

Abstract
Cardiovascular complications have been documented in HIV-1 infected populations, and antiretroviral therapy may play a role. Nucleoside reverse transcriptase inhibitors (NRTIs) are antiretrovirals known to induce mitochondrial damage in endothelial cells, culminating in endothelial dysfunction, an initiating event in atherogenesis. Though the mechanism for NRTI-induced endothelial toxicity is not yet clear, our prior work suggested that a mitochondrial oxidative stress may be involved. To further delineate the mechanism of toxicity, endothelial cells were treated with NRTIs of varying subclasses, and the level of reactive oxygen species (ROS) and mitochondrial function were assessed. To test whether rescue of mitochondrial electron transport attenuated NRTI-induced endothelial cytotoxicity, in some cases, cells were cotreated with the electron transport cofactor coenzyme Q10 (Q10). At 4–6h, NRTIs increased levels of ROS but decreased the activities of electron transport chain complexes I–IV, levels of ATP and the NAD/NADH ratio. Moreover, nitric oxide levels were decreased, whereas endothelin-1 release was increased. Q10 abolished NRTI-induced mitochondria injury and effects on endothelial agonist production. Interestingly, in cells treated with NRTIs only, markers for mitochondrial toxicity returned to baseline levels by 18–24h, suggesting a compensatory mechanism for clearing damaged mitochondria. Using confocal microscopy, with confirmation utilizing the autophagy and mitophagy markers LC-3 and Nix, respectively, we observed autophagy of mitochondria at 8–10h after treatment. Q10 prevented NRTI-mediated increase in LC-3. These findings suggest that NRTI-induced mitophagy may be involved in NRTI-induced endothelial dysfunction and that this damage likely results from oxidant injury. Further, Q10 supplementation could potentially prevent NRTI-induced endothelial dysfunction.
Key Words: nucleoside reverse transcriptase inhibitors, antiretrovirals, endothelial dysfunction, mitochondria, reactive oxygen species, mitophagy, coenzyme Q10
Combination antiretroviral therapy including nucleoside analog reverse transcriptase inhibitors (NRTIs) has been highly successful in reducing disease-associated mortality in HIV-infected patients. However, the success of these drugs has been tempered by their association with a number of cardiovascular side effects, including atherosclerosis (Jiang et al., 2006; Lewis, 2004). Using myographic vessel reactivity studies in rodents, we previously demonstrated that NRTI treatment induces endothelial dysfunction, an initiating event in the development of atherosclerosis (Jiang et al., 2006, 2010). We and others have shown that mitochondrial dysfunction is a factor in the pathogenesis of NRTI-induced endothelial dysfunction, and that coadministration of NRTIs with antioxidants attenuates NRTI-induced increases in reactive oxygen species (ROS) and decreases in endothelial function in animals (Hebert et al., 2004; Jiang et al., 2007, 2010; Kline et al., 2009; Sutliff et al., 2002). As mitochondrial dysfunction and its subsequent ROS overproduction are associated with endothelial dysfunction (Victor et al., 2009), a mechanistic explanation for NRTI-associated vascular toxicity may be that NRTIs cause endothelial dysfunction by inducing excessive mitochondrial oxidant injury (Balaban et al., 2005; Puddu et al., 2005). However, despite numerous studies and prior reports, the mitochondrial locus of injury is as yet unclear (Jiang et al., 2007; Lewis et al., 1992; Rosca et al., 2008).
Direct effects of NRTIs on the activities of mitochondrial electron transport chain (ETC) complexes I–IV have been reported (Lund and Wallace, 2004). Based on the structural characteristics of NRTIs, it has been hypothesized that the drugs may compete for NAD binding sites on complex I (NADH dehydrogenase; Lund and Wallace, 2004). In addition, it was recently demonstrated that complex I possesses ATP binding sites that regulate the activity of the complex (Grivennikova et al., 2011). Thus, it is possible that the phosphorylated metabolites of NRTIs may compete with these binding sites as well. In support of this mechanism of action, in cardiomyocytes, NRTIs were shown to decrease the phosphorylation state of some subunits of complex I (Lund and Wallace, 2008). In this study, we investigated the mechanism of NRTI-induced mitochondrial toxicity in cultured endothelial cells and its concomitant production of vasoactive factors.
Along with complex III, complex I is a major site of ROS production, particularly when electron transfer at complex I is disrupted (Dröse and Brandt, 2012). Interestingly, complex I inhibition has been shown to increase levels of endothelin-1 (ET-1), a marker for endothelial dysfunction (Yuhki et al., 2001). Thus, our first objective of the current report was to test whether complex I might be a target of NRTI toxicity. To do this, we measured the activities of each of the ETC complexes, along with levels of NAD/NADH and ATP. In some experiments, cells were cotreated with the electron transport cofactor coenzyme Q10 (Q10). A number of studies have shown that Q10 administration can rescue cells from toxicity induced by complex I inhibitors like rotenone (Menke et al., 2003; Plecitá-Hlavatá et al., 2009). In addition, because other reports suggest that the mechanism of NRTI-induced cellular toxicity can vary among differing NRTIs (Lund et al., 2007), we selected for study NRTIs of varying subclasses—i.e., a thymidine (zidovudine, AZT) and a cytodine (lamivudine, 3TC) nucleoside analogue compared with an acyclic nucleotide analogue of adenosine (tenofovir, TEN).
Another objective of this study was to elucidate mechanisms linking NRTI-induced mitochondrial dysfunction with its resulting effects on endothelial function and cellular homeostasis. Although increased ROS production and mitochondrial dysfunction are factors known to precipitate apoptosis, we previously reported no evidence of increased apoptosis in NRTI-treated endothelial cells, despite their apparent cellular dysfunction (Jiang et al., 2007). Therefore, we hypothesized that there exists a cellular repair mechanism through which NRTI-damaged mitochondria are specifically targeted and removed, thus promoting cell survival. As such, we set out to determine whether selective mitochondrial autophagy (mitophagy), the recently discovered process by which damaged mitochondria are engulfed by autophagosomes, is a mechanism through which NRTI-damaged mitochondria are cleared from endothelial cells (Goldman et al., 2010; Gottlieb and Carreira, 2010; Mammucari and Rizzuto, 2010).
MATERIALS AND METHODS
Drugs and reagents.
AZT and 3TC were obtained from Sigma-Aldrich (St Louis, MO), and TEN was purchased from Moravek Biochemicals (Brea, CA). Small amounts of AZT and 3TC were also obtained from the NIH AIDS Research and Reference Reagent Program (Germantown, MD). Q10 was purchased from Sigma-Aldrich, and MitoSOX and MitoTracker Green (MTG) were obtained from Molecular Probes (Eugene, OR), and LysoTracker Red was purchased from Lonza (Basel, Switzerland).
Cell culture.
