI] [PubMed] [Google Scholar]
- Stone DB, Tesche CD. Topological dynamics in spike-timing dependent plastic model neural networks. Front Neural Circuits. 2013;7:1–18. doi: 10.3389/fncir.2013.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na(+)-Ca2+ exchanger. The Journal of Neuroscience. 1992;12:430–439. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhailial AR, Hertecant J, Sztriha L. Adrenoleukodystrophy: cortical hypoperfusion demonstrated with 99mTc-HMPAO SPECT. J Child Neurol. 1994;9:284–286. doi: 10.1177/088307389400900312. [DOI] [PubMed] [Google Scholar]
- Sun X, He G, Qing H, Zhou W, Dobie F, Cai F, Staufenbiel M, Huang L, Song W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci U S A. 2006;103:18727–18732. doi: 10.1073/pnas.0606298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YW, Qin LD, Zhou Y, Xu Q, Qian LJ, Tao J, Xu JR. Abnormal functional connectivity in patients with vascular cognitive impairment, no dementia: a resting-state functional magnetic resonance imaging study. Behav Brain Res. 2011;223:388–394. doi: 10.1016/j.bbr.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Suter OC, Sunthorn T, Kraftsik R, Straubel J, Darekar P, Khalili K, Miklossy J. Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer disease. Stroke. 2002;33:1986–1992. doi: 10.1161/01.str.0000024523.82311.77. [DOI] [PubMed] [Google Scholar]
- Taheri S, Gasparovic C, Huisa BN, Adair JC, Edmonds E, Prestopnik J, Grossetete M, Shah NJ, Wills J, Qualls C, Rosenberg G. Blood-brain barrier permeability abnormalities in vascular cognitive impairment. Stroke. 2011;42:2158–2163. doi: 10.1161/STROKEAHA.110.611731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Fukuyama H, Yamauchi H, Narita M, Nabatame H, Yokode M, Fujimoto N, Kita T, Murakami M. Regional cerebral blood flow abnormalities in nondemented patients with memory impairment. J Neuroimaging. 2002;12:112–118. doi: 10.1111/j.1552-6569.2002.tb00106.x. [DOI] [PubMed] [Google Scholar]
- Tarumi T, Shah F, Tanaka H, Haley AP. Association between central elastic artery stiffness and cerebral perfusion in deep subcortical gray and white matter. Am J Hypertens. 2011;24:1108–1113. doi: 10.1038/ajh.2011.101. [DOI] [PubMed] [Google Scholar]
- Tatemichi TK, Desmond DW, Prohovnik I, Eidelberg D. Dementia associated with bilateral carotid occlusions: neuropsychological and haemodynamic course after extracranial to intracranial bypass surgery. J Neurol Neurosurg Psychiatr. 1995;58:633–636. doi: 10.1136/jnnp.58.5.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M, Hiltunen M, Yang SH, Zhong Z, Shen Y, Simpkins JW, Tanzi RE. Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron. 2007;54:721–737. doi: 10.1016/j.neuron.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Grinberg LT, Attems J. Vascular dementia: different forms of vessel disorders contribute to the development of dementia in the elderly brain. Exp Gerontol. 2012;47:816–824. doi: 10.1016/j.exger.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. β-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature. 1996;380:168–171. doi: 10.1038/380168a0. [DOI] [PubMed] [Google Scholar]
- Thompson CS, Hakim AM. Living beyond our physiological means: small vessel disease of the brain is an expression of a systemic failure in arteriolar function: a unifying hypothesis. Stroke. 2009;40:e322–e330. doi: 10.1161/STROKEAHA.108.542266. [DOI] [PubMed] [Google Scholar]
- Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, Monsell SE, Kukull WA, Trojanowski JQ. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain. 2013;136:2697–2706. doi: 10.1093/brain/awt188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomimoto H, Akiguchi I, Suenaga T, Nishimura M, Wakita H, Nakamura S, Kimura J. Alterations of the blood-brain barrier and glial cells in white-matter lesions in cerebrovascular and Alzheimer’s disease patients. Stroke. 1996;27:2069–2074. doi: 10.1161/01.str.27.11.2069. [DOI] [PubMed] [Google Scholar]
- Tomlinson BE, Blessed G, Roth M. Observations on the brains of demented old people. J Neurol Sci. 1970;11:205–242. doi: 10.1016/0022-510x(70)90063-8. [DOI] [PubMed] [Google Scholar]
- Tong XK, Lecrux C, Rosa-Neto P, Hamel E. Age-dependent rescue by simvastatin of Alzheimer’s disease cerebrovascular and memory deficits. J Neurosci. 2012;32:4705–4715. doi: 10.1523/JNEUROSCI.0169-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topakian R, Barrick TR, Howe FA, Markus HS. Blood-brain barrier permeability is increased in normal-appearing white matter in patients with lacunar stroke and leucoaraiosis. J Neurol Neurosurg Psychiatr. 2010;81:192–197. doi: 10.1136/jnnp.2009.172072. [DOI] [PubMed] [Google Scholar]
- Tran EH, Hoekstra K, Van Rooijen N, Dijkstra CD, Owens T. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J Immunol. 1998;161:3767–3775. [PubMed] [Google Scholar]
- Trapp BD, Stys PK. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009;8:280–291. doi: 10.1016/S1474-4422(09)70043-2. [DOI] [PubMed] [Google Scholar]
- Tullberg M, Fletcher E, Decarli C, Mungas D, Reed BR, Harvey DJ, Weiner MW, Chui HC, Jagust WJ. White matter lesions impair frontal lobe function regardless of their location. Neurology. 2004;63:246–253. doi: 10.1212/01.wnl.0000130530.55104.b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Tomimoto H, Akiguchi I, Wakita H, Sakamoto H. Blood-brain barrier disruption in white matter lesions in a rat model of chronic cerebral hypoperfusion. J Cereb Blood Flow Metab. 2002;22:97–104. doi: 10.1097/00004647-200201000-00012. [DOI] [PubMed] [Google Scholar]
- Ueno Y, Zhang N, Miyamoto N, Tanaka R, Hattori N, Urabe T. Edaravone attenuates white matter lesions through endothelial protection in a rat chronic hypoperfusion model. Neuroscience. 2009;162:317–327. doi: 10.1016/j.neuroscience.2009.04.065. [DOI] [PubMed] [Google Scholar]
- van Norden AGW, van Uden IWM, de Laat KF, Gons RAR, Kessels RPC, van Dijk EJ, de Leeuw FE. Cerebral microbleeds are related to subjective cognitive failures: the RUN DMC study. Neurobiol Aging. 2013;34:2225–2230. doi: 10.1016/j.neurobiolaging.2013.03.021. [DOI] [PubMed] [Google Scholar]
- Verdelho A, Madureira S, Ferro JM, Baezner H, Blahak C, Poggesi A, Hennerici M, Pantoni L, Fazekas F, Scheltens P, Inzitari D LADIS study. Physical activity prevents progression for cognitive impairment and vascular dementia: results from the LADIS (Leukoaraiosis and Disability) study. Stroke. 2012;43:3331–3335. doi: 10.1161/STROKEAHA.112.661793. [DOI] [PubMed] [Google Scholar]
- Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 2011;10:241–252. doi: 10.1016/S1474-4422(10)70325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernooij MW, van der Lugt A, Ikram MA, Wielopolski PA, Vrooman HA, Hofman A, Krestin GP, Breteler MMB. Total cerebral blood flow and total brain perfusion in the general population: the Rotterdam Scan Study. J Cereb Blood Flow Metab. 2008;28:412–419. doi: 10.1038/sj.jcbfm.9600526. [DOI] [PubMed] [Google Scholar]