We chose to use human umbilical vein endothelial cells (HUVEC) because the physiology of HUVEC strongly resembles that of arterial endothelial cells and because they are a commonly used cell line for studying arterial pathophysiology (Kvietys and Granger, 1997). In addition, we have found that other human vascular endothelial cells required additional growth factors during culture such that they were typically activated prior to any treatment, resulting in increased basal ROS levels compared with those in HUVEC (data not shown). Nevertheless, to verify our findings in HUVEC, we also utilized human aortic endothelial cells (HAEC) in a number of supplementary experiments. HAEC were cultured in the same manner as HUVEC, except that in this case, the EBM contained 10% fetal bovine serum (FBS) instead of 2%. Cells were cultured with endothelial basal medium (EBM) or Dulbecco’s Modified Eagle’s Medium containing 2% FBS on fibronectin-coated dishes for ROS, ATP, enzyme activity, and biochemical assays. For live cell confocal microscopy, cells were cultured with EBM with 2% FBS on fibronectin- or gelatin-coated number 1.5 coverslips fixed to the bottom of 35-mm glass bottom dishes (MatTek, Ashland, MA). To treat cells, NRTI stock solutions were prepared in deionized, distilled water before dilution at 1:1000 in medium. Cells were incubated with ± 5µM AZT, 3TC, or TEN and, in some cases, 5µM of the appropriate NRTI plus 10µM Q10. This dose was selected as representative of the peak steady-state plasma concentration (C max) for NRTI administration in humans, shown to be in the range of 1–8µM (Chittick et al., 2006; Crémieux et al., 2001).
Mitochondrial function assays.
ROS production was measured using either the fluorescent oxidant-sensitive probe dihydroethidium (DHE) or the mitochondria-specific ROS indicator MitoSOX. After NRTI treatment, cells were incubated with 5µM DHE or 5µM MitoSoc for 15min and then were washed with warm PBS. DHE fluorescence was measured at Ex 530/Em 590nm, and MitoSOX fluorescence was detected at Ex 510/Em 580nm.
The lactate/pyruvate ratio was determined by independently measuring levels of lactate and pyruvate using colorimetric assay kits from Biovision (Mountain View, CA). Levels of ATP were assessed using luminescence-based assay kit obtained from Promega (Madison, WI). The NAD+/NADH ratio was assayed using a kit from AAT Bioquest (Sunnyvale, CA) that utilized an enzyme cycling reaction to monitor the formation of NAD+.
Mitochondrial ETC activities in HUVEC treated with NRTIs were assessed using colorimetric methods. For these measurements, large numbers of cells were required. Thus, cells were grown in 15-cm cell culture dishes until confluent and then were treated with NRTIs for up to 24h. For each experimental measurement, ~3–4×107 cells from 4–5 dishes were combined, and mitochondria were isolated using a kit from Pierce/Thermo Scientific (Rockford, IL). Mitochondrial complex I activity was determined by assessing 2, 6-dichlorophenol-indophenol reduction, as determined by decreases in absorbance at 600nm over 4min. To analyze the activity of complexes II and III, the reduction of cytochrome c was monitored by measuring increases in its absorbance at 550nm over 4min. Using a kit purchased from Biochain (Hayward, CA), complex IV activity was determined by monitoring the decrease in absorbance at 550nm produced by the cytochrome c oxidase-mediated oxidation of ferrocytochrome c to ferricytochrome c.
Measurement of cellular oxygen consumption.
In experiments aimed at measuring oxygen consumption, HUVEC were cultured in 15-cm fibronectin-coated petri dishes. Upon confluence, the cells were treated with NRTIs for 6h. After treatment, the cells were trypsinized, cells collected from five dishes were combined for each sample, and an equivalent number of cells in 2ml cell suspensions (roughly 3.75×107 cells) were transferred into a chamber maintained at 37°C. The cells were gently stirred using a magnetic stir bar. Oxygen concentrations in the chamber were recorded using a YSI Clark-type O2 electrode interfaced to a YSI Biological Oxygen Monitor, and data were acquired using Winview CP software (Super Logics, Waltham, WA). The decrease in oxygen concentration in the chamber was monitored in each sample over a period of 15–20min, and the rate of oxygen consumption in each sample was calculated.
Assays for endothelial production of vasoactive factors.
ET-1 levels were measured in spent media using an ELISA kit from Assay Designs (Ann Arbor, MI). Nitric oxide (NO) levels were measured using the chemiluminescent Sievers Nitric Oxide Analyzer 280i interfaced to NOAnalysis Liquid Software from GE (Boulder, CO), as described by Venkatesh et al. (2010).
Confocal microscopy.
Cells undergoing mitophagy exhibit increased lysosomal and autophagosomal activity specifically directed at mitochondria. Consequently, fluorescent probes of acidic organelles have become a popular method to examine mitophagy by fluorescence microscopy (Kim and Lemasters, 2010; Kim et al., 2007; Rodriguez-Enriquez et al., 2006). The red-fluorescing LysoTracker Red (LTR) probe is taken up by acidic organelles like lysosomes and autophagosomes, whereas the green-fluorescing mitochondrial probe MTG is taken up by mitochondria and becomes covalently bound to mitochondrial proteins. During mitophagy, mitochondria are selectively targeted for destruction and are engulfed by lysosomes, which mature into autophagosomes that degrade their internal contents. As such, we used confocal microscopy in live cells stained with MTG and LTR and identified mitophagy by the colocalization of acidic lysosomes and autophagosomes with mitochondria (Elmore et al., 2001; Rodriguez-Enriquez et al., 2006).
MTG and LTR incubations began 1h prior to the end of AZT treatment, so as to image the cells immediately following the end of treatment. HUVEC were loaded with both MTG (50nM) and AZT for 30min. Cells were then washed twice with fresh media and were loaded with LTR (50nM) and AZT for 30min. For imaging, the cells were washed twice more, after which new medium containing no AZT, MTG, or LTR was added. MTG and LTR incubations were performed in the dark at 37°C with humidified air. Images were collected using a Leica (Wetzlar, Germany) TCS SP5 point scanning spectral confocal microscope with a HCX Plan Apo 63x/1.4-0.6 oil objective. Excitation of MTG at 488nm was accomplished using a multiline argon laser, and fluorescence emission was monitored through a 500–550nm filter. Excitations of LTR at 543 and 594nm were achieved using two helium/neon lasers, and fluorescence emission was measured through a 590-nm long pass filter. Fields were chosen randomly to ensure objectivity of sampling. For each sample, five fields that were ~600–1500 µm apart were selected, such that the total sampling diameter was ~8–15mm.
Lysosome number and lysosome-mitochondria colocalization were quantified using ImageJ software. For lysosome quantification, a standard threshold intensity for red fluorescence was set, and circular structures that fluoresced at or above that intensity and that were greater than 0.5 µm in diameter were counted. For quantification of colocalization of lysosomes and mitochondria, the same criteria were used, only a threshold intensity for yellow fluorescence (green merged with red) was used, and structures less than 1 µm in diameter were excluded.
Western blot analyses.
Levels of phosphorylated-Ser 1177 of endothelial nitric oxide synthase (eNOS), total levels of eNOS, and the autophagy marker LC-3 were assessed by Western blotting. Tissues were homogenized on ice in lysis buffer containing a protease inhibitor cocktail, and protein concentrations were determined using the bicinchoninic acid protein assay. Equal volumes of protein were subjected to SDS-PAGE under reducing conditions, after which the proteins were transferred to polyvinylidene difluoride membranes. Membranes were blocked with 5% milk and were incubated overnight with primary antibody against Nix (1:200; Catalog No. 9089; Cell Signaling Technologies, Danvers, MA), eNOS pS1177 (1:300; Cat. no. 9571; Cell Signaling), total eNOS (1:300; Cat. No. 610297; Cell Signaling), LC-3 (1:300; Cat. No. 4108; Cell Signaling), or GAPDH (1:1000; Santa Cruz Biotechnology, Dallas, TX). The membranes were further incubated with horseradish peroxidase–conjugated anti-rabbit IgG, and protein bands were visualized using enhanced chemiluminescence. Densitometric analysis was performed using ImageJ software (National Institutes of Health), and data were normalized to the housekeeping protein GAPDH.
Data analyses.
Statistical analysis of data was performed using GraphPad Prism software. All data were expressed as group mean ± SEM. Comparisons between control and NRTI-treated cells were conducted using one- or two-way ANOVA, as appropriate, followed by Neuman Keul’s post hoc tests. A value of p < 0.05 was accepted as statistical significance.
RESULTS
NRTIs Increase ROS Production
NRTI treatment decreased DHE fluorescence in HUVEC at 4, 6, and 8h, with a peak level of 43–54% above baseline levels achieved at 6h (Fig. 1A). No statistically significant changes in ROS levels were observed at 24h of treatment. To verify that the increased ROS was mitochondrially derived, ROS levels were also measured using the mitochondrially targeted superoxide indicator dye MitoSOX at 6h after NRTI treatment. Consistent with our findings using DHE, MitoSOX fluorescence was increased 42–52% after treatment with AZT, 3TC, and TEN (p < 0.05; Fig. 1B). Similar increases in MitoSOX fluorescence was also observed in NRTI-treated HAEC (Supplementary fig. 1A).
FIG. 1.

NRTI treatment reduces mitochondrial function and increases ROS production. (A) Production of ROS in HUVEC treated with 5µM AZT, 3TC, or TEN, measured as increases in DHE fluorescence. (B) NRTI-induced increases in mitochondrial ROS production were verified using the mitochondrially targeted fluorescence probe MitoSOX at 6h after treatment. (C) NRTI treatment decreases mitochondrial ATP levels at 8h but not at 4, 6, or 24h. Data are expressed as mean percent change compared with controls ± SEM (n = 3–4) and were analyzed using one-way (B) and two-way ANOVA (A and C). *p < 0.05 compared to controls.
NRTIs Decrease ATP Production
To examine the effects of NRTIs on mitochondrial ATP production, HUVEC were treated with 5µM NRTI for up to 24h and ATP levels were measured colorimetrically. Cells treated with 5µM AZT, 3TC, or TEN for 8h exhibited a significant decrease in mean ATP levels (p < 0.05), but no statistically significant changes were observed in cells treated for 4 or 6h. At 24h of treatment, ATP returned to baseline levels (Fig. 1C). In HAEC, NRTIs likewise decreased ATP levels at 8h after treatment, but these decreased levels were maintained at 24h and did not return to baseline levels until 36h (Supplementary fig. 1B).
NRTIs Decrease Mitochondrial Electron Transport Chain Activity
Because the most robust effect of NRTI treatment on ROS levels was found at 6h, we utilized this time point for probing mitochondrial ETC complex activity. Cells treated with all three NRTI subclasses studied here exhibited decreases in mean complex I activity compared with controls, with 5µM AZT, 3TC, and TEN treatments inducing 48, 33, and 18% decreases, respectively (p < 0.05; Fig. 2A). Mean activity of complexes II + III was also attenuated by NRTI treatment (Fig. 2B), with AZT, 3TC, and TEN inducing significant 30, 36, and 32% reductions in activity, respectively. Finally, NRTI-treated cells exhibited decreased mean activity of mitochondrial complex IV (Fig. 2C), with AZT, 3TC, and TEN treatments resulting in significant 50, 62, and 39% decreases, respectively.
FIG. 2.

Treatment with NRTIs for 6h decreases the activity of ETC complexes I (A), II + III (B), and IV (C) in HUVEC. Data are expressed as mean activity of each complex normalized to total protein ± SEM (n = 3–4). One-way ANOVA revealed a significant effect of treatment. *p < 0.05 compared with controls.
NRTIs Decrease the NAD+/NADH and Increase the Lactate/Pyruvate Ratio
The relative levels of NAD+ and its reduced form NADH serve as an additional index of ETC function. Because complex I converts NADH to NAD+, deficiencies in complex I lead to an accumulation of NADH, as well as a decrease in NAD+, resulting in a decreased NAD+/NADH ratio (Roy Chowdhury et al., 2010; Ido et al., 2001). As shown in Fig. 3A, NRTI treatment reduced the NAD+-to-NADH ratio at all time points from 0 to 24h although this effect only achieved significance at 6 and 8h.
FIG. 3.

Effects of NRTI treatment on the ratios of (A) NAD to NADH and (B) lactate to pyruvate in HUVEC. Data are expressed as the mean ratio of lactate/pyruvate and NAD/NADH, respectively, ± SEM (n = 3–4). Two-way (A) and one-way (B) ANOVA revealed a significant effect of treatment in each case. *p < 0.05 compared with controls.
As a final confirmation, we also measured the lactate/pyruvate ratio at what appeared to be a critical time point for NRTI toxicity—6h. As the oxidative metabolism of pyruvate to lactate occurs in part through the ETC complexes, the lactate-to-pyruvate ratio serves as an indicator of the integrity of oxidative phosphorylation (Clay et al., 2001). Dysfunction of the ETC leads to an increase in the amount of pyruvate and a decrease in lactate, thereby increasing the lactate/pyruvate ratio. (Chariot et al., 1994). After 6h of AZT, 3TC, or TEN treatment, an increased lactate-to-pyruvate ratio was observed (Fig. 3B; p < 0.05).
Coenzyme Q10 Rescues HUVEC From NRTI-Induced Mitochondrial Damage
To further investigate the possibility that complex I is a site of NRTI-induced mitochondrial injury, HUVEC were cotreated with NRTIs plus the ETC cofactor Q10, which scavenges “leaky” electrons at complex I. In the presence of Q10, NRTI treatment did not alter ROS or ATP production (Figs. 4A and B). In addition, Q10 alone increased the basal rate of cellular oxygen consumption, but cotreatment with AZT plus Q10 did not reduce oxygen consumption compared with treatment with Q10 alone. (Figs. 4C and D).