- Wakita H, Ruetzler C, Illoh KO, Chen Y, Takanohashi A, Spatz M, Hallenbeck JM. Mucosal tolerization to E-selectin protects against memory dysfunction and white matter damage in a vascular cognitive impairment model. J Cereb Blood Flow Metab. 2008;28:341–353. doi: 10.1038/sj.jcbfm.9600528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang BW, Lu E, Mackenzie IRA, Assaly M, Jacova C, Lee PE, Beattie BL, Hsiung GYR. Multiple pathologies are common in Alzheimer patients in clinical trials. Can J Neurol Sci. 2012;39:592–599. doi: 10.1017/s0317167100015316. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhang HY, Tang XC. Huperzine a improves chronic inflammation and cognitive decline in rats with cerebral hypoperfusion. J Neurosci Res. 2010;88:807–815. doi: 10.1002/jnr.22237. [DOI] [PubMed] [Google Scholar]
- Wardlaw JM, Doubal F, Armitage P, Chappell F, Carpenter T, Muñoz Maniega S, Farrall A, Sudlow C, Dennis M, Dhillon B. Lacunar stroke is associated with diffuse blood-brain barrier dysfunction. Ann Neurol. 2009;65:194–202. doi: 10.1002/ana.21549. [DOI] [PubMed] [Google Scholar]
- Wardlaw JM, Farrall A, Armitage PA, Carpenter T, Chappell F, Doubal F, Chowdhury D, Cvoro V, Dennis MS. Changes in background blood-brain barrier integrity between lacunar and cortical ischemic stroke subtypes. Stroke. 2008;39:1327–1332. doi: 10.1161/STROKEAHA.107.500124. [DOI] [PubMed] [Google Scholar]
- Wardlaw JM, Smith C, Dichgans M. Mechanisms of sporadic cerebral small vessel disease: insights from neuroimaging. Lancet Neurol. 2013a;12:483–497. doi: 10.1016/S1474-4422(13)70060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, Lindley RI, O’Brien JT, Barkhof F, Benavente OR STandards for ReportIng Vascular changes on nEuroimaging (STRIVE v1) Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013;12:822–838. doi: 10.1016/S1474-4422(13)70124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washida K, Ihara M, Nishio K, Fujita Y, Maki T, Yamada M, Takahashi J, Wu X, Kihara T, Ito H, Tomimoto H, Takahashi R. Nonhypotensive dose of telmisartan attenuates cognitive impairment partially due to peroxisome proliferator-activated receptor-gamma activation in mice with chronic cerebral hypoperfusion. Stroke. 2010;41:1798–1806. doi: 10.1161/STROKEAHA.110.583948. [DOI] [PubMed] [Google Scholar]
- Webb AJS, Simoni M, Mazzucco S, Kuker W, Schulz U, Rothwell PM. Increased cerebral arterial pulsatility in patients with leukoaraiosis: arterial stiffness enhances transmission of aortic pulsatility. Stroke. 2012;43:2631–2636. doi: 10.1161/STROKEAHA.112.655837. [DOI] [PubMed] [Google Scholar]
- Weil BR, Kushner EJ, Diehl KJ, Greiner JJ, Stauffer BL, DeSouza CA. CD31+ T cells, endothelial function and cardiovascular risk. Heart Lung Circ. 2011;20:659–662. doi: 10.1016/j.hlc.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Onyewuchi O, Yang S, Liu R, Simpkins JW. Increased beta-secretase activity and expression in rats following transient cerebral ischemia. Brain Res. 2004a;1009:1–8. doi: 10.1016/j.brainres.2003.09.086. [DOI] [PubMed] [Google Scholar]
- Wen Y, Yang SH, Liu R, Perez EJ, Brun-Zinkernagel AM, Koulen P, Simpkins JW. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim Biophys Acta. 2007;1772:473–483. doi: 10.1016/j.bbadis.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Wen Y, Yang S, Liu R, Simpkins JW. Transient cerebral ischemia induces site-specific hyperphosphorylation of tau protein. Brain Res. 2004b;1022:30–38. doi: 10.1016/j.brainres.2004.05.106. [DOI] [PubMed] [Google Scholar]
- Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, Böhm M, Nickenig G. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353:999–1007. doi: 10.1056/NEJMoa043814. [DOI] [PubMed] [Google Scholar]