FIG. 4.

Effects of cotreatment of HUVEC with coenzyme Q10 on NRTI-induced increases in ROS production (A) and decreases in ATP production (B). ROS levels were measured using the fluorescence indicator DHE, and ATP was assessed using a luminescence assay. Data are expressed as percent change relative to controls ± SEM (n = 3). Two-way ANOVA revealed no effect of treatment on ROS or ATP. (C) Representative trace of cellular oxygen consumption in HUVEC treated with AZT, Q10, and AZT + Q10. (D) Quantification of the effects of Q10 on NRTI-induced changes in mitochondrial oxygen consumption. Data are expressed as the mean change in oxygen concentration in the chamber in µM/min ± SEM (n = 3). Two-way ANOVA (2×2 design) revealed significant effects of treatment with AZT and treatment with Q10. *p < 0.05 compared with control cells receiving no Q10.
NRTIs Alter Endothelial Production of Vasoactive Factors
To examine the consequences of NRTI-induced mitochondrial dysfunction on endothelial agonist production, levels of two important vasoactive mediators, ET-1 and NO, were measured. As shown in Figure 5A, significant increases in mean ET-1 levels were observed for AZT-, 3TC-, and TEN-treated cells at 6, 8, 12, and 24h. In addition, levels of NO, measured as its stable metabolite nitrite, were decreased in NRTI-treated cells at 8 and 24h of treatment (Fig. 5B; p < 0.05). Finally, the ratio of phospho-Ser 1177, a critical activating residue of eNOS, to total eNOS was decreased 8h after the initiation of treatment (Figs. 5C and D; p < 0.05). Because ET-1 accumulates in medium, in order to truly understand the temporal effects of NRTI treatment on endothelial cells, we further compared NRTI-induced effects as a percent of control levels. Expressed in this way, ET-1 was significantly increased at 6–24h, but ET-1 levels in the medium were not further increased beyond that observed as 6h (Supplementary fig. 2).
FIG. 5.

Treatment of HUVEC with NRTIs induces endothelial cell injury. (A) Effects of NRTI treatment on levels of the endothelial activation marker ET-1, as measured by ELISA. Data are expressed as mean pg/ml ET-1 ± SEM for n = 3 experiments. (B) NRTI treatment decreases NO production. Data are expressed as nano moles of nitrite, the stable metabolite of NO, normalized to milligrams of protein and are denoted as averages ± SEM (n = 3–4). (C and D) Effects of NRTI treatment on the phosphorylation state of the S1177 activating residue of eNOS. A representative Western blot of phospho-eNOS (p-eNOS), eNOS, and GAPDH is shown in (C) for cells treated with NRTIs for 8h. Densitometric data shown in (D) are expressed as the ratio of pS1177 eNOS/total eNOS normalized to GAPDH ± SEM for n = 3–5 experiments. Two-way ANOVA revealed a significant effect of treatment in each case. *p < 0.05 compared with controls.
NRTI Treatment Increases Autophagy
Selective mitochondrial autophagy, or mitophagy, is a phenomenon that has only recently been characterized and is believed to play a role in the removal of damaged and dysfunctional mitochondria (Kim et al., 2007; Lemasters, 2005). To examine whether increased autophagy occurs after NRTI-induced mitochondrial dysfunction in HUVEC, levels of two forms of microtubule-associated protein 1 light chain 3 (LC3) were measured. LC3-I is a cytosolic protein that undergoes multistep processing to become LC3-II, which is incorporated into autophagosome membranes. LC3-I is localized exclusively to the cytosol, whereas LC3-II is tightly bound to the autophagosomal membrane and correlated with autophagic activity. Because LC3-I and LC3-II can easily be distinguished and quantified with Western blotting on the basis of their different mobilities in SDS-PAGE, the use of the LC3-II/LC3-I ratio has become the preferred marker to measure autophagic activity (Barth et al., 2010; Kabeya et al., 2000; Tanida, 2011). As shown in Figures 6A and B, NRTI treatment resulted in a significant increase in the ratio of LC3-II to LC3-I after 8–10h of treatment but not at 6 or 24h (p < 0.05). A similar time course for effects was observed for HAEC treated with NRTIs for 0–24h (Supplementary figs. 1C and D). Cells cotreated with NRTIs and Q10, however, did not show significant increases over control levels (Fig. 6C).
FIG. 6.

NRTI treatment of HUVEC induces autophagy. Representative Western blots (A) and quantification (B) of the shift from LC3-I to LC3-II, which is indicative of increased autophagy. Quantification data are expressed as the mean ratio ± SEM of LC3-II to LC3-I normalized to GAPDH for n = 3 experiments. Two-way ANOVA revealed a significant effect of treatment. *p < 0.05 compared with controls. (C) Quantification of Western blots of LC3 in cells cotreated with NRTIs and Q10. In this case, two-way ANOVA revealed no effect of treatment.
NRTI Treatment Induces Mitophagy
To determine whether the observed NRTI-induced increases in autophagy were mitochondria specific (mitophagy), we used MTG and LTR to label mitochondria and autophagosomes/lysosomes, respectively, and observe their colocalization with confocal fluorescence microscopy. As the mitochondria are engulfed by autophagosomes/lysosomes during mitophagy, the green fluorescence from MTG and the red fluorescence from LTR overlap, producing a yellow color (Fig. 7A). After 6h of treatment, red fluorescence from acidic autophagosomal/lysosomal structures in AZT-treated cells was threefold greater than in control cells and > twofold greater after 8h of treatment (p < 05; Fig. 7B). As shown in Figure 7C, colocalization of mitochondria and autophagosomes/lysosomes was sixfold greater in AZT-treated cells than in those of controls after 8h of treatment (p < 0.05).
FIG. 7.

Verification of a selective mitochondrial autophagy, or “mitophagy,” in NRTI-treated HUVEC. (A) Representative confocal micrographs of MTG-labeled mitochondria and LTR-labeled lysosomes/autophagosomes, indicating the occurrence of NRTI-induced selective mitophagy in HUVEC. Colocalization is indicated by overlap of green mitochondria and red lysosomes/early autophagosomes, yielding a yellow color (indicated by white arrows). (B) Quantification of increased LTR-labeled lysosomes. (C) Quantification of MTG-labeled mitochondria and LTR-labeled lysosome/early autophagosome colocalization. Data are expressed as mean fold-increase over controls ± SEM. Two-way ANOVA revealed an effect of treatment. *p < 0.05 compared with controls. A representative Western blot of protein levels of Nix and GAPDH is shown in (D) for cells treated with NRTIs for 8h. Densitometric data shown in (E) are expressed as Nix normalized to GAPDH ± SEM for n = 3 experiments. Two-way ANOVA revealed a significant effect of treatment. *p < 0.05 compared with controls.
As an additional confirmation of mitophagy, the levels of Nix protein were determined by Western blot analysis. Nix is a protein receptor that is expressed in the mitochondrial outer membrane and was recently shown to directly interact with the autophagy component LC-3 (Kanki, 2010). Nix has been shown inducible in endothelial cells (Sowter et al., 2001) and is a necessary component of mitochondrial “pruning” in tissue like heart (Dorn, 2010). At 8h after treatment with AZT, 3TC, or TEN, Nix expression was increased 30% compared with controls (Figs. 7D and E).
Q10 Abolishes NRTI-Induced Alterations in Endothelial Vasoactive Factors
To investigate a potential mechanistic link between NRTI-induced oxidative damage and endothelial cell injury, Q10 cotreatment was exploited. Although NRTI treatment increased levels of ET-1, cotreatment with NRTIs plus Q10 did not elicit any statistically significant increases in ET-1 levels (Fig. 8A). In addition, there were no differences in the ratio of phospho-S1177 of eNOS/total eNOS or nitrite release in cells cotreated with NRTIs plus Q10 compared with control cells or cells treated with Q10 alone (Figs. 4B and C).
FIG. 8.
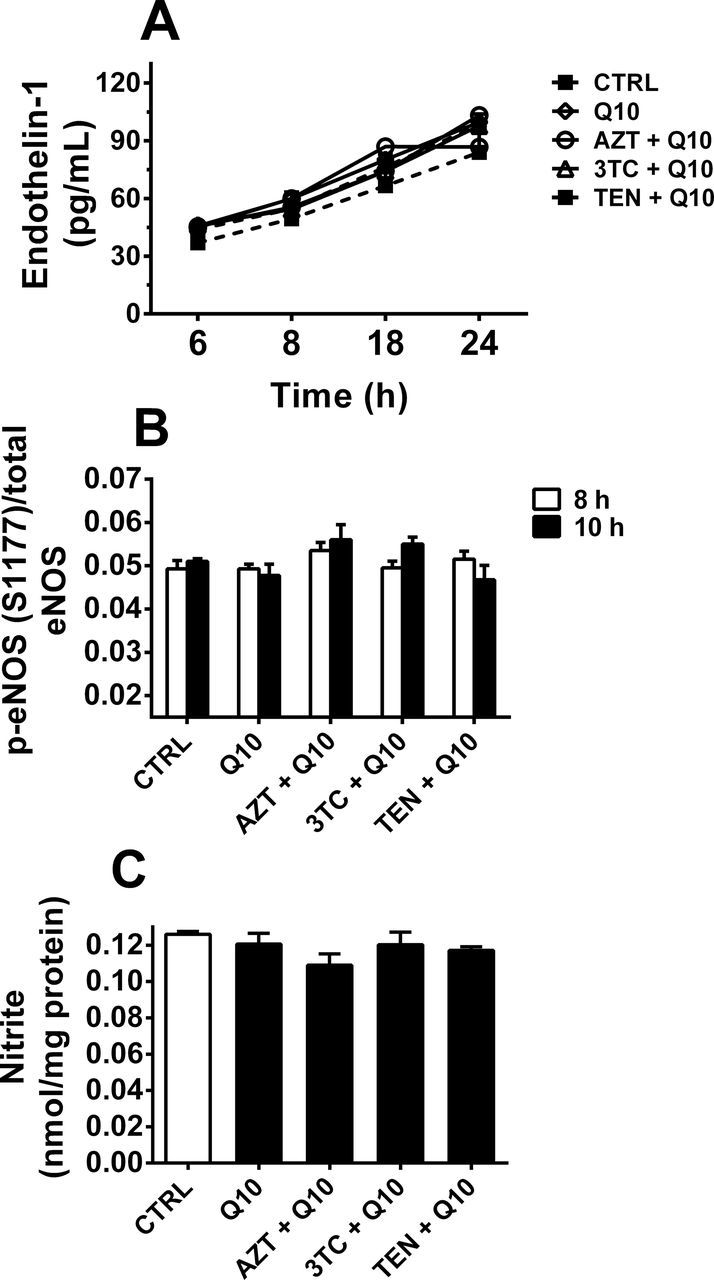
Q10 mitigates NRTI-induced endothelial injury. (A) Q10 cotreatment prevents NRTI-induced increases in ET-1 levels. Data are expressed as mean % change in ET-1 levels compared with controls for n = 4 experiments. (B) Q10 cotreatment prevents NRTI-induced decreases in phosphorylation of S1177 of eNOS in HUVEC. Data are expressed as the ratio of pS1177 eNOS/total eNOS normalized to GAPDH ± SEM for n = 4 experiments. Two-way ANOVA revealed no significant effect of treatment for either ET-1 levels or p-eNOS/total eNOS. (C) Q10 prevents NRTI-mediated effects on NO production, assessed at nitrite in the medium, in cells treated for 24h. Data are expressed as means ± SEM for n = 3 experiments. One-way ANOVA revealed no significant effect of treatment.
DISCUSSION
Though typically associated with the aged population, atherosclerosis is frequently observed in young HIV patients taking antiretrovirals (Currier et al., 2003), and a significant body of literature suggests that NRTIs can promote atherosclerosis (Friis-Møller et al., 2003; Kwong et al., 2006). Endothelial dysfunction, characterized by dysregulation of the normal physiological effects mediated by the endothelium, is a hallmark and initiating factor in atherogenesis. Although the association between NRTI treatment of rodents and endothelial dysfunction has been well described (Jiang et al., 2007; Sutliff et al., 2002), the mechanism by which this dysfunction occurs is not clear. Using murine models, we and others showed that NRTI treatment induces endothelial dysfunction, measured as decreasing responsiveness to the endothelium-dependent vasodilator acetylcholine (Jiang et al., 2006, 2010; Kline and Sutliff, 2008). In addition, treatment of endothelial cells with NRTIs increased cellular ROS production and oxidant injury (Jiang et al., 2007). Increased oxidative stress in the vasculature is known to contribute to endothelial dysfunction, and as such, NRTI-induced oxidative stress and subsequent mitochondrial dysfunction may be a mechanism underlying NRTI-induced endothelial dysfunction (Griendling and FitzGerald, 2003).
ROS are normally generated as byproducts of electron transport; however, when oxidative phosphorylation is disrupted, as occurs for NRTI treatment, higher levels of ROS are produced and mitochondrial antioxidant enzymes cannot sufficiently eliminate the accumulating oxidants. Prior work from our laboratory utilizing targeted antioxidant compounds suggested that NRTI-induced mitochondrial dysfunction contributes to the oxidative stress observed in NRTI-treated cells (Jiang et al., 2007, 2010). This study extends these findings, demonstrating that NRTI treatment of endothelial cells leads to increased ROS production, inhibition of mitochondrial ETC complex activities, and decreased production of ATP.