- Wilkins A, Majed H, Layfield R, Compston A, Chandran S. Oligodendrocytes promote neuronal survival and axonal length by distinct intracellular mechanisms: a novel role for oligodendrocyte-derived glial cell line-derived neurotrophic factor. J Neurosci. 2003;23:4967–4974. doi: 10.1523/JNEUROSCI.23-12-04967.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolburg H, Noell S, Mack A, Wolburg-Buchholz K, Fallier-Becker P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009;335:75–96. doi: 10.1007/s00441-008-0658-9. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Dementia A Public Health Priority. World Health Organization; 2012. [Google Scholar]
- Xu H, Stamova B, Jickling G, Tian Y, Zhan X, Ander BP, Liu D, Turner R, Rosand J, Goldstein LB, Furie K, Verro P, Johnston SC, Sharp F, De Carli C. Distinctive RNA expression profiles in blood associated with white matter hyperintensities in brain. Stroke. 2010;41:2744–2749. doi: 10.1161/STROKEAHA.110.591875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Craggs LJL, Watanabe A, Booth T, Attems J, Low RWC, Oakley AE, Kalaria RN. Brain Microvascular Accumulation and Distribution of the NOTCH3 Ectodomain and Granular Osmiophilic Material in CADASIL. J Neuropathol Exp Neurol. 2013;72:416–431. doi: 10.1097/NEN.0b013e31829020b5. [DOI] [PubMed] [Google Scholar]
- Yang Y, Rosenberg GA. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. 2011;42:3323–3328. doi: 10.1161/STROKEAHA.110.608257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Sadoshima S, Ibayashi S, Kuwabara Y, Ichiya Y, Fujishima M. Leukoaraiosis and dementia in hypertensive patients. Stroke. 1992;23:1673–1677. doi: 10.1161/01.str.23.11.1673. [DOI] [PubMed] [Google Scholar]
- Yarchoan M, Xie SX, Kling MA, Toledo JB, Wolk DA, Lee EB, Van Deerlin V, Lee VMY, Trojanowski JQ, Arnold SE. Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain. 2012;135:3749–3756. doi: 10.1093/brain/aws271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates KF, Sweat V, Yau PL, Turchiano MM, Convit A. Impact of metabolic syndrome on cognition and brain: a selected review of the literature. Arterioscler Thromb Vasc Biol. 2012;32:2060–2067. doi: 10.1161/ATVBAHA.112.252759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Tanaka M, Okamoto K. Immunoglobulin G induces microglial superoxide production. Neurol Res. 2002;24:361–364. doi: 10.1179/016164102101200140. [DOI] [PubMed] [Google Scholar]
- Yoshizaki K, Adachi K, Kataoka S, Watanabe A, Tabira T, Takahashi K, Wakita H. Chronic cerebral hypoperfusion induced by right unilateral common carotid artery occlusion causes delayed white matter lesions and cognitive impairment in adult mice. Exp Neurol. 2008;210:585–591. doi: 10.1016/j.expneurol.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Yu L, Boyle P, Schneider JA, Segawa E, Wilson RS, Leurgans S, Bennett DA. APOE ε4, Alzheimer’s Disease Pathology, Cerebrovascular Disease, and CognitiveChange Over the Years Prior to Death. Psychol Aging. 2013 doi: 10.1037/a0031642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Eckman C, Younkin S, Hsiao K, Iadecola C. Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor protein. J Neurosci. 1997;17:7655–7661. doi: 10.1523/JNEUROSCI.17-20-07655.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Zhao Z, Gao L, Deng J, Wang B, Xu D, Liu B, Qu Y, Yu J, Li J, Gao G. Gypenoside attenuates white matter lesions induced by chronic cerebral hypoperfusion in rats. Pharmacol Biochem Behav. 2011;99:42–51. doi: 10.1016/j.pbb.2011.03.019. [DOI] [PubMed] [Google Scholar]
- Zieren N, Duering M, Peters N, Reyes S, Jouvent E, Hervé D, Gschwendtner A, Mewald Y, Opherk C, Chabriat H, Dichgans M. Education modifies the relation of vascular pathology to cognitive function: cognitive reserve in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Neurobiol Aging. 2013;34:400–407. doi: 10.1016/j.neurobiolaging.2012.04.019. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.