Generally speaking, toxicology dogma is that excess ROS production and mitochondrial dysfunction typically promote the initiation of mitochondria-mediated apoptosis, thereby preventing the accumulation of harmful oxidants (Puddu et al., 2005). Because our prior studies indicated that NRTI treatment increases ROS production and mitochondrial dysfunction without inducing apoptosis (Jiang et al., 2007), we hypothesized that endothelial cell apoptosis is not what underlies NRTI-induced endothelial dysfunction. Although this lack of apoptosis was initially perplexing, in this study, we observed that endothelial cells (both HUVEC and HAEC) recover from NRTI-induced damage by 24–36h after treatment, as demonstrated by the rescue of a number of mitochondrial and endothelial markers (Figs. 1 and 5D; Supplementary fig. 1). We thus hypothesized that there exists an endothelial cell repair mechanism that initially allows damaged cells to recover. As evidence of this, Desai et al. (2008) found that mitochondria isolated from the livers of NRTI-treated rats had considerably different gene expression profiles than those of control animals, including an increased expression of a number of both mitochondrial- and nuclear-encoded genes involved in respiratory chain activity.
Our findings from this study suggest that a compensatory mechanism allows mitochondria to combat NRTI-induced oxidant injury. This compensation may be derived from upregulated antioxidant response pathways or alternatively the recently discovered phenomenon of mitophagy. Mitophagy, which has been documented in numerous cell types, is the selective autophagic degradation of damaged mitochondria or mitochondria producing excess ROS to prevent the accumulation of mtDNA mutations and additional cellular damage (Goldman et al., 2010; Kim et al., 2007; Lemasters, 2005). We have shown here that NRTI treatment of endothelial cells culminates in mitophagy at approximately 8h after the onset of treatment. The temporal dynamics of this are noteworthy, as ROS levels in NRTI-treated cells peak prior to this event, at ~6h after treatment. Thus, these findings support the notion that mitochondria initially function in a compensated state. Thus, the transcriptomic changes in both the nucleus and the mitochondria observed by Desai et al. (2008) may indeed reflect cellular efforts to mitigate damage. Eventually, after ROS accumulate to levels that irreparably damage the mitochondria, these cells likely undergo mitophagy, thus preventing cell death.
This study also provides mechanistic insight into the mitochondrial locus of NRTI-induced endothelial injury. The reductions in activity of all four electron transport complexes, taken together with the fact that complex I is the first step in electron transport and the concurrent decrease in the NAD+/NADH ratio, suggest that complex I may be a key site of injury. Although our observed reductions in all of complexes I, II/III, and IV are difficult to conclusively reconcile, we postulate that if electron transport at complex I is interrupted, then as a “domino effect,” perhaps the measured activities at complexes III and IV will likewise be diminished. In support of this hypothesis, cotreatment with Q10, a lipid-soluble antioxidant that has furthermore been shown to rescue electron transport at complex I, mitigates NRTI-induced increases in ROS production and decreases in ATP. In addition, Q10 attenuated NRTI-induced decreases in cellular oxygen consumption, a measure of oxidative phosphorylation. Interestingly, Q10 cotreatment also prevented NRTI-induced mitophagy, suggesting that complex I inhibition may be a factor precipitating NRTI-mediated mitophagy.
Finally, our findings provide a potential mechanism linking NRTI-induced mitochondrial damage with endothelial dysfunction. ET-1 is a marker for endothelial injury and is a potent mitogenic and vasoconstricting peptide, whereas NO is important in the maintenance of normal vascular tone. The balance between vasoconstricting factors like ET-1 and vasodilating factors like NO is critical to vascular health. Indeed, a commonly used definition of endothelial dysfunction is an imbalance between these two sets of signals (Deanfield et al., 2005; Kline et al., 2009). As such, our findings that ET-1 is increased while phosphorylation of serine 1177, the most important activating residue of the NO producer eNOS (Mount et al., 2007), is decreased in response to NRTI treatment clearly demonstrate that NRTI alters endothelial production of vasoactive factors. Additional mechanisms by which eNOS activity may be compromised include reductions in the levels of its required cofactors, such as L-arginine and tetrahydrobiopterin, and its uncoupling to produce superoxide instead of NO (Forstermann and Sessa, 2012). Although these additional mechanisms were not tested in studies presented here, our observations of reduced nitrite levels could also be explained by any one of these factors. However, we did find that Q10 prevented NRTI-mediated reductions in nitrite levels and increased release of ET-1. These findings are particularly significant, as they potentially provide a link between NRTI-induced oxidative damage, and perhaps even NRTI-mediated complex I inhibition, and endothelial dysfunction.
In summary, results presented here provide new insight into current deficiencies in knowledge concerning NRTI-induced mitochondrial dysfunction. The original hypothesis first put forth by Lewis and Dalakas in 1995 was that NRTI-mediated inhibition of the mitochondria-specific DNA polymerase γ results in reductions in mtDNA copy number over time, ultimately leading to the absence of various complexes within the ETC (Lewis and Dalakas, 1995). However, our finding that robust increases in ROS production occur after only 4h of NRTI treatment suggests that this mechanism alone does not explain NRTI-induced mitochondrial dysfunction and that a more direct mechanism of toxicity is involved. Based on findings herein, complex I may be a locus of injury. Moreover, our data suggest that immediately following this increase in mitochondrial ROS production, dysfunctional mitochondrial are removed by mitochondrial autophagy or “mitophagy.” Presumably the initial loss of mitochondria is compensated for by a stimulation of mitochondrial biogenesis. Mitochondrial biogenesis is a process initiated in part by the nuclear-encoded DNA binding transcription factors nuclear respiratory factor-1 (NRF-1) and NRF-2 (Scarpulla, 1997). These factors promote the transcription of mitochondria- and nuclear-encoded proteins. They function together with the non-DNA binding coactivator PGC-1α (peroxisome proliferator–activated nuclear receptor γ cofactor 1) and mitochondrial transcription factor A to modulate the transcription of both mitochondria- and nuclear-encoded mitochondrial genes (Wu et al., 1999). Although this cellular repair mechanism may be sufficient for rescuing endothelial dysfunction after an acute treatment, we speculate that with chronic NRTI exposure this compensation may become maladaptive, so as to elicit an endothelial dysfunction that cannot be overcome. If indeed NRTI-induced cycles of mitophagy/mitochondrial biogenesis become dysregulated, then chronic NRTI treatment should either reduce population doubling or promote the senescence of endothelial cells. Studies addressing NRTI-induced effects on endothelial senescence are thus needed. Finally, our results demonstrating that Q10 prevents NRTI-induced endothelial injury are exciting, as they imply that Q10 may be useful as a cost-effective and efficacious adjunct therapy to alleviate the vascular side effects of NRTIs. Thus, in vivo studies confirming Q10-mediated protection are warranted.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Heart Lung and Blood Institute (R01 HL082472 and 3R01HL082472-02S1 to T.R.D.).
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the laboratory of Chris Kevil, PhD, for their use of the Sievers NO Analyzer and for their expert technical assistance.
REFERENCES
- Balaban R. S., Nemoto S., Finkel T. (2005). Mitochondria, oxidants, and aging. Cell. 120, 483–495 [DOI] [PubMed] [Google Scholar]
- Barth S., Glick D., Macleod K. F. (2010). Autophagy: Assays and artifacts. J. Pathol. 221, 117–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chariot P., Monnet I., Mouchet M., Rohr M., Lefaucheur J. P., Dubreuil-Lemaire M. L., Chousterman M., Gherardi R. (1994). Determination of the blood lactate:pyruvate ratio as a noninvasive test for the diagnosis of zidovudine myopathy. Arthritis Rheum. 37, 583–586 [DOI] [PubMed] [Google Scholar]
- Chittick G. E., Zong J., Blum M. R., Sorbel J. J., Begley J. A., Adda N., Kearney B. P. (2006). Pharmacokinetics of tenofovir disoproxil fumarate and ritonavir-boosted saquinavir mesylate administered alone or in combination at steady state. Antimicrob. Agents Chemother. 50, 1304–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay A. S., Behnia M., Brown K. K. (2001). Mitochondrial disease: A pulmonary and critical-care medicine perspective. Chest 120, 634–648 [DOI] [PubMed] [Google Scholar]
- Crémieux A. C., Katlama C., Gillotin C., Demarles D., Yuen G. J., Raffi F. (2001). A comparison of the steady-state pharmacokinetics and safety of abacavir, lamivudine, and zidovudine taken as a triple combination tablet and as abacavir plus a lamivudine-zidovudine double combination tablet by HIV-1-infected adults. Pharmacotherapy. 21, 424–430 [DOI] [PubMed] [Google Scholar]
- Currier J. S., Taylor A., Boyd F., Dezii C. M., Kawabata H., Burtcel B., Maa J. F., Hodder S. (2003). Coronary heart disease in HIV-infected individuals. J. Acquir. Immune Defic. Syndr. 33, 506–512 [DOI] [PubMed] [Google Scholar]
- Deanfield J., Donald A., Ferri C., Giannattasio C., Halcox J., Halligan S., Lerman A., Mancia G., Oliver J. J., Pessina A. C., et al. (2005). Endothelial function and dysfunction. Part I: Methodological issues for assessment in the different vascular beds: A statement by the Working Group on Endothelin and Endothelial Factors of the European Society of Hypertension. J. Hypertens. 23, 7–17 [DOI] [PubMed] [Google Scholar]
- Desai V. G., Lee T., Delongchamp R. R., Leakey J. E., Lewis S. M., Lee F., Moland C. L., Branham W. S., Fuscoe J. C. (2008). Nucleoside reverse transcriptase inhibitors (NRTIs)-induced expression profile of mitochondria-related genes in the mouse liver. Mitochondrion. 8, 181–195 [DOI] [PubMed] [Google Scholar]
- Dorn G. W., 2nd (2010). Mitochondrial pruning by Nix and BNip3: An essential function for cardiac-expressed death factors. J. Cardiovasc. Transl. Res. 3, 374–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröse S., Brandt U. (2012). Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 748, 145–169 [DOI] [PubMed] [Google Scholar]
- Elmore S. P., Qian T., Grissom S. F., Lemasters J. J. (2001). The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 15, 2286–2287 [DOI] [PubMed] [Google Scholar]
- Friis-Møller N., Weber R., Reiss P., Thiébaut R., Kirk O., d’Arminio Monforte A., Pradier C., Morfeldt L., Mateu S., Law M., et al. (2003). Cardiovascular disease risk factors in HIV patients–association with antiretroviral therapy. Results from the DAD study. AIDS. 17, 1179–1193 [DOI] [PubMed] [Google Scholar]
- Goldman S. J., Taylor R., Zhang Y., Jin S. (2010). Autophagy and the degradation of mitochondria. Mitochondrion. 10, 309–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb R. A., Carreira R. S. (2010). Autophagy in health and disease. 5. Mitophagy as a way of life. Am. J. Physiol. Cell Physiol. 299, C203–C210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griendling K. K., FitzGerald G. A. (2003). Oxidative stress and cardiovascular injury: Part I: Basic mechanisms and in vivo monitoring of ROS. Circulation. 108, 1912–1916 [DOI] [PubMed] [Google Scholar]
- Grivennikova V. G., Gladyshev G. V., Vinogradov A. D. (2011). Allosteric nucleotide-binding site in the mitochondrial NADH:ubiquinone oxidoreductase (respiratory complex I). FEBS Lett. 585, 2212–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert V. Y., Crenshaw B. L., Romanoff R. L., Ekshyyan V. P., Dugas T. R. (2004). Effects of HIV drug combinations on endothelin-1 and vascular cell proliferation. Cardiovasc. Toxicol. 4, 117–131 [DOI] [PubMed] [Google Scholar]
- Ido Y., Chang K., Woolsey T. A., Williamson J. R. (2001). NADH: Sensor of blood flow need in brain, muscle, and other tissues. FASEB J. 15, 1419–1421 [DOI] [PubMed] [Google Scholar]
- Jiang B., Hebert V. Y., Li Y., Mathis J. M., Alexander J. S., Dugas T. R. (2007). HIV antiretroviral drug combination induces endothelial mitochondrial dysfunction and reactive oxygen species production, but not apoptosis. Toxicol. Appl. Pharmacol. 224, 60–71 [DOI] [PubMed] [Google Scholar]
- Jiang B., Hebert V. Y., Zavecz J. H., Dugas T. R. (2006). Antiretrovirals induce direct endothelial dysfunction in vivo. J. Acquir. Immune Defic. Syndr. 42, 391–395 [DOI] [PubMed] [Google Scholar]
- Jiang B., Khandelwal A. R., Rogers L. K., Hebert V. Y., Kleinedler J. J., Zavecz J. H., Shi W., Orr A. W., Dugas T. R. (2010). Antiretrovirals induce endothelial dysfunction via an oxidant-dependent pathway and promote neointimal hyperplasia. Toxicol. Sci. 117, 524–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T. (2010). Nix, a receptor protein for mitophagy in mammals. Autophagy. 6, 433–435 [DOI] [PubMed] [Google Scholar]
- Kim I., Lemasters J. J. (2010). Mitochondrial degradation by autophagy (mitophagy) in GFP-LC3 transgenic hepatocytes during nutrient deprivation. Am. J. Physiol. Cell Physiol. 300, C308–C317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I., Rodriguez-Enriquez S., Lemasters J. J. (2007). Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 462, 245–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline E. R., Bassit L., Hernandez-Santiago B. I., Detorio M. A., Liang B., Kleinhenz D. J., Walp E. R., Dikalov S., Jones D. P., Schinazi R. F., et al. (2009). Long-term exposure to AZT, but not d4T, increases endothelial cell oxidative stress and mitochondrial dysfunction. Cardiovasc. Toxicol. 9, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline E. R., Sutliff R. L. (2008). The roles of HIV-1 proteins and antiretroviral drug therapy in HIV-1-associated endothelial dysfunction. J. Investig. Med. 56, 752–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvietys P. R., Granger D. N. (1997). Endothelial cell monolayers as a tool for studying microvascular pathophysiology. Am. J. Physiol. 273(6 Pt 1), G1189–G1199 [DOI] [PubMed] [Google Scholar]
- Kwong G. P., Ghani A. C., Rode R. A., Bartley L. M., Cowling B. J., da Silva B., Donnelly C. A., van Sighem A. I., Cameron D. W., Danner S. A., et al. (2006). Comparison of the risks of atherosclerotic events versus death from other causes associated with antiretroviral use. AIDS. 20, 1941–1950 [DOI] [PubMed] [Google Scholar]
- Lemasters J. J. (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 8, 3–5 [DOI] [PubMed] [Google Scholar]
- Lewis W. (2004). Cardiomyopathy, nucleoside reverse transcriptase inhibitors and mitochondria are linked through AIDS and its therapy. Mitochondrion. 4, 141–152 [DOI] [PubMed] [Google Scholar]
- Lewis W., Dalakas M. C. (1995). Mitochondrial toxicity of antiviral drugs. Nat. Med. 1, 417–422 [DOI] [PubMed] [Google Scholar]
- Lewis W., Gonzalez B., Chomyn A., Papoian T. (1992). Zidovudine induces molecular, biochemical, and ultrastructural changes in rat skeletal muscle mitochondria. J. Clin. Invest. 89, 1354–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund K. C., Peterson L. L., Wallace K. B. (2007). Absence of a universal mechanism of mitochondrial toxicity by nucleoside analogs. Antimicrob. Agents Chemother. 51, 2531–2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund K. C., Wallace K. B. (2004). Direct, DNA pol-gamma-independent effects of nucleoside reverse transcriptase inhibitors on mitochondrial bioenergetics. Cardiovasc. Toxicol. 4, 217–228 [DOI] [PubMed] [Google Scholar]
- Lund K. C., Wallace K. B. (2008). Adenosine 3’,5’-cyclic monophosphate (cAMP)-dependent phosphoregulation of mitochondrial complex I is inhibited by nucleoside reverse transcriptase inhibitors. Toxicol. Appl. Pharmacol. 226, 94–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammucari C., Rizzuto R. (2010). Signaling pathways in mitochondrial dysfunction and aging. Mech. Ageing Dev. 131, 536–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke T., Gille G., Reber F., Janetzky B., Andler W., Funk R. H., Reichmann H. (2003). Coenzyme Q10 reduces the toxicity of rotenone in neuronal cultures by preserving the mitochondrial membrane potential. Biofactors. 18, 65–72 [DOI] [PubMed] [Google Scholar]
- Mount P. F., Kemp B. E., Power D. A. (2007). Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J. Mol. Cell. Cardiol. 42, 271–279 [DOI] [PubMed] [Google Scholar]
- Plecitá-Hlavatá L., Jezek J., Jezek P. (2009). Pro-oxidant mitochondrial matrix-targeted ubiquinone MitoQ10 acts as anti-oxidant at retarded electron transport or proton pumping within Complex I. Int. J. Biochem. Cell Biol. 41, 1697–1707 [DOI] [PubMed] [Google Scholar]
- Puddu P., Puddu G. M., Galletti L., Cravero E., Muscari A. (2005). Mitochondrial dysfunction as an initiating event in atherogenesis: A plausible hypothesis. Cardiology. 103, 137–141 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Enriquez S., Kim I., Currin R. T., Lemasters J. J. (2006). Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy. 2, 39–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosca M. G., Vazquez E. J., Kerner J., Parland W., Chandler M. P., Stanley W., Sabbah H. N., Hoppel C. L. (2008). Cardiac mitochondria in heart failure: Decrease in respirasomes and oxidative phosphorylation. Cardiovasc. Res. 80, 30–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy Chowdhury S. K., Sangle G. V., Xie X., Stelmack G. L., Halayko A. J., Shen G. X. (2010). Effects of extensively oxidized low-density lipoprotein on mitochondrial function and reactive oxygen species in porcine aortic endothelial cells. Am. J. Physiol. Endocrinol. Metab. 298, E89–E98 [DOI] [PubMed] [Google Scholar]
- Scarpulla R. C. (1997). Nuclear control of respiratory chain expression in mammalian cells. J. Bioenerg. Biomembr. 29, 109–119 [DOI] [PubMed] [Google Scholar]
- Sowter H. M., Ratcliffe P. J., Watson P., Greenberg A. H., Harris A. L. (2001). HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 61, 6669–6673 [PubMed] [Google Scholar]
- Sutliff R. L., Dikalov S., Weiss D., Parker J., Raidel S., Racine A. K., Russ R., Haase C. P., Taylor W. R., Lewis W. (2002). Nucleoside reverse transcriptase inhibitors impair endothelium-dependent relaxation by increasing superoxide. Am. J. Physiol. Heart Circ. Physiol. 283, H2363–H2370 [DOI] [PubMed] [Google Scholar]
- Tanida I. (2011). Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 14, 2201–2214 [DOI] [PubMed] [Google Scholar]
- Venkatesh P. K., Pattillo C. B., Branch B., Hood J., Thoma S., Illum S., Pardue S., Teng X., Patel R. P., Kevil C. G. (2010). Dipyridamole enhances ischaemia-induced arteriogenesis through an endocrine nitrite/nitric oxide-dependent pathway. Cardiovasc. Res. 85, 661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor V. M., Rocha M., Solá E., Bañuls C., Garcia-Malpartida K., Hernández-Mijares A. (2009). Oxidative stress, endothelial dysfunction and atherosclerosis. Curr. Pharm. Des. 15, 2988–3002 [DOI] [PubMed] [Google Scholar]
- Wu Z., Puigserver P., Andersson U., Zhang C., Adelmant G., Mootha V., Troy A., Cinti S., Lowell B., Scarpulla R. C., et al. (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 98, 115–124 [DOI] [PubMed] [Google Scholar]
- Yuhki K. I., Miyauchi T., Kakinuma Y., Murakoshi N., Maeda S., Goto K., Yamaguchi I., Suzuki T. (2001). Endothelin-1 production is enhanced by rotenone, a mitochondrial complex I inhibitor, in cultured rat cardiomyocytes. J. Cardiovasc. Pharmacol. 38, 850–858 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.